
Syndrom Dravetové: těžká myoklonická epilepsie v časném dětství – kazuistiky
Dravet Syndrome: Severe Myoclonic Epilepsy in Infancy – Case Reports
Dravet syndrome (DS) is classified as a rare progressive epileptic encephalopathy. Seizure onset is in the first year of life in thus far normally developing children. Typically, prolonged generalised convulsive seizures occur. Subsequently, other types of seizures are seen, accompanied by deterioration of psychomotor development. At present, detection of a specific mutation may confirm the clinical syndrome. 70–80% of patients have mutation in SCN1A gene, 5% in PCDH19 gene. Rarely, mutations in the GABARG2 gene and SCN1B gene are detected. Early diagnosis of DS is very important from the therapeutical point of view. Two case reports of patients with typical clinical course of DS and genetically detected mutation in SCN1A gene are presented.
Key words:
Dravet syndrome – myoclonic epilepsy – epilepsy – therapy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autori:
P. Danhofer 1; O. Horák 1; L. Fajkusová 2; J. Pavloušková 2; H. Ošlejšková 1
Pôsobisko autorov:
LF MU a FN Brno
Klinika dětské neurologie, Centrum pro epilepsie Brno
1; LF MU a FN Brno
Centrum molekulární biologie a genové terapie Interní hematoonkologické kliniky
2
Vyšlo v časopise:
Cesk Slov Neurol N 2014; 77/110(2): 243-246
Kategória:
Kazuistika
Súhrn
Syndrom Dravetové se řadí mezi vzácné progresivní epileptické encefalopatie. Klinická manifestace je v prvním roce života u dosud normálně se vyvíjejících kojenců. Typicky začíná febrilními protrahovanými konvulzivními záchvaty, později se objevují i jiné typy záchvatů a dochází k psychomotorické deterioraci. Diagnóza se dá stanovit na podkladě klinického obrazu, v současnosti je možné u převážné většiny pacientů i potvrzení genetické. U 70– 80 % pacientů nalézáme mutaci v genu SCN1A, u 5 % je detekována mutace v PCDH19 genu, vzácně pak v GABARG2 a SCN1B genu. Časná diagnostika pacientů je velmi žádoucí vzhledem k terapeutickým specifikům nemoci. Jsou prezentovány kazuistiky dvou pacientů s typickým průběhem DS a geneticky potvrzenou mutací v SCN1A genu.
Klíčová slova:
Dravetové syndrom – myoklonická epilepsie – epilepsie – terapie
Úvod
Syndrom Dravetové (DS), těžká myoklonická epilepsie v časném dětství, se řadí mezi vzácné progresivní epileptické encefalopatie [1]. Diagnostika a syndromologické zařazení se dá provést na podkladě klinického obrazu, v současné době je možné diagnózu potvrdit i geneticky. Cílem této publikace je upozornit na včasné rozpoznání pacientů s DS. Pacienti s podezřením na DS by měli být co nejdříve odesláni do specializovaných center ke genetické diagnostice a specifické terapii. Včasná diagnostika DS a jeho intenzivní léčba může vést k redukci protrahovaných epileptických záchvatů, které se nepřímo mohou spolupodílet na míře psychomotorické retardace u pacientů s DS.
