
Imunopatogeneze sepse
Immunopathogenenis of sepsis
Sepsis represents a major unresolved problem of current medicine. The important characteristic of sepsis is the interaction between the two subjects – the macro and the microorganism. The concept of a hyperinflammatory syndrome, dominant for two decades, has been challenged and our current understanding is that sepsis represents a dynamic syndrome characterized by many often antagonistic phenomena. Inflammation which characterizes sepsis does not act as a primary physiological compensatory mechanism but rather oscillates between the phases of a hyperinflammatory response and anergy or immunoparalysis. Understanding the pathogenesis of sepsis means understanding the immunopathological processes that characterize the interaction between the macro and the microorganism. The characteristics of the macroorganism - the genetic predisposition, the role of the innate and adaptive immunity systems, the “high“ or “low“ host response concerning the intensity of inflammation – form one side of the coin. The role of the microorganism is also important. The different ability of the species to produce pro - and antiinflammatory cytokines, their role in the innate immunity system and their different ability to escape the surveillance of the immune system form the second part in the pathogenesis of sepsis. The outcome of a patient is therefore the result of a very heterogenous and dynamic set of interactions and these complicated interactions have not been fully elucidated and understood yet.
Key words:
sepsis – macroorganism – microorganism – inflammation – innate immunity – immunoparalysis – apoptosis
Authors:
Průcha Miroslav 1; Fedora Michal 2
Authors‘ workplace:
Oddělení klinické biochemie, hematologie a imunologie, Nemocnice Na Homolce, Praha
1; Klinika dětské anesteziologie a resuscitace LF MU a Fakultní nemocnice Brno
2
Published in:
Anest. intenziv. Med., 19, 2008, č. 5, s. 269-278
Category:
Intesive Care Medicine - Review Article
Overview
Sepse představuje jeden z velkých, dosud nevyřešených problémů současné medicíny. Zásadní charakteristikou sepse je interakce dvou subjektů – makro - a mikroorganismu. Koncepce hyperzánětlivé odpovědi, dlouhá léta dominantní, byla v průběhu posledních let změněna v tom smyslu, že sepse představuje dynamický proces, který je charakterizován řadou často antagonistických jevů. Zánět, charakterizující sepsi, zde nenaplňuje svou primárně kompenzační fyziologickou roli a buď na jedné straně přestřeluje do fáze hyperzánětlivé odpovědi, nebo na straně druhé do anergie – imunoparalýzy. Objasnění patogeneze sepse představuje pochopení imunopatologických procesů, které charakterizují interakci makro - a mikroorganismu. Charakteristika makroorganismu – jeho genetické predispozice, úloha vrozeného a adaptačního systému imunity s charakteristikou pacienta jako „high“ nebo „low“ respondéra při rozvoji zánětu, popis výkonných efektorových mechanismů imunity s amplifikací signálu pro humorální a buněčnou imunitu, úloha apoptózy a nekrózy – tvoří jednu stranu mince. Důležitá je také úloha mikroorganismu. Rozdíly jednotlivých infekčních agens ve schopnosti indukce pro - a protizánětlivých cytokinů, jejich fyziologická role v přirozené antiinfekční imunitě, rozdílná schopnost unikat z dozoru imunitního systému, tvoří druhou důležitou stránku, která se uplatňuje v patogenezi sepse. Klinický výsledek u pacienta se sepsí je důsledkem této interakce, která je nesmírně heterogenní, dynamická a jejíž plné objasnění je dosud nevyřešeno.
