
Elevated Levels of the Polo Kinase Cdc5 Override the Mec1/ATR Checkpoint in Budding Yeast by Acting at Different Steps of the Signaling Pathway
Checkpoints are surveillance mechanisms that constitute a barrier to oncogenesis by preserving genome integrity. Loss of checkpoint function is an early event in tumorigenesis. Polo kinases (Plks) are fundamental regulators of cell cycle progression in all eukaryotes and are frequently overexpressed in tumors. Through their polo box domain, Plks target multiple substrates previously phosphorylated by CDKs and MAPKs. In response to DNA damage, Plks are temporally inhibited in order to maintain the checkpoint-dependent cell cycle block while their activity is required to silence the checkpoint response and resume cell cycle progression. Here, we report that, in budding yeast, overproduction of the Cdc5 polo kinase overrides the checkpoint signaling induced by double strand DNA breaks (DSBs), preventing the phosphorylation of several Mec1/ATR targets, including Ddc2/ATRIP, the checkpoint mediator Rad9, and the transducer kinase Rad53/CHK2. We also show that high levels of Cdc5 slow down DSB processing in a Rad9-dependent manner, but do not prevent the binding of checkpoint factors to a single DSB. Finally, we provide evidence that Sae2, the functional ortholog of human CtIP, which regulates DSB processing and inhibits checkpoint signaling, is regulated by Cdc5. We propose that Cdc5 interferes with the checkpoint response to DSBs acting at multiple levels in the signal transduction pathway and at an early step required to resect DSB ends.
Published in the journal:
Elevated Levels of the Polo Kinase Cdc5 Override the Mec1/ATR Checkpoint in Budding Yeast by Acting at Different Steps of the Signaling Pathway. PLoS Genet 6(1): e32767. doi:10.1371/journal.pgen.1000763
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000763
Summary
Checkpoints are surveillance mechanisms that constitute a barrier to oncogenesis by preserving genome integrity. Loss of checkpoint function is an early event in tumorigenesis. Polo kinases (Plks) are fundamental regulators of cell cycle progression in all eukaryotes and are frequently overexpressed in tumors. Through their polo box domain, Plks target multiple substrates previously phosphorylated by CDKs and MAPKs. In response to DNA damage, Plks are temporally inhibited in order to maintain the checkpoint-dependent cell cycle block while their activity is required to silence the checkpoint response and resume cell cycle progression. Here, we report that, in budding yeast, overproduction of the Cdc5 polo kinase overrides the checkpoint signaling induced by double strand DNA breaks (DSBs), preventing the phosphorylation of several Mec1/ATR targets, including Ddc2/ATRIP, the checkpoint mediator Rad9, and the transducer kinase Rad53/CHK2. We also show that high levels of Cdc5 slow down DSB processing in a Rad9-dependent manner, but do not prevent the binding of checkpoint factors to a single DSB. Finally, we provide evidence that Sae2, the functional ortholog of human CtIP, which regulates DSB processing and inhibits checkpoint signaling, is regulated by Cdc5. We propose that Cdc5 interferes with the checkpoint response to DSBs acting at multiple levels in the signal transduction pathway and at an early step required to resect DSB ends.
Introduction
Saccharomyces cerevisiae cells suffering a double stranded DNA break (DSB) activate a robust Mec1-dependent checkpoint response when DSB ends are processed to expose single-stranded DNA (ssDNA), and progression through the cell cycle is arrested prior to anaphase. Several well conserved factors are recruited at the DSB lesion, and contribute to the activation of a signaling pathway based on sequential phosphorylation events driven by the upstream kinases Tel1/ATM and Mec1/ATR which, in turn, activate the transducer kinases Rad53/Chk2 and Chk1 [1],[2]. The checkpoint response is influenced at several levels by kinases such as CDK1, CKII and Polo-like Cdc5, all involved in promoting key events throughout an unperturbed cell cycle, supporting the notion that the cellular response to DNA damage is tightly linked to cell cycle events [3]. The intensity of the DSB-induced checkpoint response correlates to the amount of the ssDNA that is accumulated at DSB lesions [4]. 5′-to-3′ nucleolytic processing of DNA ends is dependent upon several factors, including CDK1 and the nucleases Mre11, Sae2, Dna2 and Exo1 [5]. Moreover, the checkpoint is a reversible signaling pathway which is turned off when DNA lesions are repaired, thus permitting the resumption of cell cycle progression [6]. Different types of phosphatases (Pph3, Ptc2 and Ptc3) dephosphorylate and inactivate Rad53 and other checkpoint kinase targets [7]. Further, mutations in several DNA repair genes, including SAE2, KU70/80, RAD51, RDH54, SRS2, affect the inactivation of the DSB-induced checkpoint response [7],[8]. These observations suggest that the attenuation, as well the activation, of the checkpoint pathway are related to the metabolism of DSB ends, in a way that is not yet completely understood. It is also known that the checkpoint response can be attenuated when an irreparable DNA lesion is formed in the cell, leading to adaptation to DNA damage. Checkpoint inactivation during recovery and adaptation to DNA damage is a phenomenon described also in higher eukaryotes [6]. The functional role of adaptation is not completely understood; however, it was suggested that it may be partly responsible for chromosomal rearrangements, genome instability and tumorigenesis [6],[9]. Interestingly, the well conserved family of Polo-like kinases (Plks) has been involved in checkpoint adaptation and/or recovery both in budding yeast and vertebrates [10]. Cdc5 is the only polo kinase expressed in yeast, whereas higher eukaryotes usually express three or four Plks [11]. However, only Plk1, which is the most extensively studied, is a true mitotic kinase homolog to the Drosophila Polo kinase [11]. In yeast, CDC5 is an essential gene and the point mutation cdc5-ad (a Leucine-to-Tryptophan substitution at residue 251, within the kinase domain) causes the inability to adapt to one irreparable DSB lesion and to turn off Rad53 kinase [12],[13]. However, cdc5-ad cells can recover from checkpoint when the DSB is repaired, suggesting that adaptation and recovery are two genetically separate processes [14]. A corresponding cdc5-ad mutation in Plks has not yet been isolated in mammals; however, it was found that Plk1 depletion severely blocks checkpoint recovery and adaptation [10],[15],[16], and rapidly causes cell death in cancer cells [17],[18]. Based on the fact that the DNA damage checkpoint pathway is well conserved in all the eukaryotes, it is reasonable to expect that the functional role of Cdc5 in budding yeast and of Plk1 during adaptation (and perhaps in recovery) may be conserved. Polo-like kinases contain in the C-terminal region of the protein a polo box which mediates the interaction of Plks with substrates previously phosphorylated by CDK or MAPK kinases [19]. Indeed, Cdc5 targets multiple substrates during an unperturbed cell cycle [20] and could functionally interact with several checkpoint proteins as well. In vertebrates, polo kinases regulate the DNA damage checkpoint acting on multiple factors. They phosphorylate Claspin [21]–[24], a Chk1 kinase regulator, and the Fanconi-Anemia protein FANCM [25], promoting their degradation and checkpoint inactivation. Further, Plk1, Plk3 and Plk4 interact with and phosphorylate Chk2, the ortholog of Rad53 in human cells, likely influencing its activity [26]–[28]. Interestingly, yeast Cdc5 is phosphorylated and inhibited in a Mec1- and Rad53-dependent manner [29], and several studies indicate that in mammals Plk1 activity is inhibited by ATM/ATR-signaling in response to DNA damage [30]–[33]. Further, the DNA damage checkpoint regulates Plk1 protein stability in response to DNA damage in mitosis [34]. It was also shown that Aurora kinase A phosphorylates and re-activates Plk1 to promote recovery from DNA damage [35]. Altogether, these informations suggest that the DNA damage checkpoint inhibits Plk1, thus contributing to block cell cycle progression in response to DNA damage; however, the re-activation of Plk1 is a crucial event of a feedback regulatory loop in the inactivation of the DNA damage checkpoint during recovery and adaptation.