Kazuistika 1
U první pacientky, aktuálně 2,5leté, se záchvaty objevily ve čtyřech měsících věku. V rodinné anamnéze bez pozoruhodností, v graviditě bez komplikací, perinatální průběh bez rizik s normální poporodní adaptací. Motorický vývoj byl lehce disharmonický s dobrým efektem rehabilitace dle Vojty. Ve čtyřech měsících věku se objevil první afebrilní protrahovaný pravostranný hemikonvulzivní záchvat. V témže měsíci pak 50minutový epileptický status s levostrannými křečemi při febrilním infektu. Provedená vyšetření (MR mozku, odběr mozkomíšního moku, EEG a odběr krve a moči k vyšetření dědičných poruch metabolizmu) byla negativní. Byla zahájena terapie fenobarbitalem v dávce 4,5 mg/ kg/ den. V dalším průběhu ale záchvaty trvaly s frekvencí několika paroxyzmů za měsíc. Objevovaly se konvulzivní záchvaty se střídavou lateralizací, často vázány na febrilní infekt, pacientka byla velmi citlivá i na teplé počasí, v létě se stav výrazně zhoršil, délka záchvatů byla od 5 do 40 min. Vzhledem k závažnosti a délce trvání záchvatů byla pacientka opakovaně hospitalizována na jednotce intenzivní péče, opakovaně vyžadovala i hospitalizaci na ARO s nutností intubace. Od devíti měsíců věku se začaly objevovat i protrahované záchvaty charakteru areaktivity s celkovou hypotonií a deviací hlavy a bulbů (stranově střídavě, převážně ale doprava) – záchvaty hypomotorické s verzivní složkou. Tyto záchvaty byly přítomny s frekvencí několika za měsíc. V EEG se objevilo zpomalení a dezorganizace základní aktivity do pásma delta, ojediněle i ostré vlny pod F4. V dalším průběhu byly nasazeny postupně valproát, levetiracetam a klonazepam, vždy jen s přechodným efektem a vymizením záchvatů na několik měsíců. Ve dvou letech věku byla přímou DNA analýzou potvrzena mutace c.408C>A, p.(Cys136*) v genu SCN1A v heterozygotní formě. Identifikovaná mutace nebyla v literatuře ani databázi HGMD (Human Gene Mutation Database, http:/ / www.hgmd.cf.ac.uk/ ac/ index.php) popsána. Byla provedena detekce mutace u rodičů a dle předpokladu bylo zjištěno, že se jedná o mutaci de novo. Postupně byla zahájena redukce fenobarbitalu a levetiracetamu, pacientka poté zůstala na kombinaci valproát a klonazepam, který byl zaměněn za klobazam. Ve dvou letech věku bylo zahájeno nasazování stiripentolu v dávce 50 mg/ kg/ den s parciálním efektem. Došlo ke snížení frekvence záchvatů generalizovaných konvulzivních na cca jeden za měsíc, především ale byly výrazně kratší v trvání maximálně do 3 min, záchvaty hypomotorické dále rodiče nereferovali. Ve 2,5 letech věku se objevil nový typ záchvatů, atypické absence s EEG korelátem generalizovaných komplexů hrot vlna o frekvenci 3,5 Hz s počátkem temporálně vpravo. V EEG se nově objevila interiktální epileptiformní patologie vpravo temporoparietálně. Nyní pacientka zahajuje ketogenní dietu. Aktuálně ve 2,5 letech věku je v objektivním neurologickém nálezu lehká centrální hypotonie, pacientka již sama obchází kolem nábytku, jemná motorika je lehce nedokonalá. Psychický vývoj je disharmonický, odpovídá 12– 15 měsícům věku. Dominuje slabší schopnost komunikace a řečového vývoje s atypiemi v sociálním chování (menší zastoupení reciprocity), vývoj však jak po motorické, tak i psychické stránce pozvolna pokračuje.
Kazuistika 2
Druhý pacient je čtyřletý chlapec. V rodinné anamnéze bez pozoruhodností, těhotenství probíhalo bez komplikací, porod byl spontánní v termínu bez perinatálních komplikací s normální poporodní adaptací. Až do klinické manifestace epileptických záchvatů byl psychomotorický vývoj normální. Záchvaty se objevily v sedmi měsících věku s konstantní vazbou na febrilní infekty. Klinicky se jednalo o konvulzivní záchvaty se střídavou lateralizací v trvání obvykle do 5 min, nejdelší záchvat byl 10minutový. Frekvence záchvatů byla jeden za měsíc. Pacient byl opakovaně hospitalizován v okresní nemocnici, kde bylo provedeno CT mozku a MR mozku s normálním nálezem, vstupní EEG negativní. Dále byl proveden odběr séra a moči na vyšetření dědičných poruch metabolizmu s negativním nálezem. V krevním obraze zjištěna lehká hypochromní anémie, na kardiologii inkompletní blok pravého Tawarova raménka. Vzhledem k četnosti záchvatů byl nasazen valproát, poté přidán klonazepam. I přes dvojkombinaci léků se záchvaty objevovaly dále s frekvencí 1– 2 za měsíc, vždy s vazbou na febrilní infekt. Ke stávající dvojkombinaci léků byl přidán levetiracetam a pacient byl odeslán k vyšetření na Kliniku dětské neurologie LF MU a FN Brno, kde byla zahájena dispenzarizace. V dalším období, ve věku od 13 měsíců do dvou let byl chlapec klinicky kompenzován. Poté se objevily záchvaty opět, s četnější frekvencí a nově i bez vazby na horečku. Trvají záchvaty charakteru generalizovaných nebo stranově střídajících lateralizovaných křečí v délce do 5 min, dále se objevuje nový typ záchvatů bez křečí s areaktivitou a oroalimentárními automatizmy v trvání do 10 min. Záchvaty se objevují s frekvencí 1– 2 za měsíc. V EEG zachycen opakovaně negativní nález, od tří let věku se vyskytují ojedinělé ostré vlny v bifrontální lokalizaci s pravostrannou převahou. Vzhledem k neúčinnosti stávající medikace byla zahájena redukce levetiracetamu, aktuálně je chlapec na dvojkombinaci valproátu s klobazamem, který byl nasazen výměnou za klonazepam, a nasazuje se stiripentol.
