
Prehľad inhibítorov MMP a TACE hydroxamátového typu a ich terapeutický potenciál
Review of hydroxamic matrix metalloproteinase inhibitors and their therapeutic potential
The hydroxamic acid moiety is the key structural element for the potency of the inhibitors against metalloprotease enzymes. However, the selectivity of the inhibitors towards various metalloproteases depends on the structures of the templates or scaffolds as well as the variations of the substituents. A combination of the traditional substrate- and mechanism-based approaches and the new technologies, such as the structural-based drug design as well as combinatorial chemistry, will be essential for the future development of specific protease inhibitors. Success in a clinical trial of new hydroxamic acid-based inhibitors is expected soon.
Key words:
hydroxamates – MMP – TACE – metalloenzymes – metalloproteinases
Autori:
J. Šille; V. Garaj; M. Šramko; M. Remko
Pôsobisko autorov:
Univerzita Komenského Bratislava, Farmaceutická fakulta, Katedra farmaceutickej chémie, SR
Vyšlo v časopise:
Čes. slov. Farm., 2009; 58, 95-102
Kategória:
Přehledy a odborná sdělení
Súhrn
Hydroxámová funkčná skupina je významným štruktúrnym prvkom pre účinok inhibítorov na metaloproteinázy. Selektivita inhibítorov k jednotlivým metaloproteinázam závisí nielen od štruktúry nosnej časti molekuly, ale aj od rôznych substituentov. Kombinácia tradičných prístupov založených na štruktúre a mechanizmoch účinku s novými metódami, akými sú racionálne projektovanie liečiv a kombinatorická chémia, bude nevyhnutná pre ďalší rozvoj špecifických inhibítorov proteáz. V blízkej budúcnosti môžeme očakávať úspech pri klinickom skúšaní nových inhibítorov hydroxámového typu.
Kľúčové slová:
hydroxamáty – MMP – TACE – metaloenzýmy – metaloproteinázy
Úvod
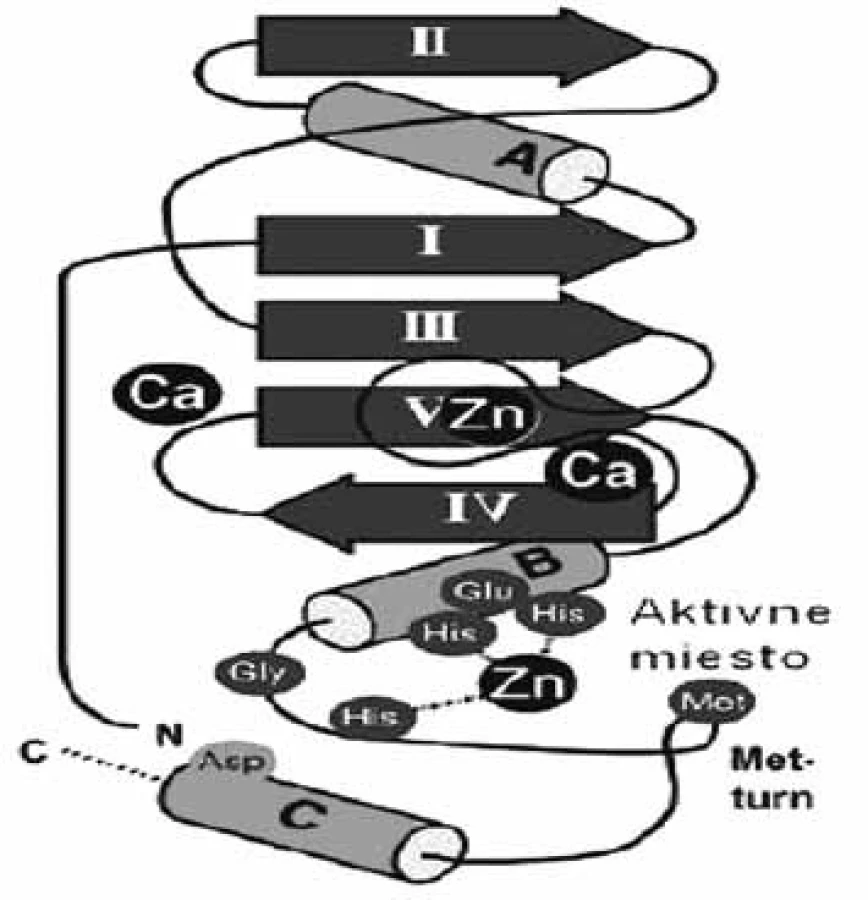
Súbor enzýmov „metzincínu“ sa skladá z veľkého počtu zinočnatých metaloproteáz, ktoré majú značne podobné sekvencie. Patria sem štyri rozdielne skupiny metaloproteáz (astacíny, adamalyzíny, matrixíny a bakteriálne serralyzíny), ktoré spája: 1. spoločný segment sekvencie HEXXHXXGXXH/D (obr. 1), ktorá obsahuje v aktívnom mieste tri ligandy zinočnatého katiónu a 2. zachovaná metionínová funkčná skupina za tretím zinočnatým ligandom, ktorá je zodpovedná za podobnú konformáciu ß slučky u metzincínov (Met-turn) 1). Enzýmy týchto štyroch rodín sa rozlišujú podľa skupín, ktoré nasledujú po treťom ligande, histidíne 2). Na základe porovnávania 3D štruktúr jednotlivých enzýmov z rôznych rodín sa zistilo, že aj napriek malej podobnosti sekvencie majú podobnú topológiu. Podobnosť štruktúry je oveľa markantnejšia v okolí aktívneho miesta enzýmov, napriek tomu, že rozdiely v regiónoch, ktoré viažu substrát majú špeciálnu úlohu pri modelovaní selektívnych inhibítorov. Najvýznamnejšie rozdiely sú v primárnej časti aktívneho miesta, v mieste, ktoré spája ß-slučku s α-závitnicovou časťou katalytickej domény, nazvanej ako S1’ slučka 3). Dĺžka a zloženie aminokyselín slučky sa výrazne líši u rôznych rodín metzincínov, ale len nepatrne medzi členmi rovnakej rodiny. Celková variabilita S1’ domény je základným kľúčom vývoja selektívnych inhibítorov týchto enzýmov 4, 5).