Therefore, the activity of Plks must be finely regulated during the DNA damage checkpoint response, and it is worth mentioning that the expression of a constitutively active Plk1 protein variant overrides the G2/M arrest induced by DNA damage [30]. Indeed, Plks are frequently overexpressed in tumor cells with uncontrolled proliferation and genome instability [36]–[39], and high level of Plk1 is predictive of a bad prognosis in several cancers [40]–[44].
To further characterize the functional link between Plks and the DNA damage checkpoint and, possibly, to understand why Plks are frequently overexpressed in cancer cells, we used budding yeast as a model system to study DNA damage related events in the presence of high levels of Cdc5.
Here, we show that overproduction of Cdc5 impairs the Mec1-signaling pathway in response to an inducible DSB lesion, altering phosphorylation of Ddc2, Rad9, Rad53 and other Mec1 targets. We also found that elevated levels of Cdc5 slow down DSB ends processing, although it does not prevent the formation of ssDNA, which triggers the recruitment of checkpoint factors. Consistently, we observed that overexpression of Cdc5 does not alter the loading of the apical Mec1 kinase checkpoint complex and recruitment of the checkpoint mediator Rad9, but surprisingly it physically interact with the checkpoint inhibitor Sae2, inducing its hyper-phosphorylation and an increased and persistent binding onto a DSB lesion.
We propose that high levels of polo kinase Cdc5 override Mec1-induced checkpoint response to DSB lesions, likely by regulating multiple factors, previously phosphorylated by CDK1, involved in both DSB processing and checkpoint signaling. Our work may represent a simple model to further understand why polo kinases are frequently overexpressed in cancer cells.
Results/Discussion
Elevated levels of Cdc5 override Mec1 signaling
DNA damage checkpoints represent a barrier to oncogenesis; in fact, loss of these surveillance mechanism is a characteristic of early tumor development [45]. Several evidences indicate that Plks are targets of the DNA damage checkpoint in all the eukaryotes [29]–, suggesting a functional model in which the DNA damage checkpoint inhibits Plks to maintain a cell cycle block at the metaphase to anaphase transition. Indeed, numerous cancer cells have been reported to display overexpression of Plks, and this may contribute to their transformed phenotype [36]–[39].
In budding yeast, overproduction of the polo kinase Cdc5 in cdc13-1 mutant cells with uncapped telomeres has been reported to override the checkpoint-dependent cell cycle block in the G2 phase of the cell cycle [46],[47]. We found that overproduction of Cdc5 impairs the replication checkpoint, which delays S phase in the presence of the alkylating agent MMS (methylmetane sulfonate, Figure 1A). Indeed, Figure 1A shows that MMS treated wild type cells accumulate in S phase for a very long period (1C<DNA<2C), while Cdc5 overproducing cells rapidly go through the replication phase and reach a G2/M DNA content (2C). Moreover, the DNA damage-induced phosphorylation of Rad53 is essentially abolished in Cdc5 overproducing cells treated with zeocin, an agent causing DSBs (Figure 1B).

We have to assume that, although the DNA damage checkpoint inhibits Cdc5 [29],[46], contribuiting to block cell cycle in the presence of DNA damage, when CDC5 is placed under the control of the GAL1 promoter, the DNA damage-induced inhibition on overproduced Cdc5 is not complete. This is likely due to the elevated Cdc5 levels, which are higher than the endogenous amount (see also Figure S1), leading to the override of the checkpoint response. Indeed, it was previously shown that the overproduction of Cdc5, which is a finely regulated protein [29], causes severe phenotypes during an unperturbed cell cycle [48]–[51].
In order to expand the analysis on the crosstalk between polo kinases and checkpoint pathways, and possibly to understand why overexpression of Plks is often found in tumor cells characterized by uncontrolled proliferation and genome instability, we analysed the effects of elevated Cdc5 levels on the DSB-induced checkpoint cascade in S. cerevisiae. We took advantage of a standard yeast genetic system (JKM background) in which one irreparable DSB can be induced at the MAT locus by expressing the site-specific HO nuclease [8]. We overexpressed wild-type CDC5 and the two cdc5-ad and cdc5-kd mutant alleles (adaptation-defective and kinase-dead alleles, respectively [51]) from the galactose-inducible promoter and examined Rad53 phosphorylation and in situ auto-phosphorylation activity, which are routinely used as markers of DNA damage checkpoint activation [52]. To prevent variations due to cell cycle differences, we first arrested cells with nocodazole in mitosis, a cell cycle stage in which the DSB-depended checkpoint can be fully activated [12], and subsequently added galactose to induce Cdc5 overproduction and HO-break formation, while maintaining the cell cycle block. Figure 2A shows the FACS profiles of the cell cultures. We observed that overproduction of Cdc5 impairs the accumulation of hyper-phosphorylated Rad53 forms and prevents Rad53 auto-phosphorylation activity in response to DSB formation (Figure 2B). Interestingly, overproduction of the protein variants Cdc5-kd or Cdc5-ad did not significantly interfere with Rad53 phosphorylation and activation, suggesting that the kinase activity of Cdc5 and its capacity to interact with specific target(s) are required to override the DSB-induced Rad53 activation.

