
Stanovení metoprololu a jeho metabolitu α-hydroxymetoprololu v séru metodou HPLC s fluorescenční detekcí
Determination of metoprolol and its metabolite α-hydroxymetoprolol in serum by HPLC method with fluorescence detection
High-performance liquid chromatography method has been developed with a view of its future use in the phenotyping of CYP2D6 enzyme by metoprolol as a probe drug. Metoprolol, α‑hydroxymetoprolol and the internal standard nadolol were extracted from serum with dichloromethane alkalinized with 1 mol/l NaOH. Chromatographic separations were performed on the reversed-phase column SupercosilTM LC-18 (15 cm × 3 mm, 5 μm) with the mobile phase containing acetonitril : methanol : water : triethylamine (15 : 5 : 80 : 0.1, pH 3.0) at a flow rate of 0.7 ml/min. Fluorescence detection (FL) was made at 230 nm (excitation) and 300 nm (emission). The total analysis time was 12 min. The retention time for α-hydroxymetoprolol, nadolol and metoprolol were 2.04, 3.02 and 9.04 min, respectively. The intra-assay and inter-assay precisions (coefficients of variation) were less than 7.2 %, and recovery values were found to be within 98.2–103.0 %. The calibration curve of the method was linear over a concentration range of 25–500 ng/ml (r = 0.999) for both compounds. The limit of detection was 5 ng/ml and the limit of quantification was 25 ng/ml for both metoprolol and α-hydroxymetoprolol. The reported method could be suitable for measurements of metoprolol and α-hydroxymetoprolol in serum from patients with hypertension, IHD and other illnesses.
Key words:
phenotyping CYP2D6 – metoprolol – α-hydroxymetoprolol – HPLC-FL
Autori:
I. Peřinová; J. Ďuricová; H. Brozmanová; I. Kacířová; M. Grundmann
Pôsobisko autorov:
Ostravská univerzita Ostrava, Ústav klinické farmakologie FN a Zdravotně-sociální fakulty
Vyšlo v časopise:
Čes. slov. Farm., 2008; 57, 254-259
Kategória:
Původní práce
Súhrn
V práci je popsána metoda vysokoúčinné kapalinové chromatografie, která byla zavedena pro účely fenotypizace enzymu CYP2D6 metoprololem jako substrátovou látkou. Metoprolol, α-hydroxmetoprolol a nadolol (vnitřní standard) byly extrahovány ze séra dichlormethanem s přídavkem 1 mol/l NaOH. Látky byly separovány na koloně s reverzní fází SupercosilTM LC-18 (15 cm × 3 mm, 5 μm) mobilní fází o složení acetonitril : methanol : voda : triethylamin (15 : 5 : 80 : 0,1; pH 3,0) při průtoku 0,7 ml/min. Fluorescenční detekce (FL) probíhala při vlnové délce 230 nm (excitační) a 300 nm (emisní). Celková doba analýzy byla 12 min. Retenční časy byly pro α-hydroxymetoprolol (h-MET) 2,04 minut, pro nadolol (IS) 3,02 minut a pro metoprolol (MET) 9,04 minut. Přesnosti v sérii a mezi sériemi vyjádřené jako relativní směrodatné odchylky byly nižší než 7,2 % a recovery se pohybovalo v rozmezí 98,2–103,0 %. Kalibrační křivka byla lineární v rozmezí 25–500 ng/ml (r = 0,999) pro obě látky. Detekční limit byl stanoven pro metoprolol a α-hydroxymetoprolol na 5 ng/ml a kvantifikační limit na 25 ng/ml. Prezentovaná metoda může být vhodná pro analýzu metoprololu a α-hydroxymetoprololu v séru u pacientů s hypertenzí, ICHS i jiných onemocnění.
Klíčová slova:
fenotypizace CYP2D6 – metoprolol – α-hydroxymetoprolol – HPLC-FL
Úvod
Metoprolol, (2RS)-3-(isopropylamino)-1-[4-(2-methoxyethyl)fenoxy]propan-2-ol (obr. 1), je lipofilní β1 – selektivní antagonista adrenergních receptorů používaný v léčbě kardiovaskulárních i nekardiovaskulárních onemocnění.
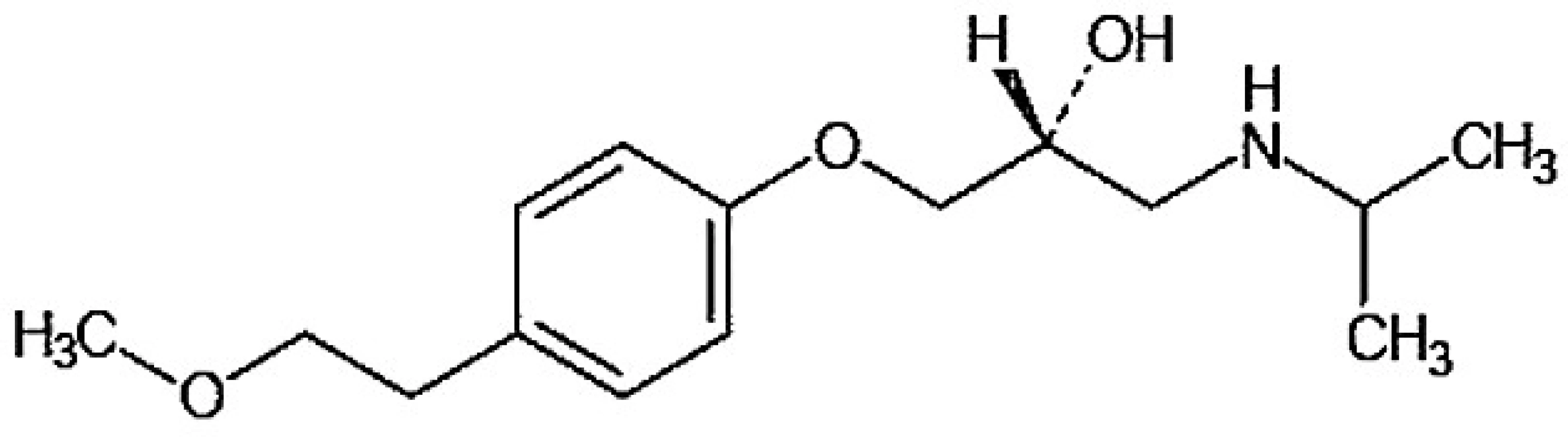
Je extenzivně metabolizován v játrech na několik metabolitů, které postrádají klinicky významný β-blokující účinek 1). Hlavními metabolickými cestami jsou O dealkylace (65 %), deaminace (10 %) a alifatická hydroxylace (10 %) 2) (obr. 2).