Metzincínové proteázy sú schopné štiepiť veľké množstvo bunkových a mimobunkových substrátov, ako aj mimobunkových matrixových substrátov, kolagény, prokolagény, proteoglykány, cytokíny a ich ligandy, chemokíny a elastíny. Takýmto spôsobom obmieňajú štruktúru a funkciu tkanív vo fyziologických a patologických stavoch 6).
Matrixová metaloproteináza
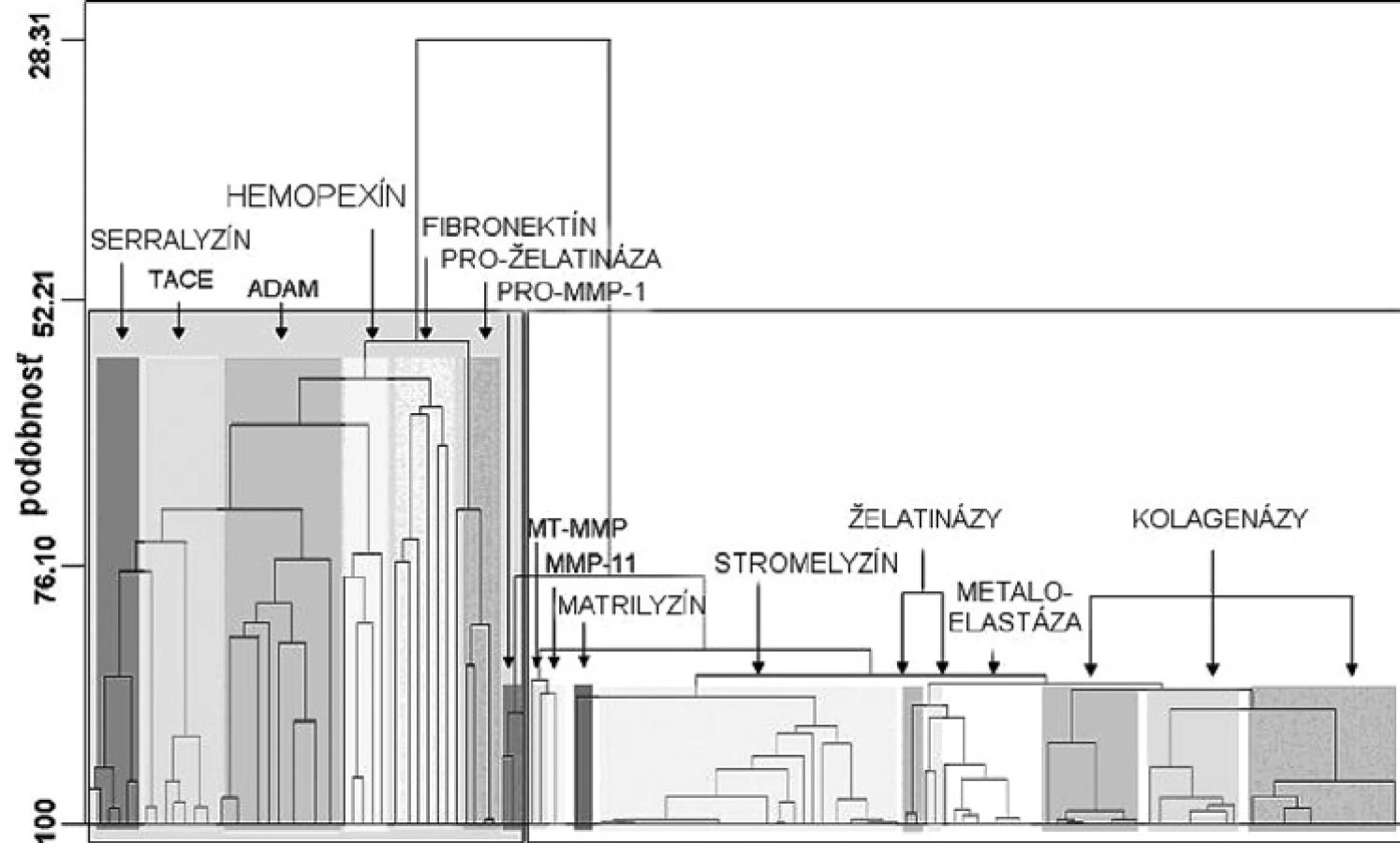
Matrixové endopeptidázy (MMP) tvoria veľkú skupinu neutrálnych endopeptidáz, ktoré degradujú prvky mimobunkového matrixu a podporujú premodelovanie tkanív 7). Všetky aktívne miesta enzýmov MMP obsahujú spoločný segment sekvencie aminokyselín HexGHxxGxxH, ktorý sa skladá z troch histidínov koordinujúcich katalytický zinočnatý katión, päťkrát stočený ß-skladaný list (jeden paralelný a štyri antiparalelné) a tri α-závitnice v endopeptidázovom vačku (obr. 2) 8). Enzýmy MMP sa skladajú zo štyroch odlišných domén: N-terminálna pro-doména, ktorá je rozštiepená počas aktivácie; katalytická doména, ramenový región; C-terminálna hemopexínová doména 9). Katalytická doména je stočená do sférickej jednotky s približným priemerom 35 Å a obsahuje dva tetraedrálne koordinované zinočnaté katióny: „štruktúrny“ a katalytický ión. Katalytická doména okrem zinočnatých iónov obsahuje aj 1–3 vápenaté ióny 10). Enzýmy MMP patria do rodiny matrixínov a na základe katalytickej domény a ďalších štruktúrnych/funkčných motívov (signálny peptid, propeptid, štiepne miesto konvertáz, fibronektínový modul, C-koniec atď.) sa rozdeľujú do štyroch hlavných skupín: 1. archetypické, 2. matrilyzíny, 3. želatinázy a 4. enzýmy aktivované konvertázou (obr. 3). Archetypické enzýmy MMP majú organizovanú katalytickú doménu, ktorá sa skladá zo signálneho peptidu, z N-koncového pro-peptidu a z hemopexínovej C-koncovej domény, ktorá je dôležitá pre substrátovú špecifickosť a interakciu s tkanivovým MMP inhibítorom. Do tejto skupiny patrí deväť členov MMP (kolagenázy: MMP-1, MMP-8, MMP-13; stromelyzíny: MMP-3, MMP-10; metaloelastázy: MMP-12; enamelyzíny: MMP-20, MMP-19, MMP 27). Druhá skupina je reprezentovaná matrilyzínmi (MMP-7, MMP-26), v ktorých chýba C-terminálny hemopexínový koniec a katalytická doména, je veľmi malá. Tretiu skupinu tvoria želatinázy s troma modulmi fibronektínového typu II, ktoré sú poskladané do domén na väzbu kolagénu. Tieto domény urýchľujú degradáciu kolagénu a želatíny. Štvrtú skupinu (konvertázou aktivované MMP) charakterizuje bázická zložka v propeptidovej časti, ktorú štiepi furínová proteáza. Medzi proteíny štvrtej skupiny patria tri vylučované enzýmy MMP, šesť enzýmov MMP membránového typu (MT) a dva atypické transmembránové MMP enzýmy typu II (MMP-23A, MMP-23B) 11).