In vertebrates, polo kinases regulate the DNA damage checkpoint response by affecting the signal transduction pathway at different levels; interestingly, Chk2, the homologue of Rad53 in human cells, interacts with and is phosphorylated by the polo kinases Plk1, Plk3 and Plk4 [26]–[28].
Therefore, we tested whether the overproduction of Cdc5 might override Rad53 activation by targeting directly the Rad53 protein and/or by acting on other upstream checkpoint factors.
We failed to co-immunoprecipitate Rad53 and Cdc5, when expressed at endogenous levels or by using the polo box of Cdc5 in a standard GST pull down assay; however, we retrieved Rad53 with overproduced Cdc5 (Figure S2). Considering such physical interaction, we analyzed how overproduction of Cdc5 might affect the events leading to full activation of Rad53, which involves a two steps-based mechanism: an in trans phosphorylation event mediated by PIKKs, followed by auto-phosphorylation [53]. In theory, Cdc5 might affect any of these events required to activate Rad53. We analysed the effect of Cdc5 overexpression on the PIKKs-dependent phosphorylation of Rad53 by taking advantage of the catalytically inactive rad53-K227A mutant. Such protein can be phosphorylated in trans by the upstream kinases, but does not undergo auto-phosphorylation in the presence of DNA damage [52], allowing us to separate and discriminate the two steps.
In nocodazole blocked cells, induction of a single irreparable HO cut induced Mec1-dependent phosphorylation of the Rad53-K227A protein variant (Figure 3A). As expected, the corresponding phosphorylated bands of Rad53-K227A protein were not visualized by western blot using the monoclonal antibody (Mab.F9) which is specific for the auto-phosphorylated and active Rad53 isoform [54]. Moreover, the same phospho-specific antibody did not significantly detect Rad53 in wild type cells responding to DSB when Cdc5 is overproduced, confirming the results of the in situ kinase assay (Figure 2B). A residual shifted band of Rad53, visualized in CDC5 overexpressing cells through the highly sensitive Mab.EL7 antibody (both in Figure 2B and Figure 3A, and in other figures below), could reflect low levels of Rad53 activation not detected by the antibody against the active form; this is consistent with the residual Rad53 activity in the in situ analysis in Figure 2B. In any case, it is unlikely that this remaining Rad53 activity is sufficient to maintain a full checkpoint response, since overproduction of Cdc5 functionally overrides the cell cycle block in the presence of DNA damage.
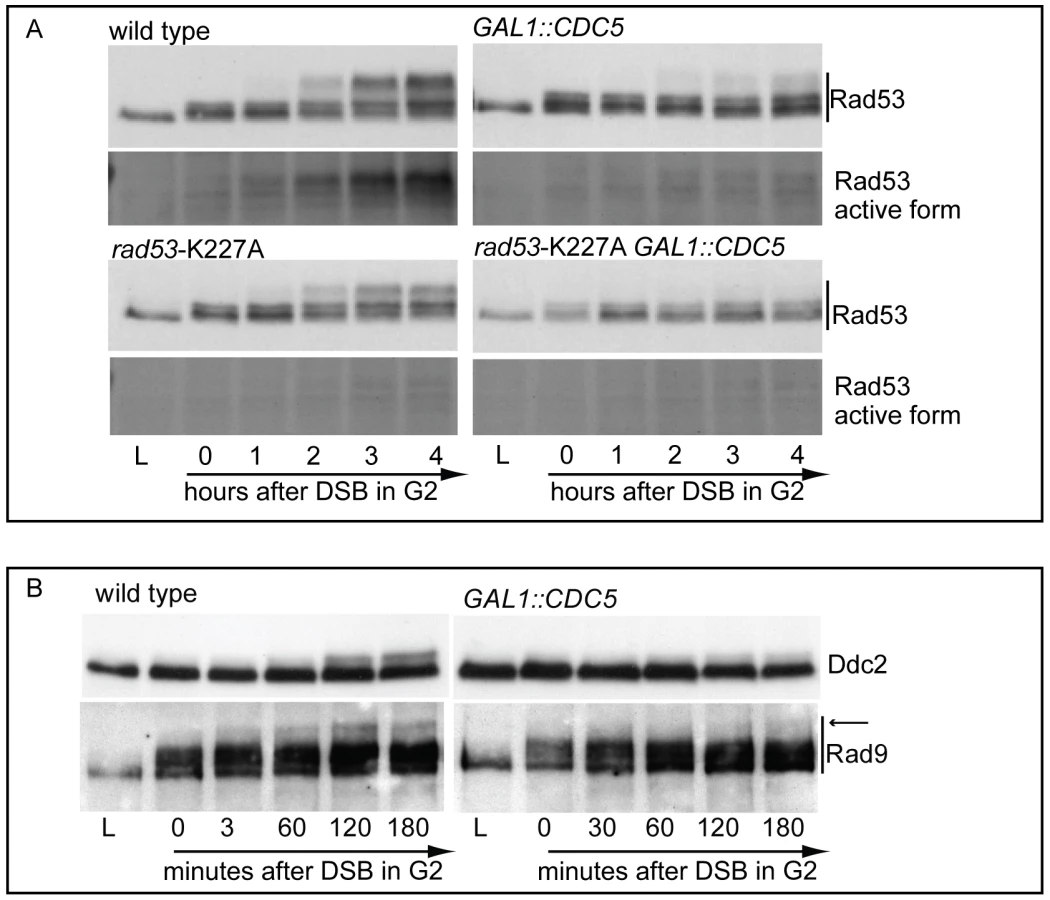
Significantly, Cdc5 overproduction abolished DSB-induced in trans phosphorylation of the Rad53-K227A variant (Figure 3A). This result rules out the hypothesis that Cdc5 may override the DSB-induced checkpoint acting only on the auto-phosphorylation step of Rad53 activation, and suggests that CDC5 overexpression likely impairs the Mec1-dependent in trans phosphorylation and activation of Rad53.