Ve 2,5 letech věku byla přímou DNA analýzou u pacienta prokázána mutace c.1989delT, p.(Phe663Phefs*9) v genu SCN1A v heterozygotní formě. Identifikovaná mutace nebyla v literatuře ani databázi HGMD popsána. V objektivním neurologickém nálezu lze sledovat centrální hypotonii s bederní hyperlordózou, je schopen samostatné chůze s valgózním postavením kolen a bérců. V psychologickém profilu dominuje hyperaktivita a porucha koncentrace, slabší sociální kontakt při zachovalé dovednosti jeho navázání. Intelekt celkově v pásmu normy rozkolísaný především pro slabou koncentraci.
Diskuze
Syndrom Dravetové byl poprvé popsán v roce 1978 profesorkou Charlotte Dravet ve Francii [2]. Dle ILAE klasifikace epileptických syndromů z roku 1989 se řadí mezi epilepsie a epileptické syndromy nezařaditelné jako ložiskové či generalizované [3]. Dle revize této klasifikace z roku 2010 patří mezi elektroklinické syndromy časného dětství [4].
V souladu s ILAE revizí epileptických syndromů 2010 se syndrom Dravetové z etiologického hlediska řadí mezi epilepsie na genetickém podkladě. Převážná část pacientů jsou nositelé mutace v genu pro alfa1 podjednotku sodíkového kanálu (gen SCN1A), tato mutace se vyskytuje u 70– 80 % pacientů [5]. Dochází zde ke snížení excitability GABAergních interneuronů v neokortexu a hipokampu [6], což má za následek snížení výbojů v těchto inhibičních interneuronech. Dojde tak ke zvýšení excitability v neuronální síti a rozvoji SCN1A epileptické encefalopatie.
Mutace v SCN1A genu jsou odpovědné za rozvoj velmi variabilního fenotypového vyjádření, tzv. GEFS+ spektra. Na „benigním“ konci spektra se vyskytují pacienti se syndromem GEFS+ (febrilní záchvaty + afebrilní generalizované tonicko‑klonické záchvaty), na druhém konci spektra se nachází DS. Mutace u pacientů s DS jsou ve 40 % tzv. truncating mutace, které způsobují předčasný vznik terminačního kodonu, čímž vedou k předčasnému ukončení translace, a nebo v dalších 40 % případů se jedná o tzv. missense mutace [7]. Za rozvoj GEFS+ je odpovědná spíše missense mutace v genu SCN1A, nicméně genotypová‑ fenotypová korelace není konzistentní. Předpokládá se, že pokud se missense mutace nachází v pór formující oblasti SCN1A genu, rozvine se DS, pokud je mutace mimo tuto oblast, fenotypově se rozvine spíše GEFS+ syndrom [7].
Genetika DS je ještě komplexnější. Vzácně byly identifikovány u pacientů s DS i mutace v genu GABARG2 [8] a SCN1B [9].
Zhruba 5 % pacientů nese mutaci v genu PCDH19 s X‑ vázanou dědičností [10]. Jedná se o gen kódující protocadherin 19, jehož funkce nebyla dosud plně objasněna, ale předpokládá se jeho zapojení v neuronálních sítích. Mutace způsobuje epilepsii limitovanou na ženy s mentální retardací (Epilepsy limited to Females with Mental Retardation, EFMR). Charakteristickým rysem je zde menší výskyt epileptických statů a jen zřídka se objevují myoklonické záchvaty. U 45 % pacientů je mentální retardace jen mírného stupně, časté jsou autistické rysy.