Enzýmy MMP sa podieľajú na mnohých patologických ako aj fyziologických procesoch. Strata riadenia aktivity a expresie MMP je spojená s množstvom chorobných procesov, ako sú nádorové metastázy, reumatoidná artritída (RA), skleróza multiplex, ochorenie fixačného aparátu zubov a ischemická choroba srdca. Napriek enormnej snahe vyvinúť efektívny inhibítor MMP (MMPI) je v súčasnosti na trhu len jediné liečivo (Periostat) na liečbu choroby fixačného aparátu zubov 12). Nanešťastie väčšina sľubných počiatočných klinických štúdií rôznych typov MMPI zlyhala v pokročilých fázach klinického skúšania hlavne kvôli nežiaducim muskuloskeletárnym účinkom. To, že tieto inhibítory neprešli klinickými skúšaniami môžeme pripísať extrémne komplexnej biológii MMP, ktorá doteraz nie je úplne objasnená. Napríklad niektoré MMP podporujú progresiu nádorov, kým iné izoformy majú obrannú úlohu pri angiogenéze a metastáze nádorov. Na základe fyziologických funkcií enzýmov môžeme rozdeliť MMP do troch kategórií: 1. cieľový enzým (target) – podporuje tvorbu nádorov (je nutná inhibícia enzýmu, kvôli farmakologickej spätnej regulácii); 2. anti-cieľový enzým (anti-target) – má esenciálnu úlohu v normálnych bunkách a ich inhibícia nie je žiadaná, nakoľko môže spôsobovať nežiaduce účinky a zrušenie výhod inhibície; 3. proti-cieľový (counter-target) – funkcia MMP nesúvisí s chorobnými procesmi, ale jeho ovplyvnenie môže zmeniť biologickú funkciu MMP a vyvolávať vedľajšie účinky 11).
Inhibítory MMP
Inhibítory MMP (MMPI) môžeme z biologického hľadiska rozdeliť na široko-spektrálne a selektívne inhibítory. Predpokladalo sa, že široko-spektrálne inhibítory by mohli byť výhodnou voľbou pri liečbe rakoviny a špecifické inhibítory kolagenáz (MMP-1, MMP-8, MMP-13) pri liečbe artritíd. Klinické štúdie širokospektrálnych inhibítorov ukázali, že inhibítory ako marimastat majú závažné muskuloskeletárne vedľajšie účinky. Na základe výsledkov vznikla hypotéza, podľa ktorej sa musí zachovať aktivita izoenzýmu MMP-1, aby sa zabránilo vedľajším účinkom 13).
Inhibítory MMP sa chemicky delia podľa mechanizmu inhbície na komplexotvorné, nekomplexotvorné a mechanické (inhibítor indukuje konformačnú zmenu mikroprostredia zinočnatého iónu). Komplexotvorné inhibítory môžeme na základe funkčnej skupiny, ktorá sa viaže na zinok ďalej rozdeliť do nasledujúcich skupín: A. sukcinyl hydroxamáty, B. nepeptidové sukcinyl hydroxamáty, C. deriváty sulfónamidových hydroxamátov, D. iné ako hydroxamátové inhibítory (napr. deriváty karboxylových kyselín, tiolov; zlúčeniny fosforu), E. rôzne prírodné produkty (tetracyklíny; katechín, pyknidón, deriváty futoenónu; skupina pseudopeptidov; nikotínamid, rifampicín) 4).