The residual Rad53 phosphorylation and activity in the presence of high levels of Cdc5 might suggest that the upstream Mec1 kinase, which is mainly responsible of the Rad53 activation in the presence of a single DSB in wild type cells [55], is strongly but not fully inhibited. Alternatively, Mec1 may still be functional as a kinase, but impaired in fully trans-activating Rad53. To test more directly the activity of the upstream kinase Mec1, we analysed the phosphorylation state of its interacting subunit Ddc2, the ortholog of human ATRIP, and that of the checkpoint mediator Rad9, which are known to be directly phosphorylated by Mec1 [1]. Cells were arrested with nocodazole and CDC5 overexpression and induction of a single unrepairable DSB were induced by galactose addition (Figure 3B). Western blot analysis indicate that phosphorylated isoforms of Ddc2 and hyper-phosphorylated Rad9 (indicated by the arrow in Figure 3) accumulated after the formation of the HO cut in wild type cells, as expected; however, overexpression of Cdc5 reduced the DSB-induced hyper-phosphorylated form of both Ddc2 and Rad9, suggesting that the activity of Mec1 kinase is strongly impaired in the presence of high level of Cdc5. A careful analysis of the blot shown in Figure 3B or in analogous experiments indicates that reduced levels of phosphorylated Rad9 isoforms are present in CDC5 overexpressing cells, suggesting that Mec1 could still retain a flebile activity toward Rad9 and Rad53, as discussed above. In addition, it is known that Rad9 is a target of multiple kinases [56] and we cannot rule out the possibility that the residual phosphorylation of Rad9 observed in cells with elevated levels of Cdc5 may be due to other kinase(-s), including Cdc5 itself.
Taken together the results shown in Figure 1, Figure 2, and Figure 3 indicate that Cdc5 activity overrides the DSB-induced checkpoint by influencing an early step of the Mec1 signaling pathway, likely reducing the functionality of Mec1 activity. However, it is possible that Cdc5 may target multiple substrates, including the Mec1 interactor Ddc2, the checkpoint mediator Rad9, whose role in promoting Mec1-to-Rad53 signaling is well established, and Rad53 itself, thus counteracting the checkpoint signaling pathway at several levels.
High levels of Cdc5 affect DSB resection
Robust Mec1 and Rad53 activation is not triggered by the DSB itself, but requires multiple interconnected events following the formation of the lesion, including the generation of nucleolytic-dependent 5′-to-3′processing of the DNA ends and recruitment of various DNA repair and checkpoint factors onto the long stretches of the generated ssDNA [4].
Therefore, we investigated whether Cdc5 may control Mec1 signaling by affecting DSB processing. We measured the kinetic of ssDNA formation after a single unrepairable DSB in cells overexpressing CDC5. Cells were arrested in mitosis, to prevent cell cycle-dependent effects on resection [57], and samples were collected at various time points after HO nuclease induction (Figure 4). The kinetic of production of ssDNA regions in genomic DNA was tested by the loss of restriction sites distal to the HO-cut site which leads to the accumulation of undigested ssDNA fragments detectable with a strand-specific probe after alkaline electrophoresis (see the scheme of the unprocessed and processed DNA locus in Figure 4A). CDC5 overexpressing cells reproducibly exhibited a slower DSB resection, measured by the kinetic of appearance of DNA fragments, which correlated with a reduced phosphorylation of Rad53 (Figure 4B–4D). However, we found that, although the kinetic of DSB ends resection was delayed, high levels of Cdc5 do not prevent the generation of a long ssDNA track (25 kb) which is required to repair the DSB in a specific yeast genetic background [14] by the single-strand annealing process (Figure S3).

We previously identified a role for the checkpoint mediator Rad9 in inhibiting the kinetic of DSB ends resection, likely by generating a non-permissive chromatin configuration around the DSB and/or interfering with the action of nucleases [58]. Therefore, we analyzed the Rad9 contribution in delaying DSB processing in CDC5 overexpressing cells. Wild-type or rad9Δ cells, with or without GAL1::CDC5, were arrested in mitosis by nocodazole treatment and the same experiment described in Figure 4B was performed. We found that the kinetic of appearance of ssDNA fragments was accelerated in rad9Δ strains, despite the high levels of Cdc5 kinase (Figure 5A and 5B). Moreover, the faster DSB resection in CDC5 overexpressing rad9Δ cells also correlated with a modest increase in Ddc2 phosphorylation (Figure 5C); however, the phosphorylated state of Ddc2 did not reach the same level found in wild-type and rad9Δ cells, suggesting that overproduction of Cdc5 impaired Mec1-dependent signaling also in a rad9Δ background. These results suggest that elevated levels of Cdc5 may slow down DSB processing through the action of the Rad9-dependent barrier on resection [58], likely targeting Rad9 itself or other factors involved in this mechanism. Interestingly, many of the proteins involved in DSB ends processing (i.e. Rad9, Dna2, Xrs2 and Sae2) are phosphorylated by CDK1 [59],[60] and inspection of their protein sequence reveals that they may be potential targets of Cdc5.

Hence, Cdc5 may influence the DSB response acting on multiple factors, affecting DSB processing and Mec1-signaling; moreover, the possibility that Cdc5 might specifically regulate Rad53 by influencing its interaction with the checkpoint mediator Rad9 cannot be excluded.
Recruitment of checkpoint factors in CDC5-overexpressing cells
Since high levels of Cdc5 did not prevent the generation of long ssDNA regions but inhibit Mec1-signaling, we tested, by chromatin immunoprecipitation (ChIP), whether overexpression of CDC5 affected the recruitment of checkpoint factors onto the HO-induced DSB lesion in nocodazole-arrested cells. Sheared chromatin from formaldehyde crosslinked cells taken at different time-points after galactose addition was immunoprecipitated to recover checkpoint proteins (i.e. Ddc2, Ddc1, Dpb11, Rad9) carrying the MYC or HA epitope tags at their carboxyl-terminal end. Quantitative multiplex PCR was then used to monitor co-immunoprecipitation of DNA fragments located either 66 kb centromere-proximal to the MAT locus (CON) or 1 kb away from the HO-cut site (DSB) (Figure 6A).