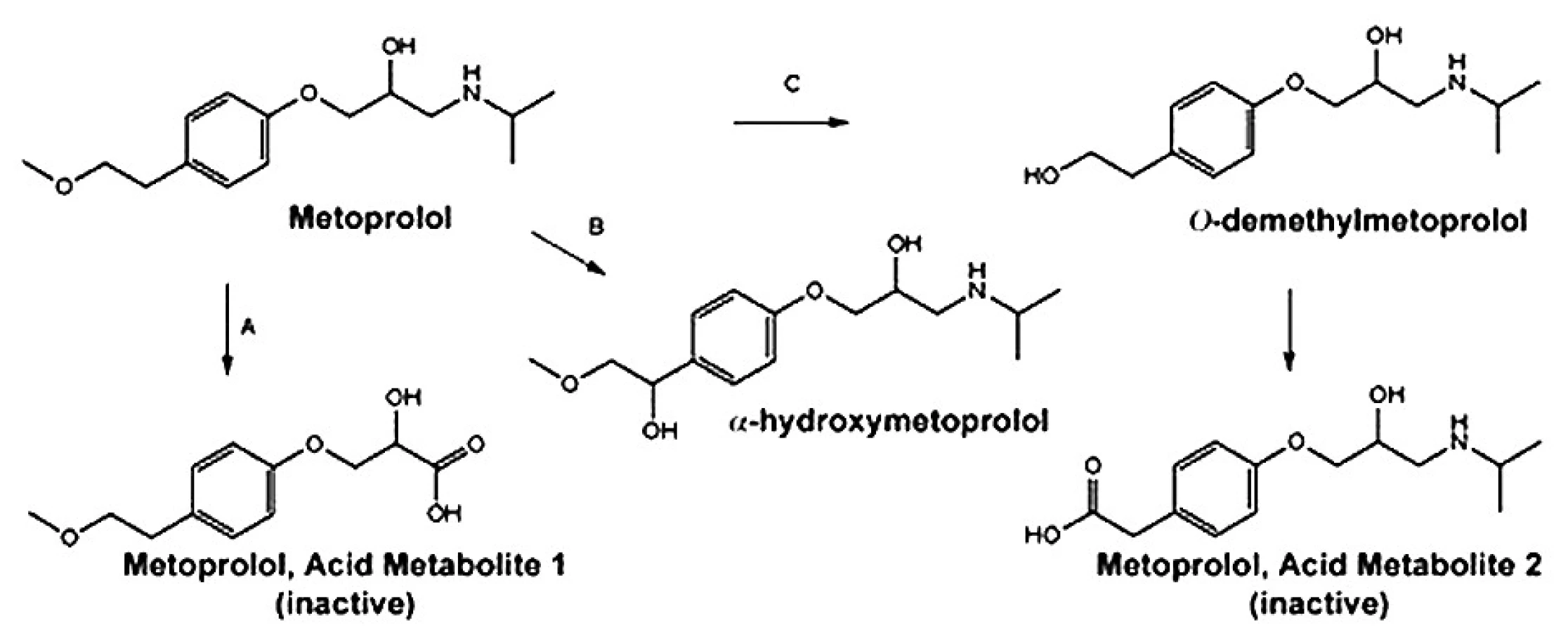
Tvorba metabolitu α-hydroxymetoprololu je exkluzivně zprostředkována cytochromem P450 2D6 (CYP2D6). CYP2D6, metabolizující přibližně 25 % všech užívaných léčiv, je známý vysokou interindividuální variabilitou. Tato variabilita je dána zejména faktory genetickými, přidruženými nemocemi a v menší míře i faktory zevního prostředí. Metoprolol, spolu s dalšími látkami, se využívá jako specifický substrát pro stanovení aktuální aktivity CYP2D6 enzymu (fenotypizace). Metabolický poměr α-hydroxymetoprolol/metoprolol určí příslušný fenotyp jedince 3).
Bylo popsáno několik metod na stanovení metoprololu společně s jeho metabolitem α-hydroxymetoprololem. Jedná se především o metody kapalinové chromatografie s UV detekcí 5, 6) nebo FL detekcí 7–17) při různých vlnových délkách. Látky byly extrahovány z biologického materiálu (plazmy, séra nebo moči) pomocí extrakce na pevné fázi 7–9) nebo extrakce kapalinou (diethyletherem, dichlormethanem, chloroformem, ethylethanolátem nebo kombinací) při alkalickém pH 5 ,10–14). Jako vnitřní standard byl použit pindolol 10, 15), phenacetin 5), alprenolol 8, 13), guanoxan 7) nebo dextromethorfan 9, 14). Mobilní fázi tvořila směs voda : acetonitril 5, 8, 10, 11, 13, 16, 17), voda : acetonitril:methanol 7) nebo voda : acetonitril : tetrahydrofuran 9, 14). Analýza probíhala na kolonách s reverzní fází 5–7, 10, 11, 13, 16, 17), na kyano-koloně 8) nebo na phenyl-koloně 12, 14, 15). Doba analýzy se pohybovala ve většině prací od 20 do 35 minut, méně pak do 15 min 9, 13, 14). Jen výjimečně se metoprolol a α-hydroxymetoprolol stanovuje pomocí kapalinové chromatografie s hmotnostní detekcí 18) nebo plynové chromatografie s hmotnostní detekcí 19, 20).
Cílem práce bylo zavést, optimalizovat a validovat HPLC metodu na stanovení metoprololu a α-hydroxymetoprololu pro účely fenotypizace enzymu CYP2D6 metoprololem jako substrátovou látkou. Požadavkem byla rychlá a selektivní metoda s jednokrokovou extrakcí malého množství séra, vhodná pro rutinní analýzu pacientských vzorků.
Pokusná část
Chemikálie a roztoky
Substance metoprololu (metoprolol tartarát) a nadololu byly získány od firmy Sigma, α-hydroxymetoprolol byl získán od firmy Astra Zeneca. Dichlormethan (LiChrosolv®, pro kapalinovou chromatografii), acetonitril a methanol (LiChrosolv®, gradient grade for liquid chromatography) byly zakoupeny od firmy Merck, voda (Chromasolv® Plus HPLC) a triethylamin (TEA – triethylamine, 99%) od firmy Sigma-Aldrich. 1 mol/l NaOH (Sodium hydroxide Solution for HPCE) byl získán od firmy Fluka.
Přístrojové vybavení a chromatografické podmínky
HPLC zařízení firmy Spectra Physics Analytical se skládalo z čerpadla (Spectra SYSTEM P1500), smyčky pro manuální nástřik a fluorescenčního detektoru (Spectra SYSTEM FL 2000). Byla použita kolona firmy Supelco, SupercosilTM LC-18 (15 cm × 3 mm, 5 μm). Mobilní fázi tvořila směs acetonitril : methanol : voda : TEA (15 : 5 : 80 : 0,1; pH 3,0), průtok mobilní fáze byl 0,7 ml/min. Fluorescenční detektor byl nastaven na 230 nm (excitační) a 300 nm (emisní) vlnové délky. Celková délka analýzy byla 12 min.