Keď ešte neexistovalo mnoho poznatkov o biologickej úlohe MMP v patofyziológii rakoviny a zápalu, chelatačné inhibítory hydroxamátového typu dominovali vo vývoji nových potenciálnych liečiv. Hydroxamáty vo forme aniónov vytvárajú komplexy so zinočnatým iónom aktívneho miesta, kde sa ako ligandy viažu prostredníctvom dvoch atómov kyslíka 14).Zlyhanie klinických skúšok betimastatu a marimastatu prinútilo vedeckú komunitu, aby prehodnotila situáciu vývoja ďalších inhibítorov hydroxamátového typu 15). Príčinou masívneho zlyhávania bol nedostatok spoľahlivých klinických skúšok a slabý farmakologický profil MMPI 16).
V dnešnej dobe je veľkým problémom index selektivity. Hlavným dôvodom neschopnosti prvotných inhibítorov hydroxamátového typu selektívne inhibovať enzýmy MMP bola príliš veľká kapacita funkčnej skupiny kyseliny hydroxámovej viazať zinočnatý ión, ktorá prevyšuje slabšie, ale špecifické nekovalentné interakcie inhibítora so substrátovými regiónmi enzýmu. Napriek týmto faktom sa mnoho vývojových skupín snaží racionálne optimalizovať štruktúru MMPI hydroxamátového typu využitím rozdielov medzi špecifickými interakčnými miestami MMP (obr. 4). Zatiaľ sa však dosiahol len čiastočný úspech (parciálne selektívne inhibítory pre malú podskupinu MMP). Prvé inhibítory MMP napodobňovali sekvenciu aminokyselín štiepneho miesta kolagénu (obr. 5). V substrátom riadenom projektovaní inhibítorov je funkčná skupina, zodpovedná za väzbu na zinočnatý ión, pripojená na peptidový skelet, ktorý kopíruje časť štiepnej sekvencie. Rozlišujeme tri typy inhibítorov MMP: inhibítory, v ktorých sa peptidový skelet nachádza napravo aj naľavo od funkčnej skupiny zodpovednej za väzbu na zinočnatý ión, a inhibítory, v ktorých peptidová časť smeruje buď doľava, alebo do prava (obr. 5).


Výskumná skupina British Biotech vyvinula sériu selektívnych MMPI modifikáciou nosnej štruktúry, marimastatu. V štruktúre sa najprv nahradila terc-butyl glycínová P2’ jednotka za piperidín (viď obrázok 6 šablónu nosnej štruktúry I). Zavedenie N-metyl sulfónamidovej skupiny do alfa polohy voči hydroxámovej funkčnej skupine prinieslo enzýmovú selektivitu založenú na charaktere sulfonylového substituenta. Zvýšenie aktivity sa môže dosiahnuť integrovaním cyklopentyl metylovej skupiny v oblasti P1’. Zlúčenina I je selektívny inhibítor MMP-1, MMP-8 a MMP-13 s dobrou perorálnou dostupnosťou 17).

Hannesianova skupina uskutočnila porovnávacie štúdium kotviacich programov na dostupných 3D štruktúrach MMP získaných NMR a röntgenovou difrakciou, a ukázali, že program Autodock spoľahlivo reprodukuje spôsob väzby inhibítorov pozorovaný v kryštálových štruktúrach 18). Na základe výsledkov táto skupina pripravila sériu N-arylsulfónových S-alkyl homocysteín hydroxámových kyselín s nanomolárnou a subnanomolárnou inhibičnou aktivitou voči rôznym MMP 19). Obmena P1, P1’, P2’oblastí výrazne ovplyvňuje účinnosť a selektivitu MMPI, napríklad zlúčenina II je 2900 krát silnejším inhibítorom MMP-9 ako MMP-1.
Spoločnosť Pfizer objavila skupinu bifenylsulfónových derivátov a karboxylových analógov ako selektívnych inhibítorov MMP-2, MMP-3 a MMP-13 s mikromolárnou aktivitou voči MMP-1, MMP-7 a MMP-9 20). Z tejto série (PD 166793) mala najlepšie farmakokinetické vlastnosti zlúčenina III 21). Ďalšia skupina z firmy Pfizer (Robinson a spol.) objavila potenciálne pyrolidínové inhibítory MMP-13 štruktúrne riadeným projektovaním liečiv, využitím homológového modelovania enzýmu, pričom vychádzali zo štruktúry MMP-8 22) (nosná štruktúra IV).
Barta a kol. (Pharmacia) zistili, že nové arylhydroxámové sulfónamidy, ktoré šetria MMP-1, sú nanomolárnymi inhibítormi MMP-2 a MMP-13 23). Analýza kryštálovej štruktúry komplexu zlúčeniny V s izoenzýmom MMP-8 ukázala, že selektivita k MMP-1 sa dá dosiahnuť úpravou skupiny P1’ na požadovanú dĺžku, tak aby vznikla stérická interferencia s arginínom MMP-1, ktorá je pozičným ekvivalentom leucínu v MMP-8.
Fray a kol. z firmy Pfizer (UK) objavili sukcinyl hydroxámové selektívne inhibítory MMP–3 24, 25). Zistili, že afinita inhibítorov k MMP-2 sa dá drasticky znížiť modifikáciou P3’ skupiny (zlúčenina VI). Ďalšia skupina vo firme Celltech (Baxter a kol.) objavila nové arylsulfónové hydroxamáty (VII), ktoré sú vynikajúcimi inhibítormi MMP-2, MMP-3, MMP-8 a MMP-9, pričom vykazujú len minimálnu inhibičnú aktivitu k MMP-1 26).