Ddc2 and Ddc1 association at the DSB was not significantly affected in CDC5 overexpressing cells blocked by nocodazole treatment (Figure 6B and 6C). The Mec1 interacting factor Ddc2 and Ddc1, one of three subunits of the stable PCNA-like 9-1-1 checkpoint complex, are recruited early onto a DSB lesion [61]–[63]. We, therefore, assume that Cdc5 overproduction does not prevent the recruitment of upstream checkpoint protein complexes onto damaged DNA. This observation also confirms that elevated levels of Cdc5, while delaying resection, do not prevent the generation of ssDNA (see Figure 4, Figure 5, and Figure S3) which is required for the recruitment of checkpoint factors [4].
Similarly, we found that overproduction of Cdc5 did not prevent the localization near the DSB of Dpb11 (Figure 6D), the yeast ortholog of TopBP1, which, together with the 9-1-1 complex, stimulates the Mec1 kinase activity [64].
Moreover, when we tested by ChIP analysis the binding of the checkpoint mediator Rad9, we found that also its localization onto the DSB was not altered in CDC5 overexpressing cells (Figure 6E).
Taken together, the ChIP analyses of checkpoint factors at a DSB site indicate that high levels of Cdc5 kinase do not significantly interfere with the binding of checkpoint proteins to a processed DSB.
We then tested the DSB binding of Sae2, which is a protein regulated by CDK1 [60] and PIKKs [65] after DNA damage and is involved in DSB processing [5] and checkpoint inactivation [66],[67]. Surprisingly, while in wild-type cells Sae2 loading was not significantly enriched at the HO-cut site (Figure 7A), likely because of its dynamic and transient binding to DSBs [67], Sae2 localization near the break greatly increased in CDC5 overexpressing cells (Figure 7A). To test whether Cdc5 may specifically target Sae2 influencing its binding onto DSBs, we analysed the level and modification of Sae2 by western blotting following DSB formation. In nocodazole-blocked cells, induction of the HO cut caused PIKKs-dependent phosphorylation of Sae2 at the same time-points at which Rad53 phosphorylation was observed (Figure 7B). Interestingly, although high levels of Cdc5 impair Rad53 phosphorylation, they seem to cause hyperphosphorylation of Sae2. Infact, in CDC5 overexpressing cells we observed the appearance of a ladder of slower migrating forms of Sae2 (Figure 7B), which are abolished by in vitro treatment with λ phosphatase (Figure 7C), indicating that they are due to phosphorylation events of Sae2. We then found that overproduction of Cdc5 induces Sae2 hyper-phosphorylation in untreated cells and in nocodazole-blocked cells without the HO-cut formation (Figures 7D), supporting the idea that Sae2 might be a direct target of Cdc5. Indeed, as mentioned above, Sae2 protein sequence reveals several sites that could be bound and/or phosphorylated by Cdc5 (Figure 8A). The C-terminus of Cdc5, like other Polo-like kinases, contains a phospho-serine/phospho-threonine binding domain called the Polo-box Domain (PBD) [19]. The PBD is known to bind Plk substrates after they have been “primed” by a preliminary phosphorylation by another protein kinase [19]. Interestingly, the putative PBD binding motif of Sae2 has been previously shown to be phosphorylated by CDK1 [60], making it a perfect candidate for mediating the interaction between Sae2 and Cdc5. Indeed, by a 2-hybrid assay we found that the PBD of Cdc5 interacts with Sae2 (Figure 8B), and a recombinant GST-PBD fusion protein, purified from E. coli, precipitated Sae2-3HA from yeast extracts (Figure 8C).

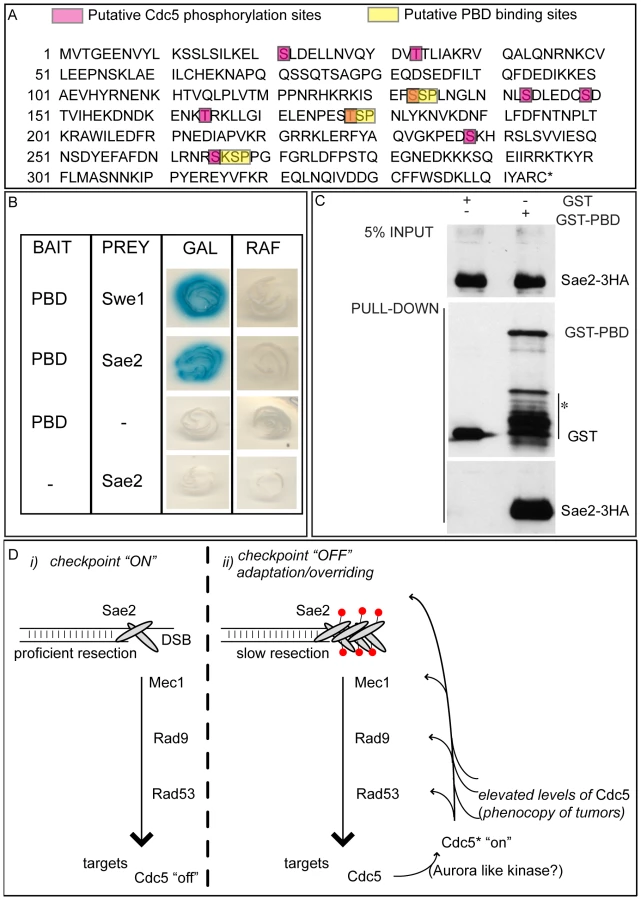
Taken together, the results shown in Figure 7 and Figure 8 indicate that Cdc5, through its PBD, interacts with Sae2, causing its hyper-phosphorylation and accumulation at the DSB (see also a model in Figure 8D). It is interesting to point out that CtIP, the functional ortholog of Sae2 in human cells, was found to be associated to chromatin following DNA damage and its chromatin binding is promoted by phosphorylation and ubiquitination [68]. Indeed, recent evidences indicate that CtIP and Ctp1 (the CtIP counterpart in S. pombe [69]), are recruited to DSB sites through their interaction with Nbs1 [70]–[72], a subunit of Mre11 complex, and BRCA1 [73],[74]. Moreover, CtIP is phosphorylated and regulated by CDK1 [74],[75]. In yeast, Sae2 is involved both in promoting an early step of DSB ends resection [5] and in inactivating checkpoint signaling during recovery and adaptation [66],[67], although the exact role of Sae2 in these processes is not yet fully understood. Interestingly, the overproduction of Sae2 also causes the overriding of the Mec1-signaling [66], while deletion of SAE2 gene prevents switching off of the checkpoint [65],[66].