Klíčová slova:
sepse – makroorganismus – mikroorganismus – zánět – vrozená imunita – imunoparalýza – apoptóza
Úvod
Sepse představuje jeden z největších současných problémů medicíny. Během posledních dvou desetiletí incidence sepse stoupá, ve Spojených státech připadá 240 případů sepse na 100 000 obyvatel [1], na léčbu se vydá více než 16 miliard dolarů. Česká jednodenní studie o prevalenci sepse z roku 2003 prokázala prevalenci sepse 34,2% a těžké sepse 16,8% [2]. Z práce publikované v roce 2001 [3], která zahrnula 847 nemocnic a přes 6,6 milionu pacientů, vyplývá, že incidence těžké sepse v USA je 3,0/1000 obyvatel a 2,26/100 propuštění z nemocnice. Incidence se zvyšuje s věkem více než stonásobně (0,2/1000 dětí a 26,2/1000 u dospělých nad 85 let). Počet hospitalizací s těžkou sepsí se v letech 1993–2003 téměř zdvojnásobil, počet pacientů s těžkou sepsí mezi všemi septickými pacienty kontinuálně narůstá a představuje 43,8 % všech pacientů se sepsí [4].Problémem zůstává vysoká mortalita sepse (28,6 %) i těžké sepse (37,8 %). Mortalita sepse stoupá s věkem (od 10 % u dětí až 38,4 % u dospělých nad 85 let) a je srovnatelná s mortalitou akutního infarktu myokardu [5]. Mortalita těžké sepse vyjádřená k počtu obyvatel dokonce stoupá: z 30,3/100000 obyvatel v roce 1993 na 49,7/100000 obyvatel v roce 2003. Ještě nepříznivější data ukazuje analýza pacientů se septickým šokem [6]. Jeho incidence je 58% u pacientů s těžkou sepsí, což ve Spojených státech představuje cca 436 tisíc pacientů ročně. Mortalita septického šoku sice postupně klesá, přesto zůstává i v dnešní době neakceptovatelně vysoká – téměř 50%.
V patogenezi sepse se významným způsobem uplatňují dysregulace mechanismů vrozené i adaptivní imunity. Jednou z úloh imunitního systému je zabezpečení homeostázy. Mechanismy vrozené a adaptivní imunity zabezpečují odpověď makroorganismu na infekční agens. V případě sepse je tato homeostáza podstatným způsobem narušena. Lokalizovaná zánětlivá odpověď přerůstá do odpovědi systémové, která je výrazem selhání imunologických kompenzačních mechanismů. Dysbalance, respektive nedostatečnost imunologických mechanismů, je jedním z důležitých faktorů v patogenezi sepse. Tato práce sumarizuje současné poznatky o imunopatogenetických mechanismech sepse.
Definice sepse, SIRS a PIRO systému
Jedním z důležitých mezníků v chápání etiopatogeneze sepse byla konsenzuální konference v roce 1991 s definováním pojmů: syndrom systémové zánětlivé odpovědi (SIRS), sepse, těžká sepse, septický šok, bakterémie [7]. Zásadním přelomem byla změna hodnocení odpovědi makrorganismu na podnět infekčního i neinfekčního charakteru. Konkrétním vyjádřením byla definice syndromu systémové zánětlivé odpovědi – SIRS. Představovala nové chápání interakcí pro - a protizánětlivých mechanismů a mediátorů uplatňujících se v rámci odpovědi makroorganismu na infekci, trauma, závažný operační výkon. Nový termín, syndrom systémové zánětlivé odpovědi, zahrnoval širokou etiologickou škálu: kromě infekce také neinfekční příčiny systémové zánětlivé odpovědi – popáleniny, operační trauma, pankreatitidu a další. Definice SIRS nabídla koncept účasti a významu endogenních zánětlivých mediátorů u syndromu zánětlivé odpovědi infekční i neinfekční etiologie.
PIRO koncept
Klinická praxe ukázala, že kritéria SIRS mají vysokou senzitivitu, ale příliš nízkou specificitu. Skupiny pacientů definované podle konsenzu z roku 1991 byly velice heterogenní a této heterogenitě se přičítal vliv na špatné klinické výsledky s použitím imunomodulační terapie. Vznikla proto snaha tuto heterogenitu odstranit, přičemž východiskem se staly nové metodologie s detekcí nových biomarkerů, jakými jsou např. prokalcitonin, studování genetických polymorfismů atd. Cílem je rozšířit diagnostické možností u pacientů se sepsí a jejich staging, jako je tomu např. u onkologických nemocných a používaného TNM systému. V roce 2001 byla přijata tzv. PIRO klasifikace [8]. PIRO systém znamená: P – Predisposition, I – Infection, R – Response a O – Organ dysfunction.