Firma Abbott sa snažila vyvinúť efektívne inhibítory MMP-9 a MMP-2, ktoré by sa mohli použiť na terapiu nádorov. Pripravili sériu N-formylhydroxylamínov, vysoko selektívnych inhibítory MMP-2 a MMP-9 v porovnávaní s inhibíciou MMP-1 27). Látka VIII (ABT 518), ktorá výrazne inhibovala rast nádorov na zvieracích modeloch, sa dostala do prvej fázy klinického testovania.
ADAM
Rodinu ADAM (A desintegrin a metaloproteáza) tvorí približne 40 proteínov, pričom 30 z nich je exprimovaných v ľudskom organizme a polovica týchto enzýmov má aj proteolytickú aktivitu 28). ADAM sú integrované membránové proteíny typu I, mnoho týchto enzýmov je súčasťou ektodoménového uvoľňovania membránových proteínov 29). Najviac preštudovaným členom je ADAM 17, proteáza, ktorá uvoľňuje zápalový cytokín faktor-α nekrotizujúci nádor (TNF-α) z jej membránového prekurzoru. ADAM-17 je hlavnou TNF-α konvertázou (TACE) a zodpovedá za riadenie hladiny solubilnej TNF-α v krvi pri zápalových ochoreniach 30). Vďaka výsledkom liečby artritídy TNF-špecifickými protilátkami sa väčšina klinických štúdií s TACE sústredila na tento typ ochorenia 31, 32). Na nešťastie sa kvôli hepatotoxicite dostali do druhej fázy klinického skúšania len dve látky 33, 34). O osude TACE inhibítorov rozhodne klinický vývoj vysoko selektívnych inhbítorov s vhodnými fyzikálno-chemickými vlastnosťami.
Inhibítory TACE
Počas hľadania selektívnych inhibítorov TACE bolo hlavnou snahou zabrániť možnej inhibícii MMP, aby sa predišlo vedľajším účinkom spojeným s inhibíciou MMP a len malá pozornosť sa venovala diskriminácií ďalších členov ADAM a ADAMTS (desintegrin a metaloproteináza s trombo-spondínovým motívom). Prvá úspešná práca o vývoji selektívnych inhibítorov TACE bola uverejnená v roku 2001 skupinou DuPont. Vložením vhodného spojovacieho reťazca medzi skupiny P1 a P2’ širokospektrálneho MMPI, marimastatu (obr. 7), vylepšili fyzikálno-chemické vlastnosti inhibítora 35). Spojovací reťazec vytvára makrocyklický skelet inhibítora a napodobňuje proteín. Zvýšenú selektivitu voči prasačiemu TACE (pTACE) oproti MMP dosiahli na základe SAR (structure-activity relationship) štúdií bočného reťazca inhibítora P1’. Interakcie bifenylovej funkčnej skupiny s P1’ kavitou TACE sú uprednostňované pred interakciami s MMP (zlúčenina IX, obr. 8) 36).


Výskumníci z firmy Bristol-Myers Squibb objavili ß laktámové hydroxámové kyseliny 37). ß-laktámovú kostru vytvorili na základe kľúčových interakcií široko-spektrálnych inhibítorov marimastatu a CGS27023A (zlúčenina X, obr. 7) s izoenzýmom MMP-3. Analyzovaním kryštálových štruktúr MMP a homológového modelu TACE (pripraveného podľa atrolyzínu, hadieho jedu z adamalyzínovej skupiny) sa identifikovala unikátna S1’ kavita s prehnutým tvarom, ktorá sa dá ideálne využiť na modelovanie selektívnych inhibítorov 38). Po zavedení (2 metyl-chinolín-4-yl)metoxy substituenta v para polohe fenylového jadra (zlúčenina XI, obr. 8) sa získali zo širokospektrálnych MMP/TACE inhibítorov vysoko selektívne pTACE inhibítory (IK682, zlúčenina XII). IK682 je extrémne silným inhibítorom ľudskej TACE s dobrou orálnou dostupnosťou 39). Modifikáciou štruktúry IK682 získali analóg XIII (obr. 7), ktorý sa dostal do druhej fázy klinických štúdií.
Racionálnou obmenou hlavného skeletu MMPI CGS27023A (X) a kombináciou s (2-metyl-chinolín-4 yl)metoxy skupinou v polohe P1’ (determinant pre TACE selektivitu oproti MMP) vyvinuli deriváty cyklických sukcinátov 40, 41). Zlúčenina IM491 (piperidín dikarboxamidový derivát, XIV, obr. 9) sa vyznačuje nanomolárnou pTACE inhibičnou aktitivitou, dobrou membránovou permeabilitou a šetrí enzýmy MMP- 1,-2 a -9. Aby sa zabránilo vzniku toxických metabolitov, ktoré vznikajú hydrolýzou anilidovej väzby, anilidová väzba sa nahradila amidovou 42). V ďalšej sérii nasyntetizovaných amidov (XV, XVI)sa sukcinyl hydroxámový skelet vymenil za ß-hydroxámovú kyselinu. Zlúčeniny tejto série mali výbornú TACE selektivitu a dobré farmakokinetické vlastnosti. ß, ß-tetrahydropyránový derivát XV je špecifickým subnanomolárnym inhibítorom pTACE bez výraznejšej inhibície väčšej skupiny MMP (mierna selektivita oproti MMP-3 a -7) a ADAMTS. Piperidinylový analóg XVI je tiež selektívnym inhibítorom, ale má zvýšenú afinitu voči MMP-14 a 15 v porovnaní s XV (obr. 9) 43). Metabolická nestabilita fenoxy väzby a slabá, ale nie zanedbateľná afinita k MMP-3, -7, -8 a 12 izoformám priviedol vedcov k tomu, aby ďalej optimalizovali P1’ bočný reťazec 44, 45). Zlúčeniny XVII a XVIII (obr. 9) sú najzaujímavejšími predstaviteľmi novej generácie. Majú excelentný inhibičný a farmakokinetický profil. Zlúčenina XVII sa nachádza v predklinickej fáze vývoja RA.