One possible working model (Figure 8D), which needs to be verified, predicts that the increased and persistent binding of Sae2 to a DSB, induced by overproduction of Cdc5, may affect both DSB resection and Mec1-signaling. It is tempting to speculate that even physiological levels of Cdc5 may regulate Sae2 during recovery and adaptation, contributing to switch off the checkpoint signal. It is also possible that Sae2 is regulated by Cdc5 only when this kinase is expressed at elevated levels, leading to the checkpoint overriding. Indeed, such situation is frequently observed in cancer cells, when Plks are overexpressed [36]–[39], suggesting that what we found in yeast may represent a model for a pathological condition in human cells. Future works, requiring the analysis of sae2 mutations in the sites regulated by Cdc5, may help to discriminate between the two possibilities.
In conclusion, in the present study we further explored the role of the polo kinase Cdc5 in attenuating the DNA damage checkpoint in budding yeast. We found that overproduction of Cdc5 affects different parameters of the cellular response to an inducible DSB: i) it overrides Mec1 signaling and prevents the phosphorylation of various Mec1 targets (Rad53, Rad9, Ddc2); ii) it causes a slower resection of DSB ends in a RAD9-dependent manner; iii) it binds Sae2 protein, causing its hyper-phosphorylation and leading to its increased and persistent binding onto DSB.
The emerging scenario suggests that Cdc5 may target multiple factors involved in various aspects of the cellular response to DSB lesions and DNA damage checkpoint signaling. Indeed, Cdc5 is a fundamental regulator of cell cycle progression and targets many proteins throughout a normal cell cycle [20]. Most of the Cdc5 substrates are proteins previously phosphorylated by CDK1, which is the principal regulator of the DSB-induced response, regulating DSB processing, recombination and checkpoint signaling [57]. Here we found that high levels of Cdc5 separately affected Mec1 signaling and DSB processing, leading us to speculate that Cdc5 may regulate multiple targets in response to DNA damage, including factors phosphorylated by CDK1. In support of such hypothesis, Plks phosphorylate, in vertebrates, several proteins involved in various aspects of the DNA damage response, such as FANCM [25], Claspin [21]–[24], Chk2 [26]–[28], MCM5 [76], MCM7 [77] and others. Moreover, our findings on the functional role of Cdc5 in responding to a DSB in yeast rise the possibility that Plks may also regulate CtIP.
Recently, a proteome-wide screening led to the identification of novel Cdc5 targets in a normal cell cycle [20]; we believe that a similar approach is promising to identify Cdc5 targets regulated in response to DSBs. Good experimental evidence indicates that the functional role of Cdc5 in the DNA damage response is evolutionary conserved and the outputs of such a screening may provide important information for new cancer therapy strategies, targeting Plks and their substrates with specific tools.
Materials and Methods
Yeast strains and plasmids
Strains are listed in Table S1. All the strains were constructed during this study, and all were derivatives of JKM (MATα, hmldelta::ADE1, hmrdelta::ADE1 ade1-100, trp1delta::hisG, leu2-3, leu2-112, lys5, ura3-52, ade3::GAL::HO), with the exception of strain Y38, which was generated from strain Y5 (YMV80, matΔ::hisG1, hmlΔ::ADE, hmrΔ::ADE1, lys5, ura3-52, leu2::HOcs, ade3::GAL::HO, his-URA3-5′Δleu2-is4). To construct strains, standard genetic procedures for transformation and tetrad analysis were followed. Y38 and Y210 were obtained by integration of ApaI-digested plasmid pJC57 (pGAL1::CDC5-3HA) at the URA3 locus. Y215 was derived by integration of ApaI-digested pJC59 (pGAL1::CDC5-3myc) at URA3 locus. Y220 was obtained by integration of ApaI-digested plasmid pJC62 (pGAL1::cdc5-K110A-3HA) at URA3 locus. Y222 was obtained by integration of ApaI-digested plasmid pJC69 (pGAL1::cdc5-L251W-3HA) at URA3 locus. Deletions and tag fusions were generated by the one-step PCR system [78]. The yeast two-hybrid assay was performed using the B42/lexA system with strain EGY48 (Mata his3 ura3 trp1 6lexAOP-LEU2; lexAOP-lacZ reporter on plasmid pSH18-34) as the host strain [79]. Bait plasmid pEG202-PBD340–705 for the two-hybrid assay, expressing lexA fusion with polo box domain of Cdc5, was obtained by amplifying the corresponding coding sequence of CDC5 gene (aa 340 to 705) from genomic DNA and ligating the resulting fragment into pEG202 (kind gift from R. Brent). Prey plasmids pJG4-5-Swe1173–400 and pJG4-5-SAE2, expressing B42 activating domain fusions, were obtained by amplifying the corresponding coding sequence of SWE1 (aa 173 to 400) and SAE2 (full length) from genomic DNA and ligating the resulting fragments into pJG4-5.
Western blot analysis
The TCA protein extraction and the western blot procedures have been previously described [29]. Rad53, Rad9, Sae2-HA, Ddc2-HA, Ddc1-myc, Dpb11-myc, Cdc5-HA, Cdc5-myc were analysed using specific monoclonal or polyclonal antibodies: anti-Rad53 Mab.EL7 and Mab.F9 monoclonal [54], anti-HA 12CA5 monoclonal, anti-myc 9E10 monoclonal, anti-Rad9 polyclonal (a kind gift from N Lowndes's lab).
In situ auto-phosphorylation assay
It was performed as previously described [52].
Immunoprecipitation analysis
Yeast whole cell extracts were prepared by FastPrep (MP Biomedicals) in NP-40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mm Tris (pH 7.5), 1 mM dithiothreitol (DTT), 60 mM β-glycerophosphate, 1 mM NaVO3, cocktail proteases inhibitors (Roche)). HA-tagged proteins were immunoprecipitated using anti HA monoclonal antibody (12CA5) conjugated to protein G Agarose.
GST pulldown assay
GST and GST-PBD were induced in BL21 E. coli cells as previously described [80] and conjugated to glutathione-Sepharose 4B beads (GSH beads, Amersham). Yeast whole cell extracts, prepared as indicated above, were incubated with GST or GST-PBD GSH beads and rotated for 1 hour at 4°C. Samples were washed three times with NP-40 buffer, boiled in SDS-based sample buffer, and analyzed by Western blotting analysis.