Příprava roztoků pro kalibraci a validaci
Zásobní roztoky metoprololu (MET), α-hydroxymetoprololu (hMET) a nadololu (vnitřní standard – IS) byly připraveny v koncentracích 100 μg/l. Ředěním zásobních roztoků methanolem byly připraveny pracovní roztoky všech tří látek o koncentraci 1000 ng/ml. Standardní a kontrolní roztoky metoprololu a α-hydroxymetoprololu byly připraveny ředěním jednotlivých pracovních roztoků na koncentrace 25, 50, 100, 250, 500 ng/ml a 40, 200, 400 ng/ml, resp. Roztok nadololu byl použit o koncentraci 1000 ng/ml. Všechny roztoky byly uchovávány při -20 °C v mrazničce.
Příprava vzorků pro analýzu
Vzorky pro kalibraci a validaci
Standardní a kontrolní vorky se připravily odpařením 100 μl jednotlivých standardních, resp. kontrolních roztoků a 50 μl vnitřního standardu. Extrakce probíhala stejně jako u neznámých vzorků.
Neznámé vzorky
K 50 μl odpařeného vnitřního standardu se přidalo 200 μl neznámého séra (nulového séra v případě standardních a kontrolních vzorků), 50 μl 1 mol/l NaOH a 1,5 ml dichlormethanu. Vzorky se 1 min extrahovaly. Po 10 min centrifugaci při 3800 ot./min se odsála horní vrstva a spodní dichlormethanová vrstva se přenesla do čisté zkumavky a nechala se odpařit pod dusíkem. Odparek se rozpustil ve 20 μl methanolu a 50 μl HPLC vody, 20 μl se injikovalo na kolonu.
Validace metody
Přesnost a správnost
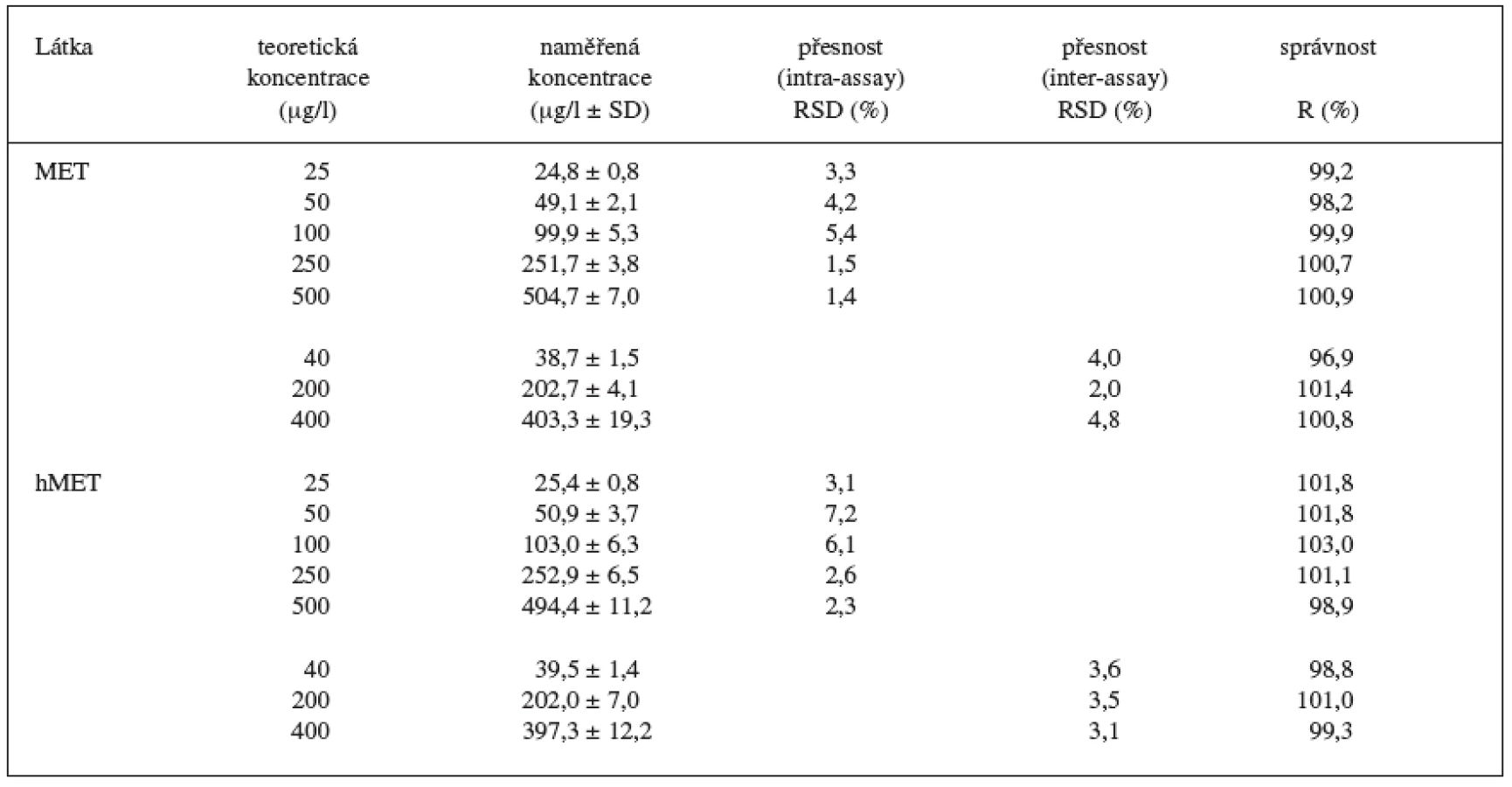
Během 3 dnů byly proměřeny standardní vzorky v 5 koncentračních hladinách (25, 50, 100, 250, 500 ng/ml, n = 6 pro každou koncentrační hladinu) a kontrolní vzorky ve 3 koncentračních hladinách (40, 200, 400 ng/ml, n = 6 pro každou koncentrační hladinu). Vzorky byly analyzovány kompletním postupem a odečteny z kalibrační křivky připravené v den analýzy. Přesnost byla vyjádřena jako relativní směrodatná odchylka (RSD) pro analýzu v sérii (intra-assay) a analýzu mezi sériemi (inter-assay). Správnost byla vyjádřena jako recovery (R).
Linearita
Proměřením náhodně vybraných vzorků (vzorky před podáním, 1, 2, 3 a 4 hodiny po podání) pacientů, kterým byl podáván metoprolol v různých dávkách v rámci antihypertenzní terapie, se získala představa o koncentračním rozmezí metoprololu a α-hydroxymetoprololu. Ze základního standardního roztoku blízkého horní hranici pracovního rozsahu se naředěním připravilo 5 standardních roztoků s klesající koncentrací analytu tak, aby došlo k pokrytí celého pracovního rozsahu měření – 25, 50, 100, 250 a 500 ng/ml pro metoprolol i α-hydroxymetoprolol. Kalibrační křivka byla zkonstruována na základě analýzy nejmenších čtverců jako poměr plochy píků jednotlivých látek k ploše píku vnitřního standardu v závislosti na koncentraci.