Mechanismy rozvoje SIRS a sepse
Syndrom systémové zánětlivé odpovědi i sepse je charakterizován zánětem a poruchou homeostázy. V obou případech se jedná o komplex reakcí vzniklý jako odpovědˇ na porušení integrity organismu. Mechanismy zánětlivé reakce jsou v případě syndromu systémové zánětlivé odpovědi neinfekční etiologie i sepse identické, rozdíl je v (ne)přítomnosti infekce. Patogeneze uvedených stavů je významným způsobem spojena s poruchou imunitních funkcí na úrovni humorální i buňkami zprostředkované imunity. Zahrnuje systémovou odpověď k infekci nebo neinfekčním podnětům, při které monocyto-makrofágový systém produkuje velké množství pro - a protizánětlivých cytokinů – tumor nekrotizující faktor-α (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-10 (IL-10) a další [9, 10, 11, 12].Sepse i SIRS jsou na svém počátku charakterizovány zvýšením koncentrace zánětlivých mediátorů v krvi. Souběžně dochází ke kompenzační protizánětlivé imunosupresivní odpovědi.
V patogenezi rozvoje sepse a SIRS se uvažuje o tzv. two-hit hypotéze. Při prvním zásahu se uplatňuje prvotní inzult (chirurgický výkon, trauma, hypoxie, šok) s následným poškozením buněčných struktur. Zánětlivá odpověď má kompenzační charakter. Druhým zásahem pak může být opakovaný chirurgický výkon, přechodná bakterémie, což má za důsledek dysbalanci kompenzační zánětlivé odpovědi s přechodem do autoagresivní destrukce. Mechanismy adaptivní imunity, které se zde uplatňují, jsou zprostředkovány především buňkami zprostředkovanou imunitou. Aktivované CD4+ T lymfocyty se dělí na dvě podskupiny s antagonistickým profilem produkovaných cytokinů – typ TH1 produkující zejména TNF-α, interferon-γ a interleukin-2 (prozánětlivé) a typ TH2 s produkcí IL-4 a IL-10 (protizánětlivé). IFN-γ a TNF-α produkované TH1 buňkami zvyšují schopnost fagocytů zabíjet infekční agens, zatímco IL-4 a IL-10 produkované TH2 buňkami podporují zejména humorální odpověď proti extracelulárním patogenům. Faktory, které rozhodují o diferenciaci T buněk, nejsou zcela známy, ale v případě infekce je důležitý typ patogenu, množství infekčního inokula a lokalizace infekce [13]. Důležitým faktorem je také poměr koncentrací IL-12 a IL-4. IL-12 je produkován makrofágy a dendritickými buňkami, IL-4 bazofily a mastocyty. Převažující typ mediátorové produkce je důležitý pro dominanci pro - nebo protizánětlivé odpovědi. Její intenzita rozhoduje rovněž o tzv. kompartmentalizaci imunitní odpovědi, zda bude zánětlivá odpověď lokalizovaná nebo systémová. Také stresová odpověď se svými mechanismy hraje důležitou roli v navození imunosuprese nejen při infekci, ale také po traumatu nebo krevní ztrátě. Regulační role osy hypothalamus-hypofýza–nadledvinka se stimulací produkce kortikosteroidů je dobře známa. Nedávné práce prokázaly vliv autonomního nervstva (sympatiku a parasympatiku) na imunitní odpověď [14, 15].Neuroendokrinní systém tak reguluje prozánětlivou roli monocytů a makrofágů (produkce TNF-α) a antigen prezentující funkci přímo i nepřímo indukcí imunomodulačních cytokinů, např. IL-10. Účinkem jeho působení dochází k deaktivaci TH1 buněk. Tyto interakce jsou důležité v zabránění excesivní zánětlivé reakci. Pokud se ale zvyšuje intenzita zánětlivé reakce – při masivní infekci, traumatu – dochází k výraznější aktivaci stresové osy, která vede k vyšší imunosupresi a tím ke zvýšenému riziku systémové infekce. Vysoké