In vitro dephosphorylation assay
Crude extracts were prepared as described [52], and resuspended in λ phosphatase buffer with or without 4000 U of λ phosphatase (Biolabs). Samples were incubated 30 min at 30°C and resuspended in Laemmli buffer.
Measurements of DNA resection and SSA at DSBs
Cells grown in YEP-raffinose 3% medium at 28°C to a concentration of 5×106 cells/ml were arrested with nocodazole (20µg/ml). A DSB was produced by adding 2% galactose and inducing the production of the HO endonuclease. The maintenance of the arrest was confirmed by FACS analysis and monitoring of nuclear division. Genomic DNA was isolated at intervals, and the loss of the 5′ ends of the HO-cleaved MAT locus was determined by Southern blotting [14],[81],[82]. To visualize the kinetics of resection, the graphs shown in Figure 4C and Figure 5B display, for each strain and for each ssDNA fragment (r1–r7), the time of the first appearance in the blot. In particular, since the appearance of a ssDNA fragment signal in the gel was due to the loss of the internal SspI sites, we represented the length of the minimal resection for each time point in the graph (see scheme in Figure 4A). All the experiments have repeated al least 3 times. In the corresponding figures, one representative example is shown with its graphic representation.
Chromatin immunoprecipitation analysis (ChIP)
ChIP analysis was performed as described previously [83],[84]. Multiplex PCRs were carried out by using primer pairs complementary to DNA sequences located 1 kb from the HO-cut site at MAT (DSB) and to DNA sequences located 66 kb from MAT (CON). Gel quantitation was determined by using the NIH Image program. The relative fold enrichments of DSB-bound protein were calculated as follow: [DSB_IP/CON_IP]/[DSB_input/CON_input], where IP and Input represent the amount of PCR product in the immunoprecipitates and in input samples before immunoprecipitation, respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HarrisonJC
HaberJE
2006 Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40 209 235
2. HarperJW
ElledgeSJ
2007 The DNA damage response: ten years after. Mol Cell 28 739 745
3. BranzeiD
FoianiM
2008 Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 9 297 308
4. LazzaroF
GiannattasioM
PudduF
GranataM
PellicioliA
2009 Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst) 8 1055 1067
5. MimitouEP
SymingtonLS
2009 Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 34 264 272
6. BartekJ
LukasJ
2007 DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 19 238 245
7. ClemensonC
Marsolier-KergoatMC
2009 DNA damage checkpoint inactivation: adaptation and recovery. DNA Repair (Amst) 8 1101 1109
8. LeeSE
PellicioliA
DemeterJ
VazeMP
GaschAP
2000 Arrest, adaptation, and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol 65 303 314
9. GalgoczyDJ
ToczyskiDP
2001 Checkpoint adaptation precedes spontaneous and damage-induced genomic instability in yeast. Mol Cell Biol 21 1710 1718
10. van VugtMA
MedemaRH
2004 Checkpoint adaptation and recovery: back with Polo after the break. Cell Cycle 3 1383 1386
11. van de WeerdtBC
MedemaRH
2006 Polo-like kinases: a team in control of the division. Cell Cycle 5 853 864
12. PellicioliA
LeeSE
LuccaC
FoianiM
HaberJE
2001 Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7 293 300
13. ToczyskiDP
GalgoczyDJ
HartwellLH
1997 CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90 1097 1106
14. VazeMB
PellicioliA
LeeSE
IraG
LiberiG
2002 Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 373 385
15. van VugtMA
BrasA
MedemaRH
2004 Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 15 799 811
16. SyljuasenRG
JensenS
BartekJ
LukasJ
2006 Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res 66 10253 10257
17. LiuX
LeiM
EriksonRL
2006 Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol 26 2093 2108
18. LaneHA
NiggEA
1996 Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol 135 1701 1713
19. LoweryDM
MohammadDH
EliaAE
YaffeMB
2004 The Polo-box domain: a molecular integrator of mitotic kinase cascades and Polo-like kinase function. Cell Cycle 3 128 131
20. SneadJL
SullivanM
LoweryDM
CohenMS
ZhangC
2007 A coupled chemical-genetic and bioinformatic approach to Polo-like kinase pathway exploration. Chem Biol 14 1261 1272
21. PeschiaroliA
DorrelloNV
GuardavaccaroD
VenereM
HalazonetisT
2006 SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 23 319 329
22. MamelyI
van VugtMA
SmitsVA
SempleJI
LemmensB
2006 Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol 16 1950 1955
23. MailandN
Bekker-JensenS
BartekJ
LukasJ
2006 Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 23 307 318
24. YooHY
KumagaiA
ShevchenkoA
DunphyWG
2004 Adaptation of a DNA replication checkpoint response depends upon inactivation of Claspin by the Polo-like kinase. Cell 117 575 588
25. KeeY
KimJM
D'AndreaAD
2009 Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev 23 555 560
26. Bahassi elM
ConnCW
MyerDL
HenniganRF
McGowanCH
2002 Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene 21 6633 6640
27. TsvetkovLM
TsekovaRT
XuX
SternDF
2005 The Plk1 Polo box domain mediates a cell cycle and DNA damage regulated interaction with Chk2. Cell Cycle 4 609 617
28. PetrinacS
GanuelasML
BonniS
NantaisJ
HudsonJW
2009 Polo-like kinase 4 phosphorylates Chk2. Cell Cycle 8 327 329
29. ChengL
HunkeL
HardyCF
1998 Cell cycle regulation of the Saccharomyces cerevisiae polo-like kinase cdc5p. Mol Cell Biol 18 7360 7370
30. SmitsVA
KlompmakerR
ArnaudL
RijksenG
NiggEA
2000 Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol 2 672 676
31. van VugtMA
SmitsVA
KlompmakerR
MedemaRH
2001 Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem 276 41656 41660
32. AndoK
OzakiT
YamamotoH
FuruyaK
HosodaM
2004 Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem 279 25549 25561
33. TsvetkovL
SternDF
2005 Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle 4 166 171
34. BassermannF
FrescasD
GuardavaccaroD
BusinoL
PeschiaroliA
2008 The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 134 256 267
35. MacurekL
LindqvistA
LimD
LampsonMA
KlompmakerR
2008 Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455 119 123
36. LuLY
YuX
2009 The balance of Polo-like kinase 1 in tumorigenesis. Cell Div 4 4