Firma Wyeth na základe SAR štúdií modifikovala sulfónové a sulfónamidové hydroxamáty, aby získala TACE a MMP inhibítory. Úpravou sulfónamidovej štruktúry v pozícii P1’, konkrétne zavedením alkinyl skupiny získali selektívne TACE inhibítory s nízkou MMP afinitou 46). Butinyl substituent naviazaný na fenoxy skupinu je schopný natiahnuť sa do S3’ polohy cez S1’ kavitu a takto kopírovať prehnutú konformáciu, ktorá nie je prijateľná pre MMP enzýmy s dlhým S1’ kanálom 47). Reprezentatívnym príkladom tejto skupiny je TACE inhibítor TMI-2 (zlúčenina XIX, obr. 10), ktorý má dobrú selektivitu a nízku afinitu k šiestim MMP a ADAM-10 a používa sa v predklinických štúdiách liečby RA 48).
Súčasné štúdie Zhu a kol. poukázali na niektoré nedostatky existujúcich farmakofórov a SAR štúdií 49). Inhibítory čiastočne hydroxamátového typu XX a XXI(obr. 10) sú vysoko selektívne inhibítory a líšia sa len stereokonfiguráciou cyklopropylového cyklu, ale iba zlúčenina XXje inaktívna voči ADAM-10. Kým hydroxamát XX svoju chinolínovú skupinu orientuje smerom k podjednotke S3’ cez plytkú S1’ kavitu, XXI je rozšírený opačným smerom (karbonylmetoxy a chinolínová skupina smerujú k S1 a S3 podjednotkám. Na základe rozdielneho tvaru S3 kavity sa môžu naprojektovať TACE inhibítory s lepším farmakologickým profilom a výraznejšou selektivitou 50).

Záver
V dnešnej dobe, ako vidíme, je najväčším problémom index selektivity. Hlavným dôvodom zlyhania prvotných inhibítorov hydroxamátového typu selektívne inhibovať enzýmy bola príliš veľká kapacita funkčnej skupiny kyseliny hydroxámovej viazať zinočnatý ión, ktorá prevyšuje slabšie, ale špecifické nekovalentné interakcie skeletu inhibítorov s postrannými reťazcami aminokyselín aktívneho miesta enzýmu. Napriek týmto faktom sa mnoho vývojových skupín snaží racionálne optimalizovať štruktúru hydroxamátového typu využitím rozdielov medzi špecifickými interakčnými miestami daných enzýmov. Súčasné štúdie poukázali na niektoré nedostatky existujúcich farmakofórov a SAR štúdií. Možnosti terapeutického využitia inhibítorov, ako ukázali aj in vitro a in vivo testy, sú naozaj široké: liečba nádorov, antiflogistický a imuno-modulačný účinok, antifibrotický a antiprotozoálny účinok, prevencia neurodegenerácie, stimulácia replikácie vírusov a prevencia hypertrofie srdca. Pomer účinnosti a toxicity inhibítorov prvej triedy počas klinických skúšok bol prijateľný. V klinických štúdiách sa najčastejšie objavovali gastrointestinálne vedľajšie nežiadúce účinky, preto môže byť vývoj inhibítorov s dobrou orálnou dostupnosťou problematický, aj napriek tomu, že nové inhibítory majú výhodné farmakokinetické vlastnosti. Objav selektívneho inhibítora hydroxamátového typu môže v budúcnosti znamenať prevratný krok v tejto oblasti.
Táto práca bola vytvorená s podporou grantu Ministerstva školstva SR (č. grantu 1/4301/07).
Došlo 14. dubna 2009 / Přijato 5. května 2009
Adresa pre korešpondenciu:
PharmDr. Július Šille Katedra farmaceutickej chémie FaF UK
Odbojárov 10, 832 32 Bratislava, Slovenská republika
e-mail: sille@fpharm.uniba.sk
Zdroje
1. Bode, W., Gomis-Rüth, F.X., Stöckler, W.: FEBS Lett., 1993; 331, 134–140.
2. Hooper, N. L.: FEBS Lett., 1994; 354, 1–6.
3. Georgiadis, D., Yiotakis, A.: Bioorg. Med. Chem., 2008; 16, 8781–8794.
4. Whitakker, M., Floyd, C. D., Brown, P., Gearing, A. J. H.: Chem. Rev., 1999; 99, 2735–2776.
5. Rao, B. G.: Curr. Pharm. Des., 2005; 11, 295–322.
6. Turk, B.: Nat. Rev. Drug. Disc., 2006; 5, 785–799.
7. Flannery, C. R.: Front. Biosci., 2006; 11, 544–569.
8. Baran, I., Varekova, R. S., Parthasarathi, L., Suchomel, S., Casey, F., Shields, D. C.: J. Chem. Inf. Model., 2007; 47, 464–474.