koncentrace kortikosteroidů a katecholaminů vedou ke zvýšené apoptóze lymfocytů a snížení funkčnosti adaptivních mechanismů imunity. Anergie je stav neodpovídavosti na antigen. T buňky jsou anergní, pokud nejsou schopny proliferace nebo sekrece cytokinů po podání antigenů. Heidecke et al. zjistili, že snížení TH1 funkce bez zvýšení TH2 cytokinové produkce odpovídá stavu anergie [16]. Tato snížená funkce T lymfocytů korelovala s mortalitou. Pacienti s traumatem, sepsí a popálení mají snížené počty cirkulujících T lymfocytů a jsou anergní [17]. Velké množství lymfocytů a epiteliálních buněk střeva hyne během sepse mechanismem apoptózy [18, 19]. Jedním z mechanismů je stresem indukované uvolnění glukokortikosteroidů [20]. Apoptické buňky navozují anergii a protizánětlivou cytokinovou produkci, zatímco buňky nekrotické působí imunostimulačně a zvyšují obranyschopnost organismu [21, 22]. Kombinace uvedených faktorů tak navozuje imunosupresi charakteristickou pro pacienty se sepsí – tabulka 1.

Zánět jako hlavní kompenzační mechanismus systému vrozené imunity a hlavní patogenetický mechanismus sepse
Hlavním patogenním mechanismem u pacientů se sepsí je zánět. Zánět představuje fyziologický kompenzační mechanismus, který propojuje systém vrozené a získané imunity, a jeho primární úlohou je zachování homeostázy, odstranění inzultů, které tuto homeostázu narušují. V případě sepse je tímto inzultem infekce. Pokud je schopnost zánětlivých mechanismů zachovat homeostázu narušena, nebo v důsledku predispozic makroorganismu reagovat na inzult odlišným způsobem – genetické polymorfismy genů pro tvorbu cytokinů, např. zánět – se stává ve svém důsledku makroorganismus poškozující. Strategie léčby sepse je založena mimo jiné na ovlivnění intenzity zánětu (rekombinantní protein C, monoklonální protilátky proti mediátorům – TNF-α). V této souvislosti je nutné zmínit výsledky studie Eichackera et al., která zkoumala souvislost mezi účinností protizánětlivé léčby u sepse a závažností klinického stavu. Provedená metaanalýza experimentálních a klinických studií prokázala, že účinnost protizánětlivé terapie závisí na závažnosti klinického stavu. Čím závažnější byl klinický stav, tím lepší byl efekt protizánětlivé terapie [23]. Práce Maciase tuto závislost neprokázala [24], ale aktuální klinické studie potvrzují spíše závěry Eichackera [25].
Zánětlivá odpověď při sepsi
Imunitní odpověd pacientů se sepsí je charakterizována hyperzánětlivou systémovou odpovědí nebo stavem imunoparalýzy, který charakterizuje nízká exprese HLA-DR na monocytech a snížená produkce TNF-αex vivo (z plné krve) po stimulaci lipopolysacharidem [26]. Hyperzánětlivou fázi charakterizuje vysoká produkce cytokinů – TNF-α, migrace inhibujícího faktoru (MIF) a (HMGB-I) proteinu (high-mobility group box I) [27, 28]. Obě fáze se vyskytují časově nezávisle na sobě [29]. Prognostický význam těchto parametrů byl opakovaně doložen v experimentálních a klinických studiích [30, 31]. Důležitou roli v procesu navození imunoparalýzy hraje IL-10, který suprimuje expresi znaků II. třídy MHC a touto cestou inhibuje antigen-specifickou proliferaci T lymfocytů. Mimoto inhibuje produkci interferonu-γ lymfocyty mechanismem závislým na monocytech [32] a produkci prozánětlivých cytokinů a chemokinů aktivovanými monocyty [33]. Jeho komplexní role z něj činí potencionální cíl pro imunoterapii sepse [34].