Detekční limit (LOD) a kvantifikační limit (LOQ)
LOD (nejnižší detekovatelná koncentrace látky) a LOQ (nejnižší koncentrace látky ve vzorku, stanovitelná s přijatelnou přesností a správností za uvedených podmínek metody) byly stanoveny na základě poměru signálu k šumu (S/N). Pro LOD byl poměr S/N stanoven na 3 : 1 a pro LOQ na 10 : 1.
Selektivita
Byla testována schopnost měřit správně a specificky stanovovanou látku v přítomnosti jiných látek, které mohou být očekávány ve vzorku. Analyzoval se vzorek séra o tzv. nulové koncentraci (krev neléčeného dárce testovaná na přítomnost HIV a HBsAg) a během 2 měsíců bylo stanoveno 98 vzorků séra pacientů léčených metoprololem při antihypertenzní terapii.
Robustnost
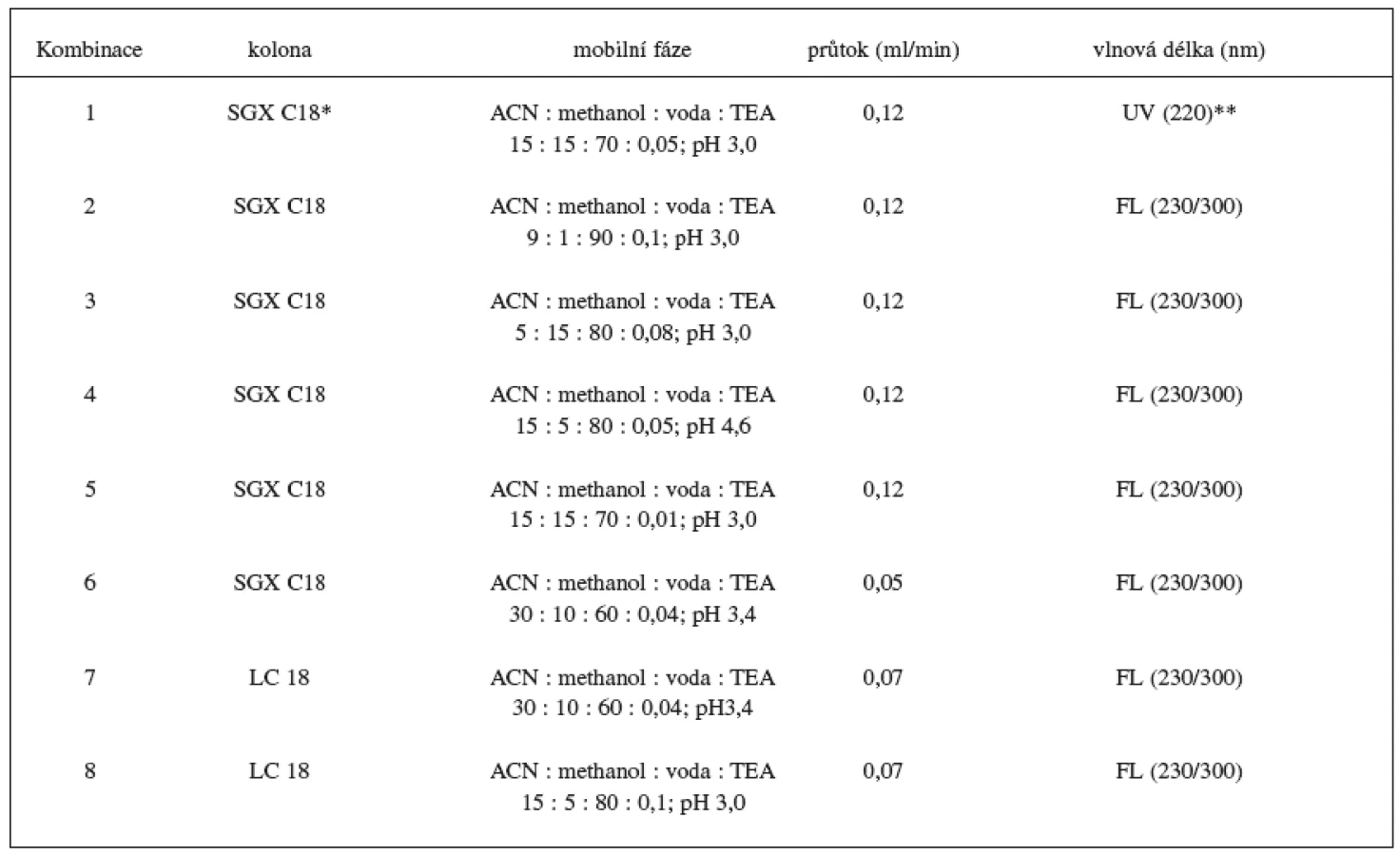
Sledovalo se jak výběr stacionární fáze a změna ve složení mobilní fáze ovlivní analýzu metoprololu a α hydroxymetoprololu. Vybrané podmínky jsou shrnuty v tabulce 1.
Výsledky
Byla zavedena, optimalizována a validována, HPLC metoda na stanovení metoprololu a α-hydroxymetoprololu v séru. Na základě předběžných studií uvedených v tabulce 1 byl vybrán jako nejvhodnější postup č. 8, HPLC metoda s fluorescenční detekcí (230 nm excitační a 300 nm emisní vlnové délky), s reverzní stacionární fází LC 18 (15 cm × 3 mm, 5 μm) a ternární mobilní fází acetonitril : methanol : voda : TEA (15 : 5 : 80 : 0,1; pH 3,0). Průtok mobilní fáze byl 0,7 ml/min. Použitím mobilní fáze s větším obsahem vodné složky (vyžadovalo větší přídavek TEA) se ve srovnání s organičtější fází mírně zhoršila citlivost, ale výrazně se zvýšila selektivita metody. Jako vnitřní standard byl nejdříve testován pindolol. Vzhledem k jeho nízké detekovatelnosti při použité vlnové délce byl pro konečnou analýzu vybrán nadolol. Optimalizován byl také extrakční postup pro přečištění vzorků séra. Pro danou metodu byla vybrána jako nejvhodnější extrakce dichlormethanem (1,5 ml) s přídavkem 50 μl 1 mol/l NaOH. Při analýze se vycházelo z malého množství séra (200 μl), což je vhodné pro rutinní měření pacientských vzorků.