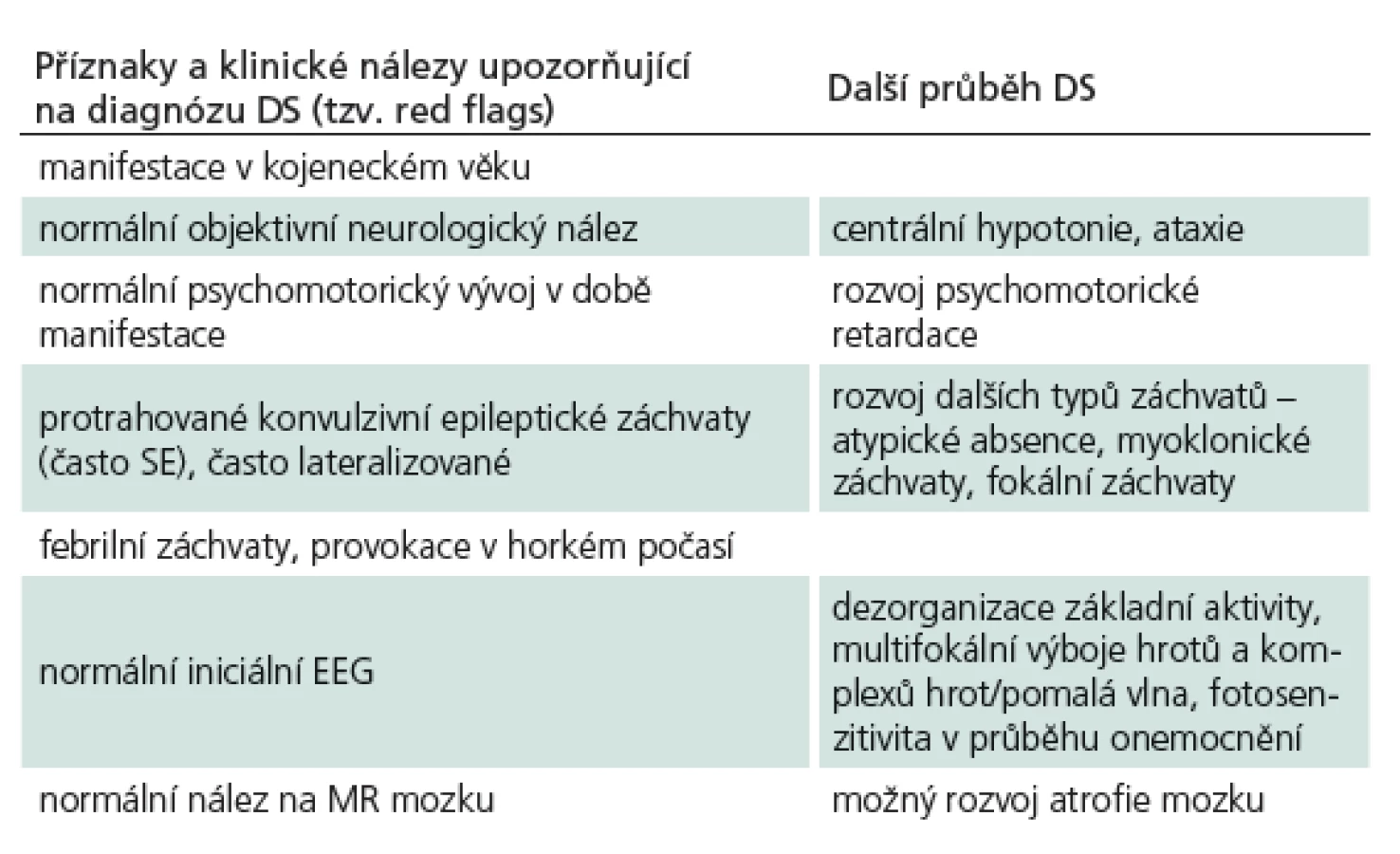
U pacienta s typickým fenotypem DS dochází k rozvoji epileptických záchvatů v kojeneckém věku s vrcholem kolem 5. měsíce u dosud normálně se vyvíjejících dětí. Incidence je udávána 1 : 30 000 dětí [1], častěji jsou postiženi chlapci v poměru 2 : 1. Objevují se tyto typy záchvatů: generalizované klonicko‑tonické záchvaty (resp. často lateralizované stranově střídající), myoklonické záchvaty, atypické absence a fokální záchvaty s poruchou vědomí. Velmi častý je výskyt status epilepticus, především v počátečním období, a to převážně u febrilních záchvatů. Tab. 1 ukazuje příznaky, kdy je třeba myslet na možnost DS (tzv. red flags).
V klinickém obraze pacientů s DS existuje jistá variabilita, mluvíme o DS‑ spektru (tab. 2) [5].