Imunoparalýza v procesu sepse
Exprese MHC znaků II. třídy na monocytech hraje důležitou roli při imunoregulačních procesech [35]. Němečtí autoři popsali souvislost mezi nízkou expresí HLA-DR znaku na CD14 pozitivních buňkách a:
- sníženou antigenní prezentací: Monocyty od pacientů s expresí HLA-DR na monocytech pod 30 % nebyly schopny prezentovat tetanický toxoid nebo anti-CD3 monoklonální protilátku HLA alogenním lymfocytům od zdravých dárců. Při dodání monocytů od zdravých jedinců se navodila již normální odpověď. Monocyty od septických pacientů s expresí HLA-DR > 40% byly schopny prezentace v obou případech [36];
- sníženou schopností vytvářet reaktivní kyslíkové radikály [37];
- alterovanou produkcí cytokinů po podání lipopolysacharidu [38, 39];
- zvýšenou letalitou těchto pacientů.
Provedené studie demonstrovaly skutečnost, že někteří pacienti se syndromem sepse přecházejí z fáze hyperzánětlivé odpovědi do fáze, kdy převažuje naopak odpověď protizánětlivá, charakterizovaná nízkou expresí HLA-DR na monocytech a sníženou produkcí prozánětlivých cytokinů – TNF-α, IL-1, IL-6, IL-8. Tato protizánětlivá fáze je nazývána CARS (compensatory anti-inflammatory response syndrom) a může přejít až do stavu imunoparalýzy. Protizánětlivá odpověď zahrnuje produkci: IL-4, IL-10, IL-11 a IL-13, transformujícího růstového faktoru-β (TGF-β), kolonie stimulujících faktorů – G-CSF a GM-CSF, solubilní formu receptorů pro TNF-α a antagonisty receptoru pro IL-1 (IL-1ra) [40, 41, 42].Tito pacienti jsou charakterizováni sníženou kapacitou monocytů produkovat TNF-αex vivo po stimulaci lipopolysacharidem, sníženou expresí HLA-DR znaku na monocytech a sníženou kapacitou antigen prezentujících buněk. Snížená exprese HLA-DR je přitom významným prediktivním parametrem pro konečný klinický výsledek u pacientů se sepsí [43, 44]. Byl zjištěn cytokin zodpovědný za deaktivaci monocytů. Jedná se o IL-10. Prokázala se časová souvislost mezi dynamikou IL-10 a TNF-α [45, 46]. IL-10 působí imunosupresivně na expresi znaků II. třídy MHC a touto cestou inhibuje antigen-specifickou proliferaci T lymfocytů. Mimoto inhibuje produkci interferonu-γ lymfocyty mechanismem závislým na monocytech [47] a produkci prozánětlivých cytokinů a chemokinů aktivovanými monocyty [48]. Dalším cytokinem, uplatňujícím se v navození imunosuprese u septických pacientů, je TGF-β (transforming growth faktor-β) produkovaný aktivovanými makrofágy. TGF-β snižuje produkci cytokinů monocyty, expresi MHC znaků II. třídy a inhibuje cytokiny indukovanou aktivaci makrofágů [49, 50, 51]. Také byly uveřejněny studie, které nepotvrdily prediktivní význam exprese HLA-DR na monocytech pro prognózu nemocných [52, 53], převažuje však názor, že tento fenomén je z hlediska monitorování imunokompetence důležitým prediktivním markerem s vazbou na výslednou mortalitu.