37. EckerdtF
YuanJ
StrebhardtK
2005 Polo-like kinases and oncogenesis. Oncogene 24 267 276
38. TakaiN
HamanakaR
YoshimatsuJ
MiyakawaI
2005 Polo-like kinases (Plks) and cancer. Oncogene 24 287 291
39. SmithMR
WilsonML
HamanakaR
ChaseD
KungH
1997 Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun 234 397 405
40. WeichertW
DenkertC
SchmidtM
GekelerV
WolfG
2004 Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer 90 815 821
41. TokumitsuY
MoriM
TanakaS
AkazawaK
NakanoS
1999 Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol 15 687 692
42. KnechtR
OberhauserC
StrebhardtK
2000 PLK (polo-like kinase), a new prognostic marker for oropharyngeal carcinomas. Int J Cancer 89 535 536
43. KneiselL
StrebhardtK
BerndA
WolterM
BinderA
2002 Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol 29 354 358
44. YamadaS
OhiraM
HorieH
AndoK
TakayasuH
2004 Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 23 5901 5911
45. BartekJ
LukasJ
BartkovaJ
2007 DNA damage response as an anti-cancer barrier: damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 6 2344 2347
46. SanchezY
BachantJ
WangH
HuF
LiuD
1999 Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286 1166 1171
47. HuF
WangY
LiuD
LiY
QinJ
2001 Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 107 655 665
48. SongS
GrenfellTZ
GarfieldS
EriksonRL
LeeKS
2000 Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Mol Cell Biol 20 286 298
49. SongS
LeeKS
2001 A novel function of Saccharomyces cerevisiae CDC5 in cytokinesis. J Cell Biol 152 451 469
50. BartholomewCR
WooSH
ChungYS
JonesC
HardyCF
2001 Cdc5 interacts with the Wee1 kinase in budding yeast. Mol Cell Biol 21 4949 4959
51. CharlesJF
JaspersenSL
Tinker-KulbergRL
HwangL
SzidonA
1998 The Polo-related kinase Cdc5 activates and is destroyed by the mitotic cyclin destruction machinery in S. cerevisiae. Curr Biol 8 497 507
52. PellicioliA
LuccaC
LiberiG
MariniF
LopesM
1999 Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18 6561 6572
53. PellicioliA
FoianiM
2005 Signal transduction: how rad53 kinase is activated. Curr Biol 15 R769 771
54. FioraniS
MimunG
CalecaL
PicciniD
PellicioliA
2008 Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7 493 499
55. MantieroD
ClericiM
LucchiniG
LongheseMP
2007 Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep 8 380 387
56. TohGW
LowndesNF
2003 Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans 31 242 246
57. IraG
PellicioliA
BalijjaA
WangX
FioraniS
2004 DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 1011 1017
58. LazzaroF
SapountziV
GranataM
PellicioliA
VazeM
2008 Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J 27 1502 1512
59. UbersaxJA
WoodburyEL
QuangPN
ParazM
BlethrowJD
2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
60. HuertasP
Cortes-LedesmaF
SartoriAA
AguileraA
JacksonSP
2008 CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455 689 692
61. KondoT
WakayamaT
NaikiT
MatsumotoK
SugimotoK
2001 Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 294 867 870
62. ZouL
ElledgeSJ
2003 Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300 1542 1548
63. LisbyM
BarlowJH
BurgessRC
RothsteinR
2004 Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118 699 713
64. Navadgi-PatilVM
BurgersPM
2009 A tale of two tails: Activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair (Amst)
65. BaroniE
ViscardiV
Cartagena-LirolaH
LucchiniG
LongheseMP
2004 The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol Cell Biol 24 4151 4165
66. ClericiM
MantieroD
LucchiniG
LongheseMP
2006 The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep 7 212 218
67. KimHS
VijayakumarS
RegerM
HarrisonJC
HaberJE
2008 Functional interactions between Sae2 and the Mre11 complex. Genetics 178 711 723
68. YuX
FuS
LaiM
BaerR
ChenJ
2006 BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 20 1721 1726
69. LimboO
ChahwanC
YamadaY
de BruinRA
WittenbergC
2007 Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 28 134 146
70. YuanJ
ChenJ
2009 N terminus of CtIP is critical for homologous recombination mediated double-strand break repair. J Biol Chem
71. LloydJ
ChapmanJR
ClappertonJA
HaireLF
HartsuikerE
2009 A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 139 100 111
72. WilliamsRS
DodsonGE
LimboO
YamadaY
WilliamsJS
2009 Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139 87 99
73. ChenL
NieveraCJ
LeeAY
WuX
2008 Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 283 7713 7720
74. YuX
ChenJ
2004 DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol 24 9478 9486
75. HuertasP
JacksonSP
2009 Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284 9558 9565
76. TrenzK
ErricoA
CostanzoV
2008 Plx1 is required for chromosomal DNA replication under stressful conditions. EMBO J 27 876 885
77. TsvetkovL
SternDF
2005 Interaction of chromatin-associated Plk1 and Mcm7. J Biol Chem 280 11943 11947
78. LongtineMS
McKenzieA3rd
DemariniDJ
ShahNG
WachA
1998 Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 953 961
79. GyurisJ
GolemisE
ChertkovH
BrentR
1993 Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75 791 803
80. MillerCT
GabrielseC
ChenYC
WeinreichM
2009 Cdc7p-dbf4p regulates mitotic exit by inhibiting Polo kinase. PLoS Genet 5 e1000498 doi:10.1371/journal.pgen.1000498
81. LeeSE
MooreJK
HolmesA
UmezuK
KolodnerRD
1998 Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94 399 409
82. ClericiM
MantieroD
LucchiniG
LongheseMP
2005 The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem 280 38631 38638
83. ViscardiV
BonettiD
Cartagena-LirolaH
LucchiniG
LongheseMP
2007 MRX-dependent DNA damage response to short telomeres. Mol Biol Cell 18 3047 3058
84. ClericiM
MantieroD
GueriniI
LucchiniG
LongheseMP
2008 The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep 9 810 818
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2010 Číslo 1
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
Najčítanejšie v tomto čísle
- A Major Role of the RecFOR Pathway in DNA Double-Strand-Break Repair through ESDSA in
- Kidney Development in the Absence of and Requires
- The Werner Syndrome Protein Functions Upstream of ATR and ATM in Response to DNA Replication Inhibition and Double-Strand DNA Breaks
- Alternative Epigenetic Chromatin States of Polycomb Target Genes