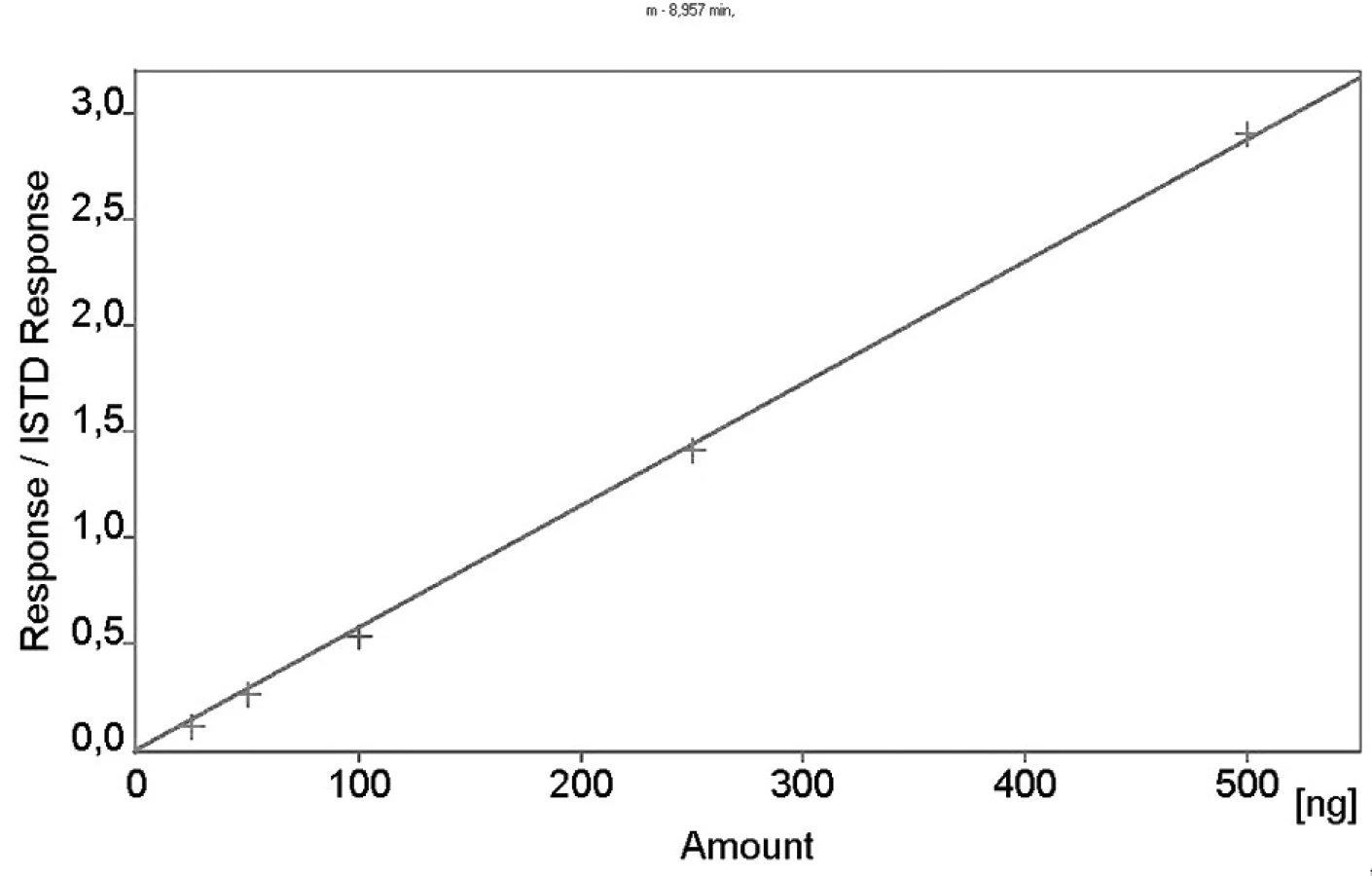
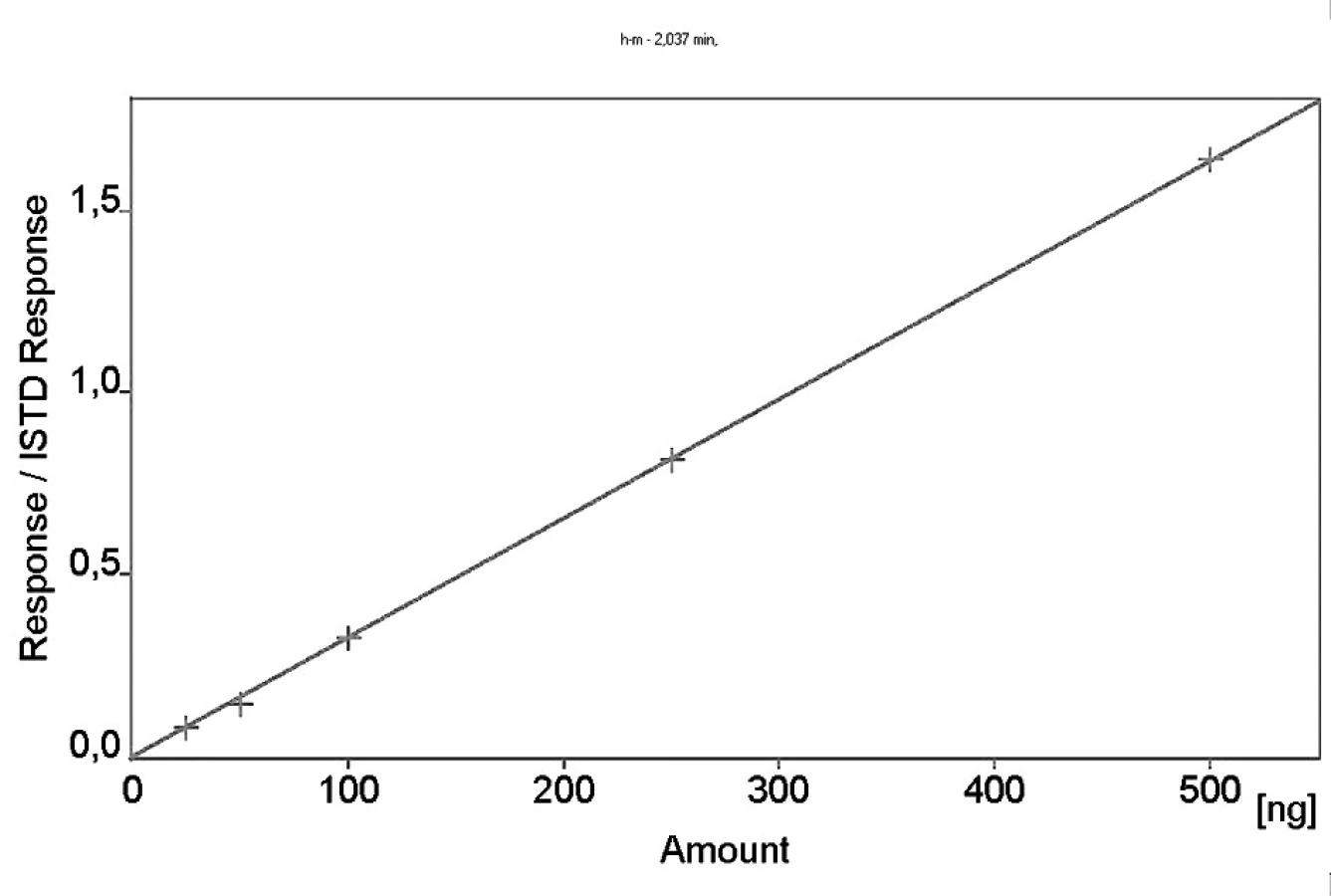
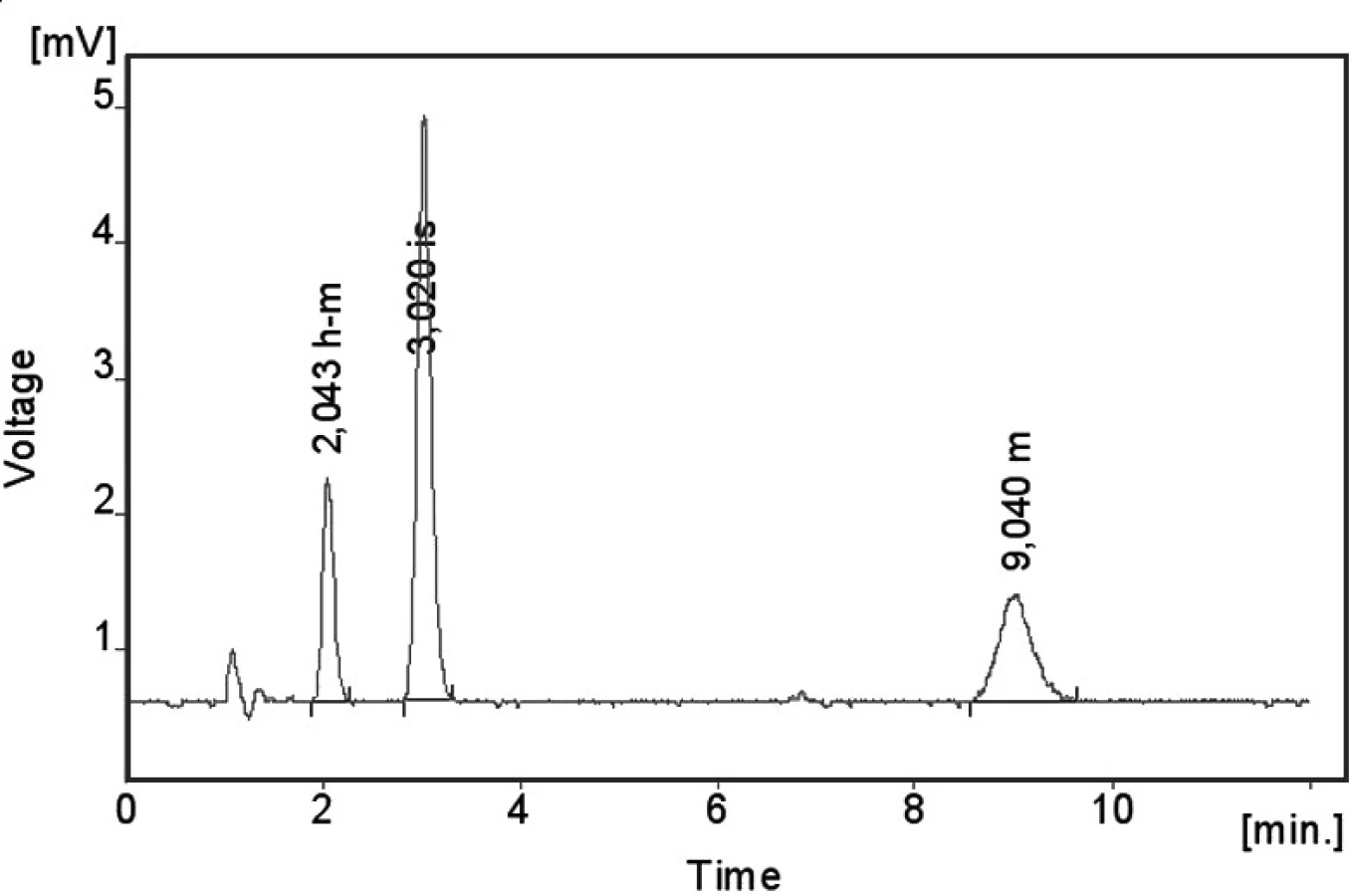
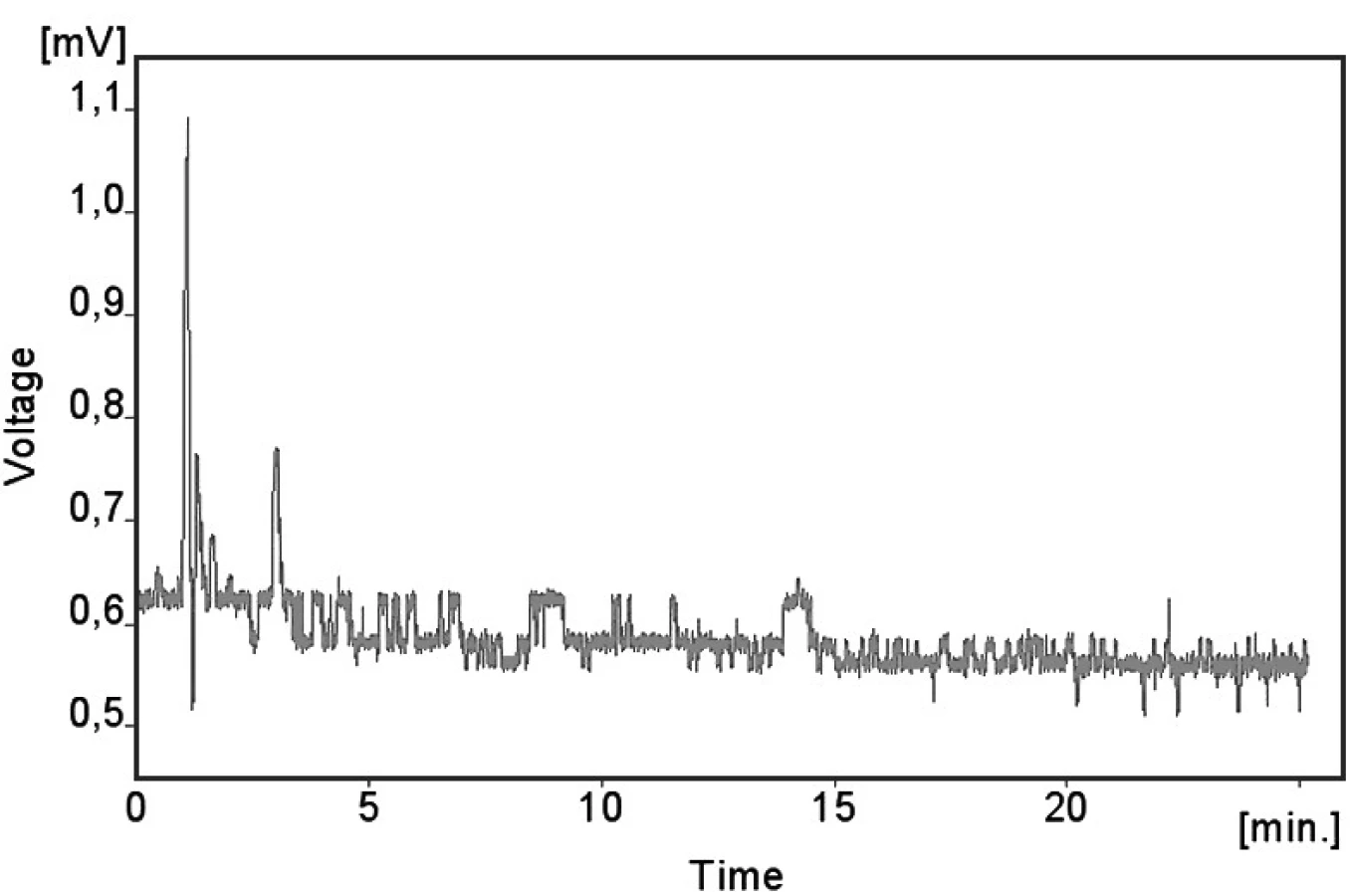
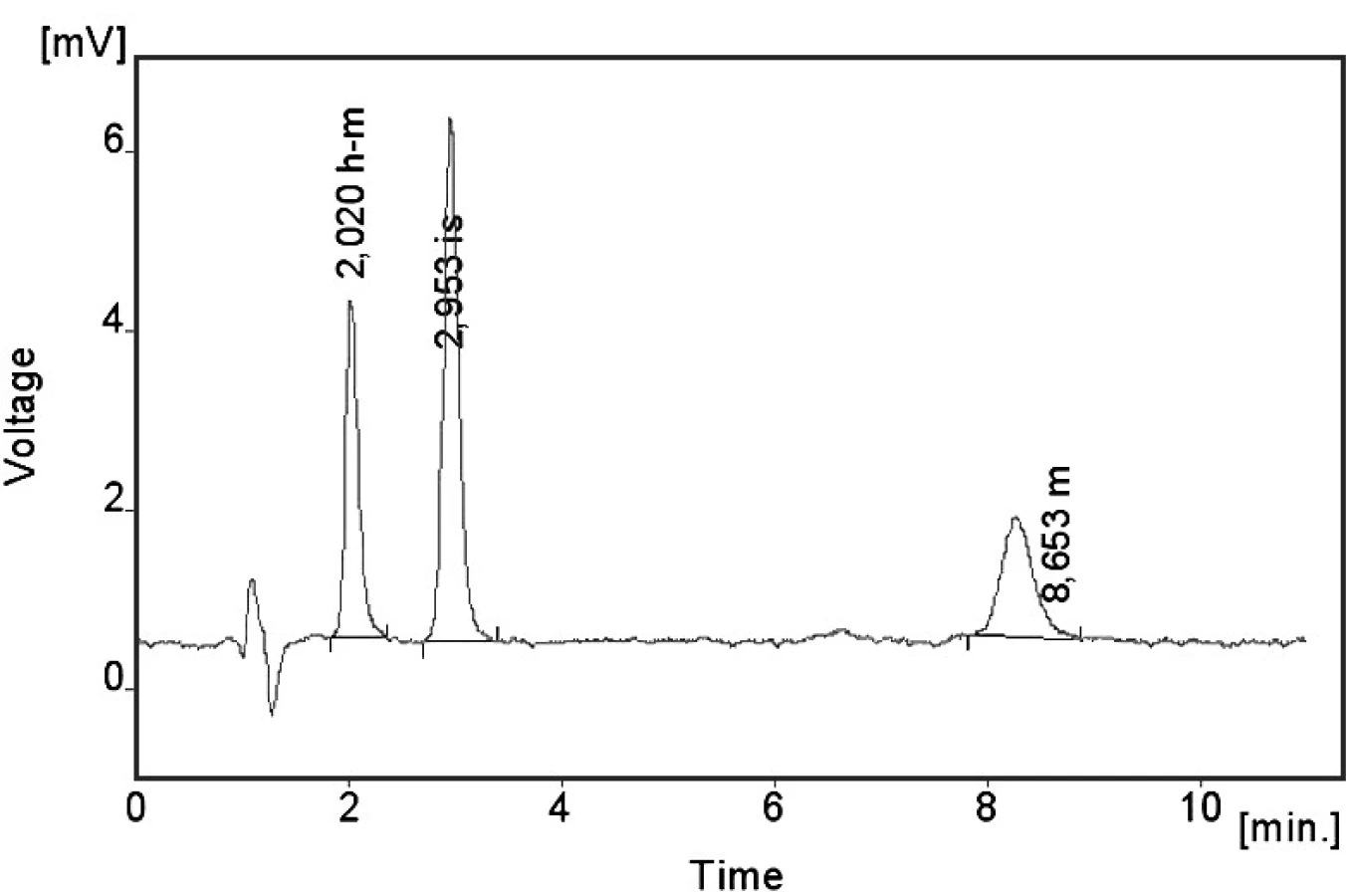
Kalibrační křivka pro metoprolol a jeho metabolit α hydroxymetoprolol byla lineární v rozmezí 25–500 ng/ml s korelačním koeficientem 0,999 pro obě látky (obr. 3 a 4). Na obrázku 5 je ukázán chromatografický záznam extrahovaného standardního vzorku metoprololu a α-hydroxymetoprololu o koncentraci 100 ng/ml. Retenční časy byly