Mikroorganismy a jejich vliv na imunitní a koagulační systém
Sepse představuje systémovou zánětlivou reakci organismu na infekci. Etiologicky se uplatňují bakteriální, virová, mykotická nebo parazitární agens. U gramnegativních bakterií je základní stavební složkou endotoxin, respektive lipid A, který je odpovědný za patogenetické účinky. Braude demonstroval, že endotoxémie může komplikovat lokální gramnegativní infekci a protilátky proti endotoxinu zeslabují jeho systémové účinky [54]. Plazmatická koncentrace endotoxinu koreluje s rozvojem MODS a letalitou. Příčinou endotoxémie může být bakteriální translokace, při které dochází k prostupu mikrobů a jejich produktů přes slizniční bariéru [55, 56]. Dalším důležitým faktorem, uplatňujícím se při navození translokace střevní flóry, je dysfunkce retikulo-endotelového systému, zejména jater [57]. Bakteriální translokace a její spojení se vznikem SIRS a sepse se považují za prokázané [58, 59]. Závislost bakteriální translokace na faktorech, jako je imunosuprese, nalezla oporu v experimentálních i klinických studiích. Zvýšená bakteriální translokace byla prokázána u athymických myší [60], závislost na provedeném chirurgickém výkonu při střevní obstrukci zase na lidském modelu [61]. Podobné účinky jako endotoxin mají struktury grampozitivních bakterií – teichoová a lipoteichoová kyselina, kapsulární lipopolysacharid a skupinově specifické karbohydráty. Jejich role u sepse však zůstává nejasná, neboť nebyla doložena přesvědčivá data o jejich přítomnosti v cirkulaci v průběhu sepse. Většina patogenetických účinků u grampozitivních bakterií je výsledkem působení exotoxinů. Tyto exotoxiny mohou působit také jako superantigeny. Superantigeny se váží přímo na MHC antigeny II. třídy exprimované na antigen prezentujících buňkách a reagují přímo s T buněčnými receptory (TCR). Příkladem je syndrom toxického šoku způsobený toxinem 1, který produkuje Staphylococcus aureus.
Účinkem výše zmíněných bakteriálních struktur na leukocyty a buňky parenchymatózních orgánů dochází k produkci prozánětlivých cytokinů. Dva hlavní cytokiny jsou důležité v rozvinutí zánětlivé kaskády indukované endotoxinem – TNF-α a IL-1β [41, 62]. Tyto cytokiny stimulují imunitní odpověď, která mj. zahrnuje: uvolnění cytokinů, aktivizaci metabolismu kyseliny arachidonové, expresi integrinů, aktivaci komplementu, produkci oxidu dusnatého. Pro navození imunitní odpovědi u gramnegativní infekce je nezbytná vazba LPS k receptoru monocytárních fagocytů – molekule CD14, což je 53kDa glykoproteinový receptor exprimovaný na povrchu myelomonocytárních buněk. Kromě této buněčné formy existují dvě solubilní formy CD14, které prostřednictvím vazby k LPS indukují rozvoj zánětlivé kaskády. Proteinem, bezprostředně odpovědným za vazbu LPS k molekule CD14, je LPS - -vázající protein (LBP), který je přítomen v plazmě. LPS uvolněný z gramnegativních bakterií je ve formě agregátů. Působením LBP je LPS transformován na monomery a tyto monoméry jsou pak neutralizovány vazbou na lipoproteiny nebo se vážou na buňce exprimovanou a solubilní formou molekuly CD14. Vznik této vazby vede k produkci prozánětlivých cytokinů. Solubilní forma CD14 (sCD14) umožňuje transport LPS k lipoproteinům vysoké denzity (HDL) s následnou neutralizací LPS. V dalším průběhu odpovědi se specificky uplatňuje receptor z rodiny TLR (toll-like receptors), konkrétně TLR-4 [63]. CD14 molekula se rovněž uplatňuje jako receptor pro peptidoglykan, který je komponentou buněčné stěny grampozitivních bakterií [64]. Stejná závislost byla prokázána pro indukci produkce TNF-α u hemoragického šoku [65]. Grampozitivní i gramnegativní bakterie i další patogeny disponují molekulárními strukturami, které jsou hlavními „terči“ při rozpoznání systémemu vrozené imunity. Tyto molekuly označujeme jako „s patogenem asociované molekulární vzory“ – PAMPs (pathogen-associated molecular patterns) [66]. Těmito strukturami je u gramnegativních bakterií lipid A, u grampozitivních bakterií peptidoglykan a teichoová kyselina. Dvouvláknová RNA je typická pro RNA viry, manany jsou charakteristické pro kvasinky. V systému vrozené imunity se při jejich rozpoznání uplatňují jako antigen prezentující buňky (APC) makrofágy a dendritické buňky (DB), které je pak předkládají T lymfocytům. Oba typy antigen prezentujících buněk disponují „receptory rozpoznávání“ – PRRs (pattern recognition receptors), které se na tyto struktury vážou. Mezi tyto receptory patří kolektiny, CD14, TLRs, manózu vázající protein, sérový amyloid P, receptory pro komplement, C-reaktivní protein, Fc receptory, CD11b,c/CD18 a DEC 205. Důsledkem těchto vazeb je přenos signálu prostřednictvím MYD88 (protein myeloid differentiation factor 88) a NFκB (nuclear factor κB) do nitra hostitelské buňky, což vede k transkripci genů pro produkci cytokinů – IL-1 a IL-12, TNF-α a B7 [66, 67, 68, 69].