9. Skiles, J.W., Monovich, L.G., Jeng, A.Y.: Annual Reports in Medicinal Chemistry, 2000; 35, 167–176.
10. Hagmann, W.K., Lark, W.K., Becker, J.W.: Annual Reports in Medicinal Chemisry, 1996; 31, 231–240.
11. Nicolotti, O., Miscioscia, T. F., Lonetti, F., Muncipinto, G., Carotti, A.: J. Chem. Inf. Model., 2007; 47, 2439–2448.
12. Hu, J., Van den Steen, P. E., Sang, Q. X., Opdenakker, G.: Nat. Rev. Drug Disc., 2007; 6, 480–498.
13. Hutchinson, J. W., Tierney, G. M., Parsons, S. L., Davis, T. R. J.: J. Bone Joint Surg. (Br), 1998; 80–B: 907–908.
14. Abbenante, G., Fairlie, D. P.: Med. Chem., 2005; 1, 71–104.
15. Peterson, J. T.: Heart Fail. Rev., 2004; 9, 63–79.
16. Zucker, S., Cao, J., Chen, W. T.: Oncogene, 2000; 19, 6642–6650.
17. Martin, F. M., Beckett, R. P., Bellamy, C. L., Courtney, P. F., Davies, S. J., Drummond, A. H., Dodd, R., Pratt, L. M., Patel, S. R., Ricketts, M. L., Todd, R. S., Tuffnell, A. R., Ward, J. W. S., Whittaker, M.: Bioorg. Med. Chem. Lett., 1999; 9, 2887–2892.
18. Hanessian, S., Moitessier, N., Therrien, E.: J. Computer-Aided Mol. Design, 2001; 15, 873–881.
19. Hanessian, S., Moitessier, N., Gauchet, C., Viau, M.: J. Med. Chem., 2001; 44, 3066–3074.
20. O’Brien, P. M., Ortwine, D. F., Pavlovsky, A. G., Picard, J. A., Sliskovic, D. R., Roth, B. D., Dyer, R. D., Johnson, L. L., Man, C. F., Hallak, H.: J. Med. Chem., 2000; 43, 156–166.
21. Parker M. H., Ortwine D. F., O’Brien P. M., Lunney E. A., Banotai C. A., Mueller W. T., McConnell P., Brouillette C. G.: Bioorg. Med. Chem. Lett., 2000; 10, 2427–2430.
22. Robinson, R. P., Laird, E. R., Blake, J. F., Bordner, J., Donahue, K. M., Lopresti–Morrow, L. L., Mitchell, P. G., Reese, M. R., Reeves, L. M., Stam, E., Yocum, S. A.: J. Med. Chem., 2000; 43, 2293–2296.
23. Barta, T. E., Becker, D. P., Bedell, L. J., De Crescenzo, G. A., McDonald, J. J., Munie, G. E., Rao, S., Shieh, H.–S., Stegeman, R., Stevens, A. M., Villamil, C. I.: Bioorg. Med. Chem. Lett., 2000; 10, 2815–2817.
24. Fray, M. J., Burslem, M. F., Dickinson, R. P.: Bioorg. Med. Chem. Lett., 2001; 11, 567–570.
25. Fray M. J., Dickinson R. P.: Bioorg. Med. Chem. Lett., 2001; 11, 571–574.
26. Baxter, A. D., Bhogal, R., Bird, J., Keily, J. F., Manallack, D. T., Montana, J. G., Owen, D. A., Pitt, W. R., Watson, R. J., Wills, R. E.: Bioorg. Med. Chem. Lett., 2001; 11, 1465–1468.
27. Wada, C. K., Holms, J. H., Curtin, M. L., Dai, Y., Florjancic, A. S., Garland, R. B., Guo, Y., Heyman, H. R., Stacey, J. R., Steinman, D. H., Albert, D. H., Bouska, J. J., Elmore, I. N., Goodfellow, C. L., Marcotte, P. A., Tapang, P., Morgan, D. W., Michaelides, M. R., Davidsen, S. K.: J. Med. Chem., 2002; 45, 219–232.
28. Seals, D. F., Courtneidge S. A.: Genes Dev., 2003; 17, 7–30.
29. Huovila A. P., Turner A. J., Pelto–Huikko M., Kärkkäinen I., Ortiz R. M.: Trends Biochem. Sci., 2005; 30, 413–422.
30. Black, R. A.: Int. J. Biochem. Cell Biol., 2002; 34, 1–5.
31. Moss M. L., Bartsch J. W.: Biochemistry, 2004; 43, 7227–7235.
32. Moss M. L., Sklair-Tavron L., Nudelman R.: Nat. Clin. Pract. Rheumatol., 2008; 4, 300–309.
33. Grootveld M., McDermott M. F.: Curr. Opin. Investig. Drugs, 2003; 4, 598–602.
34. Thabet, M. M., Huizinga, T. W.: Curr. Opin. Investig. Drugs, 2006; 7, 1014–1019.
35. Xue, C. B., He, X., Corbett, R. L., Roderick, J., Wasserman, Z. R., Liu, R. Q., Jaffee B. D., Covington, M. B., Qian, M., Trzaskos, J. M., Newton, R. C., Magolda, R. L., Wexler, R. R., Decicco, C. P.: J. Med. Chem., 2001; 44, 3351–3354.
36. Holms, J., Mast, K., Marcotte, P., Elmore, I., Li, J., Pease, L., Glaser, K., Morgan, D., Michaelides, M., Davidsen, S.: Bioorg. Med. Chem. Lett., 2001; 11, 2907–2910.