pro α-hydroxymetoprolol (h-MET) 2,04 minut, pro nadolol (IS) 3,02 minut a pro metoprolol (MET) 9,04 minut. Detekční limit (LOD – S/N = 3 : 1) byl stanoven pro metoprolol i α-hydroxymetoprolol 5 ng/ml a kvantifikační limit (LOQ – S/N = 10 : 1) 25 ng/ml. LOQ byl potvrzen analýzou 6 vzorků této koncentrace s dostatečnou přesností a správností – RSD pro MET byla 3,3 % a pro h-MET 3,1 %, R pro MET bylo 99,2 % a pro h-MET 101,8 %. Přesnost a správnost metody je shrnuta v tabulce 2. Relativní směrodatné odchylky metoprololu a α-hydroxymetoprololu pro analýzu v sérii byly ≤ 5,4 % (MET) a 7,2 % (h-MET) a pro analýzu mezi sériemi ≤ 4,8 % (MET) a 3,6 % (h MET). Recovery metoprololu bylo v rozmezí 98,2 až 100,9 % a α hydroxymetoprololu v rozmezí 98,8 až 103,0 %. Tyto hodnoty jsou přijatelné podle platných kritérií pro validační parametry chromatografických metod 21–26). Při analýze vzorku séra o nulové koncentraci nebyly nalezeny žádné interferující látky (obr. 6). Interference nenastává ani s léky, které jsou běžně podávány pacientům s hypertenzí (obr. 7).