Primární úlohou zánětlivé odpovědi je lokalizace poškození. Uvolnění zánětlivých mediátorů – leukotrienů, složek komplementu, cytokinů a tvorba antigen-protilátkových komplexů způsobuje akumulaci neutrofilů a monocytů v místě poškození. Monocyty a neutrofily lokálně uvolňují množství prozánětlivých cytokinů – – TNF-α, IL-1, IL-2, IL-6, interferon-γ a další. Vedle produkce prozánětlivých cytokinů se kompenzačně uplatňuje protizánětlivá odpověď, kterou představuje produkce IL-4, IL-10, IL-11, IL-13, solubilních TNF-α receptorů. Neutrofily, které fungují jako fagocyty, jsou ale také hlavní příčinou orgánového poškození. Vlivem zánětlivých cytokinů produkovaných neutrofily se na povrchu endotelií poškozených tkání exprimují adhezivní molekuly. Proces zahrnuje primární adhezi leukocytů zprostředkovanou selektiny L, E a P, které jsou přítomny na leukocytech, endoteliích a destičkách. Prvotní adhezivní interakce pohyb neutrofilů zpomalí, v další fázi dochází k vazbě leukocytárních integrinů na povrchový glykoprotein endoteliálních buněk ICAM-1 (intercellular adhesion molecule-1). Monocyty přilínají na cévní stěnu prostřednictvím β1-integrinů. Protějškem na endoteliích je molekula VCAM-1 (vascular adhesion molecule). Po pevném zachycení na povrchu endotelií neutrofily cévu opustí a přejdou do tkáně. Neutrofily v místě poškození odstraní mikroorganismy, eventuálně jiné cizorodé částice, a během krátké doby hynou. Samy jsou odstraněny dalšími fagocytujícími buňkami, zejména makrofágy. Přehled nejdůležitějších zánětlivých mediátorů je v tabulce 2.