![Klinické spektrum syndrom Dravetové – DS spektrum [5].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ff80e61db24a91b85a0403277749ab8c.png)
V současné době je v České republice genetické potvrzení DS možné provést v Brně a v Praze, a to na úrovni vyšetření SCN1A genu. V Brně vyšetření provádí Centrum molekulární biologie a genové terapie Interní hematoonkologické kliniky LF MU a FN Brno (kontakt: RNDr. Lenka Fajkusová, CSc., lfajkusova@fnbrno.cz). V Praze se lze obrátit na Ústav biologie a lékařské genetiky 2. LF UK a FN v Motole (kontakt: RNDr. Petra Hedvičáková, petra.hedvicakova@lfmotol.cuni.cz). V případě podezření na DS je vhodné provést genetickou konzultaci na výše uvedených pracovištích.
Vyšetření vzácnějších mutací DS probíhá na celoevropské úrovni pod záštitou projektu EuroEPINOMICS, a to v programu RES (genetics of Rare Epilepsy Syndromes). Pro zájemce odkazujeme na stránku http:/ / www.euroepinomics.org/ .
Terapeutické možnosti jsou poměrně bohaté, jejich efekt je však limitován vysokou farmakorezistencí pacientů. Lékem první volby jsou valproát a benzodiazepiny [11], především klobazam. Při neefektu těchto je indikován stiripentol. Stiripentol (Diacomit) je řazen mezi orphan‑ drug v terapii DS a je schválen jako přídatná terapie k valproátu + klobazamu. V terapii DS je v Evropě schválen od roku 2007. Dosud byly realizovány dvě randomizované placebem kontrolované studie, které sledují efekt stiripentolu jako přídatné terapie ke kombinaci VPA + CLB. Za tímto účelem byla zformována studijní skupina STICLO (STIripentol CLObazame study group) ve Francii a Itálii. Výsledky francouzské skupiny byly publikovány Catherine Chiron et al v roce 2000 [12]. Výsledky italské skupiny publikovány dosud nebyly, jsou však zahrnuty v metaanalýze [13]. Výsledky ukázaly 71 % [13], resp. 67 % [12] respondérů ve srovnání s placebem 5 % [13], resp. 9 % [12]. Další terapeutickou možností jsou topiramat [14] a levetiracetam (účinný především na myoklonické záchvaty [15]), které lze využít jako přídatné léky. Lamotrigin, karbamazepin, vigabatrin a vysoké dávky fenobarbitalu mohou záchvaty zhoršovat a je vhodné se jim v léčbě vyhnout. V neposlední řadě je nutno uvést i ketogenní dietu, která má v terapii DS relativně vysokou účinnost ve srovnání s jinými epileptickými encefalopatiemi [16]. Příznivé výsledky ukazuje v terapii DS i implantace vagového stimulátoru [17].
Prezentované kazuistiky představují geneticky potvrzené případy DS s prokázanou mutací v genu SCN1A. Klinický průběh onemocnění je zde poměrně typický. Zavádějící může být fakt, že se u pacientů dosud nevyskytly myoklonické záchvaty, které má syndrom Dravetové – jako těžká myoklonická epilepsie v časném dětství – přímo v názvu. Myoklonické záchvaty se však vyskytovat vůbec nemusí [1] nebo často se objevují až po několika letech od klinické manifestace onemocnění. Název tak může být zavádějící. Již v roce 1994 Aicardi navrhoval pro DS název „těžká polymorfní epilepsie v časném dětství“, který však v klasifikačním systému epileptických syndromů etablován nebyl.
U 70– 80 % pacientů se syndrom geneticky potvrdit podařit nemusí. Je třeba však myslet na to, že DS lze diagnostikovat již na podkladě klinického obrazu a diagnostika genetická je pouze potvrzující. Diagnóza již na základě klinického vyjádření je zásadní, poněvadž umožní nasměrovat terapii správným směrem, vyvarovat se potenciálně zhoršujících léků a zvolit naopak ty, které by mohly mít efekt na redukci záchvatů. Zásadní je především časná diagnostika syndromu. Stiripentol se ukázal efektivní v redukci především konvulzivních záchvatů, jež mohou být v počátečním stadiu protrahované až charakteru epileptických statů. Právě ty se velkou měrou spolupodílí na rozvoji neurokognitivního deficitu pacientů a regresu v psychomotorickém vývoji [11].
Pojmenování nemoci je důležité i pro rodiče pacienta. Ukončí extenzivní a často stresující došetřování příčiny epilepsie, umožní s rodiči mluvit o prognóze, vysvětlit jim možné komplikace a soustředit se více na komplexní péči zahrnující psychologa, logopeda, ortopeda, rehabilitaci a další formy podpůrné terapie, které mohou celkový stav dítěte a jeho psychomotorický vývoj povzbudit. Důležité informace mohou rodiče i odborníci získat na stránkách http:/ / www.dravetfoundation.org/ .