Diskuze
Uvedená HPLC metoda s fluorescenční detekcí je přesná, správná a selektivní metoda, vhodná pro stanovení metoprololu a α-hydroxymetoprololu v séru. Metoda vyžaduje minimální množství séra a jednoduchou úpravu vzorků. Celková doba analýzy je pouze 12 minut, což je vhodné pro rutinní používání. Na rozdíl od většiny citovaných prací, které používají dvousložkovou mobilní fázi acetonitril-voda, byla v práci použita ternární mobilní fáze. Přídavek methanolu do mobilní fáze zlepšil symetrii píků a prodloužil životnost kolony. Jako vnitřní standard byl nově použit ß blokátor nadolol, což je látka podobné chemické struktury jako metoprolol a blízké polarity jako α-hydroxymetoprolol. V České republice není nadolol k farmakoterapii používán. Nadolol byl dokonale oddělen od obou složek, vykazoval dobrou fluorescenční intenzitu při použité vlnové délce a extrahovatelnost při zvoleném postupu.
Metoda byla vyvinuta pro měření jak vzorků s nižšími koncentracemi (před podáním léku) tak vzorků s vyššími koncentracemi (po podání léku), takže by mohla být vhodná pro terapeutické monitorování metoprololu (TDM) a může být použita pro testování fenotypu enzymu CYP2D6 v séru pacientů, kteří metoprolol užívají.
Předběžně se zavedená metoda testovala také pro stanovení metoprololu a α-hydroxymetoprololu v moči. Jednalo se o vzorky ranní moče pacientů s hypertenzí. První výsledky ukazují, že po naředění vzorků moče a přečištění uvedenou extrakční procedurou bude metoda využitelná i pro stanovení fenotypu CYP2D6 v moči.
Došlo 25. srpna 2008
Přijato 10. října 2008
Adresa pro korespondenci:
Mgr. Ilona Peřinová, Ph.D.
Ústav klinické farmakologie FN a Zdravotně sociální fakulta OU
17. listopadu 1790, 708 52 Ostrava – Poruba
e-mail: ilona.perinova@fno.cz
Zdroje
1. Fux, R., Mörike, K., Pröhmer, A. M. et al.: Clin. Pharmacol. Ther., 2005; 78, 378–387.
2. Silas, J. H., Lennard, M. S., Tucker, G. T. et al.: Br. J. Clin. Pharmacol., 1984; 17,11–19.
3. Duricová, J., Grundmann, M.: Ces. slov. Farm., 2007; 56, 220–224.
4. Bodor, N., Buchwald, P.: AAPS J., 2005; 7, 820–833.
5. Ismail, R., The, L. K.: J. Clin. Pharm. Ther., 2006; 31, 99–109.
6. Godbillon, J., Duval, M.: J. Chromatogr., 1984; 309, 198–202.
7. Chiu, F. C., Damani, L. A., Li, R. C., Tomlinson, B.: J. Chromatogr. B. Biomed. Sci. Appl., 1997; 696, 69–74.
8. Fang, J., Semple, H. A., Song, J.: J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci., 2004; 809, 9–14.
9. Mistry, B., Leslie, J., Eddington, N. E.: J. Pharm. Biomed. Anal., 1998; 16, 1041–1049.
10. Sohn, D. R., Kusaka, M., Shin, S. G. et al.: Ther. Drug. Monit., 1992; 14, 184–189.
11. Li, G., Wang, R.: Chin. Med. J., 2006; 119, 2013–2017.
12. Ramenskaya, G. V., Savchenko, A. Y., Agafonov, A. A. et al.: Bull. Exp. Biol. Med., 2002; 134, 159–160.
13. Lecaillon, J. B., Godbillon, J., Abadie, F., Gosset, G.: J. Chromatogr., 1984; 305, 411–417.
14. Goryachkina, K., Burbello, A., Boldueva, S. et al.: Eur. J. Clin. Pharmacol., 2008; 64, 275–282.
15. Bramer, SL., Suri, A.: Clin. Pharmacokinet., 1999; 37, 41–51.
16. Kirchheiner, J., Heesch, C., Bauer, S. et al.: Clin. Pharmacol. Ther., 2004; 76, 302–312.
17. Rutledge, D. R., Garrick, C.: J. Chromatogr. Sci., 1989; 27, 561–565.
18. Asimus, S., Elsherbiny, D., Hai, T. N. et al.: Fundam. Clin. Pharmacol., 2007; 21, 307–316.
19. Mautz, D. S., Shen, D. D., Nelson, W. L.: Pharm. Res., 1995; 12, 2053–2056.
20. Hoffmann, K. J., Gyllenhaal, O., Vessman, J.: Biomed. Environ. Mass. Spectrom., 1987; 14, 543–548.
21. Rewiewer of Guidance. Validation of Chromatographic Methods. Center of Drug Evaluation and Research (CDER), CMC 3, 1994.
22. Guidance for Industry. Q2B Validation of Analytical Procedures: Methodology. The International Conference on Harmonisation of Thechnical Requirements for Registration of Pharmaceuticals for Human Use (ICH), 1996.
23. Thompson, M., Ellison, S. L. R., Wood, R.: Pure Appl. Chem., 2002; 74, 835–855.
24. Bievre, B. et al.: The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics, EURACHEM Guide. 1998, s. 14.
25. Brown, R., Caphart, M., Faustino, P. et al.: LCGC, 2001; 19, 74–79.
26. Causon, R.: J. Chromatogr. B. Biomed. Sci. Appl., 1997; 689, 175–180.
Štítky
Farmácia FarmakológiaČlánok vyšiel v časopise
Česká a slovenská farmacie

2008 Číslo 6
Najčítanejšie v tomto čísle
- Možnosti ovplyvnenia liberácie alaptidu z dermálnych polotuhých liekov
- Štúdium lokálnych anestetík Časť 185: Termodynamické parametre heptakaíniumchloridu v prostredí roztoku NaBr*
- Optimalizácia extrakčnej metódy na stanovenie metadónu a jeho metabolitu EDDP v moči plynovou chromatografiou
- Vliv koncentrace lipofilního nosiče na vlastnosti hydrofilně-lipofilních matricových systémů