37. Duan, J. J., Chen, L., Wasserman, Z. R., Lu, Z., Liu, R. Q., Covington, M. B., Qian, M., Hardman, K. D., Magolda, R. L., Newton, R. C., Christ, D. D., Wexler, R. R., Decicco, C. P.: J. Med. Chem., 2002; 45, 4954–4957.
38. Wasserman, Z. R., Duan, J. J., Voss, M. E., Xue, C. B., Cherney, R. J., Nelson, D. J., Hardman, K. D., Decicco, C. P.: Chem. Biol., 2003; 10, 215–223.
39. Niu, X., Umland, S., Ingram, R., Beyer, B. M., Liu, Y. H., Sun, J., Lundell, D., Orth, P.: Arch. Biochem. Biophys., 2006; 451, 43–50.
40. Cherney, R. J., Duan, J. J., Voss, M. E., Chen, L., Wang, L., Meyer, D. T., Wasserman, Z. R., Hardman, K. D., Liu, R. Q., Covington, M. B., Qian M., Mandlekar, S., Christ, D. D., Trzaskos, J. M., Newton, R. C., Magolda, R. L.,Wexler, R. R., Decicco, C. P.: J. Med. Chem., 2003; 46, 1811–1823.
41. Xue, C. B., He, X., Roderick, J., Corbett, R. L., Duan, J. J., Liu, R. Q., Covington, M. B., Qian, M., Ribadeneira, M. D., Vaddi, K., Christ, D. D., Newton, R. C., Trzaskos, J. M., Magolda, R. L., Wexler, R. R., Decicco, C. P.: Bioorg. Med. Chem. Lett., 2003; 13, 4299–4304.
42. Gilmore, J. L., King, B. W., Harris, C., Maduskuie, T., Mercer, S. E., Liu, R. Q., Covington, M. B., Qian, M., Ribadeneria, M. D., Vaddi, K., Trzaskos, J. M., Newton, R. C., Decicco, C. P., Duan, J. J.: Bioorg. Med. Chem. Lett., 2006; 16, 2699–2704.
43. Wang, X., Feuerstein, G. Z., Xu, L., Wang, H., Schumacher, W. A., Ogletree, M. L., Taub, R., Duan, J. J., Decicco, C. P., Liu, R. Q.: Mol. Pharmacol., 2004; 65, 890–896.
44. Chen, X. T., Ghavimi, B., Corbett, R. L., Xue, C. B., Liu, R. Q., Covington, M. B., Qian, M., Vaddi, K. G., Christ, D. D., Hartman, K. D., Ribadeneira, M. D., Trzaskos, J. M., Newton, R. C., Decicco, C. P., Duan, J. J.: Bioorg. Med. Chem. Lett., 2007; 17, 1865–1870.
45. Lu, Z., Ott, G. R., Anand, R., Liu, R. Q., Covington, M. B., Vaddi, K., Qian, M., Newton, R. C., Christ, D. D., Trzaskos, J., Duan, J. J.: Bioorg. Med. Chem. Lett., 2008, 18, 1958–1962.
46. Levin, J. I., Chen, J. M., Du, M. T., Nelson, F. C., Killar, L. M., Skala, S., Sung, A., Jin, G., Cowling, R., Barone, D., March, C. J., Mohler, K. M., Black, R. A., Skotnicki, J. S.: Bioorg. Med. Chem. Lett., 2002; 12, 1199–1202.
47. Levin, J. I., Chen, J. M. Cheung, K., Cole, D., Crago, C., Santos, E. D., Du, X., Khafizova, G., MacEwan, G., Niu, C., Salaski, E. J., Zask, A., Cummons, T., Sung, A., Xu, J., Zhang, Y., Xu, W., Ayral–Kaloustian, S., Jin, G., Cowling, R., Barone, D., Mohler, K. M., Black, R. A., Skotnicki, J. S.: Bioorg. Med. Chem. Lett., 2003; 13, 2799–2803 .
48. Zhang, Y., Hegen, M., Xu, J., Keith, J. C. Jr., Jin, G., Du X., Cummons, T., Sheppard, B. J., Sun, L., Zhu, Y., Rao, V. R., Wang, Q., Xu, W., Cowling, R., Nickerson–Nutter, C. L., Gibbons, J., Skotnicki, J., Lin, L. L., Levin, J.: Int. Immunopharmacol., 2004; 4, 1845–1847.
49. Zhu, Z., Mazzola, R., Sinning, L., McKittrick, B., Niu, X., Lundell, D., Sun, J., Orth, P., Guo, Z., Madison, V., Ingram, R., Beyer, B. M.: J. Med. Chem., 2008; 51, 725–736.
50. Lukacova, V., Zhang, Y., Kroll, D. M., Raha, S., Comez, D., Balaz, S.: J. Med. Chem., 2005; 48, 2361–2370.
51. Gomis-Rüth, F. X.: Molecular, Mol. Biotechnol., 2003; 24, 157–202.
52. Boliang L., Kexin Y.: Mini Rev. Med. Chem., 2003; 3, 609–620.
Štítky
Farmácia FarmakológiaČlánok vyšiel v časopise
Česká a slovenská farmacie

2009 Číslo 3
Najčítanejšie v tomto čísle
- Masti ve středověké Evropě
- Stručný úvod do problematiky obsahových látek řas a sinic – důkazy o výskytu fenolických metabolitů
- Prehľad inhibítorov MMP a TACE hydroxamátového typu a ich terapeutický potenciál
- Pracovní den sekce přírodních léčiv české farmaceutické společnosti ČLS JEP