Závěr
Syndrom Dravetové se řadí mezi prognosticky závažné epilepsie manifestující se v časném dětství. Časná a správná diagnostika je zásadní pro další terapii, která u DS představuje určitá specifika. V současné době je možná i diagnostika genetická, jež u většiny pacientů prokáže mutaci a pro diagnózu DS je potvrzující.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 5. 7. 2013
Přijato do tisku: 24. 10. 2013
MUDr. Pavlína Danhofer
Klinika dětské neurologie
LF MU a FN Brno
Centrum pro epilepsie Brno
Černopolní 9
613 00 Brno
e-mail: pavlina.cahova@seznam.cz
Zdroje
1. Panayiotopoulos CP. A clinical guide to Epileptic syndromes and their treatment. 2nd ed. London: Springer Healthcare Ltd 2002: 283– 287.
2. Dravet C. Les épilepsies graves de l’enfant. Vie Med 1978; 8: 543– 548.
3. Commission on Classification and the Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30(4): 389– 399.
4. Berg AT, Berkovic SF, Brodie MJ, Buchhlater J, Cross HJ,Van Emde Boas W et al. Revised terminology and concepts for organisation of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005– 2009. Epilepsia 2010; 51(4): 676– 685.
5. Marini C, Scheffer IE, Nabbout R, Suls A, de Jonghe P,Zara F et al. The genetics of Dravet syndrome. Epilepsia 2011; 52 (Suppl 2): 24– 29.
6. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA,Klaume F, Burton KA et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe epilepsy in infancy. Nat Neurosci 2006; 9(9): 1142– 1149.
7. Dravet C, Guerrini R. Dravet syndrome. Topics in Epilepsy series. Vol. 3. London: John Libbey Eurotext 2011: 51– 61.
8. Harkin LA, Bowser DN, Dibbens LM, Singh R, Phillips F,Wallace RH et al. Truncation of the GABA/ A- receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet 2002; 70(2): 530– 536.
9. Patino GA, Claes LR, Lopez‑ Santiago LF, Slat EA, Dondeti RS, Chen C et al. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 2009; 29(34): 10764– 10778.
10. Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O et al. Sporadic infantile epiletic encephalopathy caused by mutation in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet 2009; 5: e1000381.
11. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011; 53 (Suppl 2): 16– 18.
12. Chiron C, Marchand MC, Tran A. STICLO study group. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo‑controlled syndrome‑ dedicated trial. Lancet 2000; 356(9242): 1638– 1642.
13. Kassaï B, Chiron C, Augier S, Cucherat M, Rey E,Gueyffier F et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta‑analysis of individual patient data. Epilepsia 2008; 49(2): 343– 348.
14. Coppola G, Capovilla G, Montagnini A, Romeo A,Spanò M, Tortorella G et al. Topiramate as add‑ on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002; 49(1): 45– 48.
15. Striano P, Coppola G, Pezzella M, Ciampa C, Specchio N, Ragona F et al. An open‑ label trial of levetiracetam in severe myoclonic epilepsy in infancy. Neurology 2007; 69(3): 250– 254.
16. Caraballo RH. Nonpharmacologic treatments of Dravet syndrome: focus on ketogenic diet. Epilepsia 2011; 52 (Suppl 2): 79– 82.
17. Zamponi N, Passamonti C, Cappanera S, Petrelli C.Clinical course of young patients with Dravet syndrome after vagal nerve stimulation. Eur J Paediatr Neurol 2011; 15(1): 8– 14.
Štítky
Detská neurológia Neurochirurgia NeurológiaČlánok vyšiel v časopise
Česká a slovenská neurologie a neurochirurgie

2014 Číslo 2
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Neuromultivit v terapii neuropatií, neuritid a neuralgií u dospělých pacientů
- Antidepresivní efekt kombinovaného analgetika tramadolu s paracetamolem
- Geriatrická křehkost a léčba bolesti
Najčítanejšie v tomto čísle
- Autonomní dysreflexie – závažná komplikace u pacientů po poranění míchy
- Syndrom Dravetové: těžká myoklonická epilepsie v časném dětství – kazuistiky
- Normativní hodnoty parametrů vedení pro nervus ulnaris a nervus medianus měřené standardizovaným způsobem
- Neuromodulace