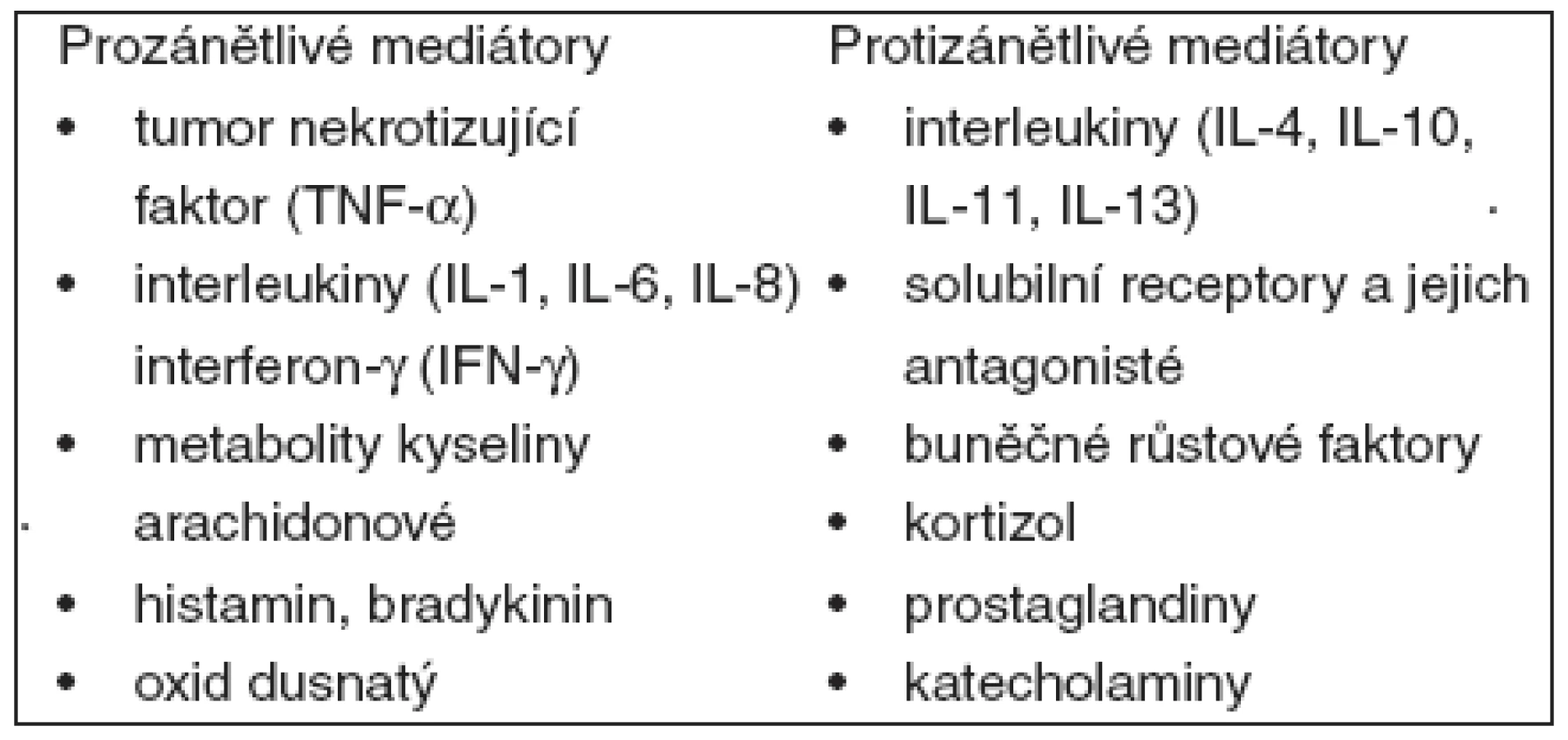
Pokud nedojde k lokalizaci etiologického agens poranění, nastává progrese prozánětlivé odpovědi s masivní produkcí prozánětlivých mediátorů, která je příčinou klinické manifestace syndromu systémové zánětlivé odpovědi. Cytokiny produkované u septických pacientů aktivují jejich koagulační systém [70, 71]. Klíčovou roli má v koagulační kaskádě tkáňový faktor (TF), který je aktivován TNF-α, IL-2, IL-6. Působení cytokinů vede k redukci fibrinolýzy, poklesu proteinu C a antitrombinu III. Aktivovaný protein C inhibuje faktory Va, VIIa a inhibitor aktivátoru plazminogenu (PAI-1). Antitrombin III inhibuje extrinsické koagulační faktory Xa, XIa, IIa a plazmin. Progredující prokoagulační stav akcentuje zánětlivou odpověď při sepsi. Významnou roli v další progresi má dysfunkce endotelu. Působením TNF-α, IL-1 a dalších prozánětlivých mediátorů dochází k významné dysfunkci endotelu, společně se změnou jeho fenotypu na protrombotický. Důsledkem je tvorba mikrovaskulárních trombů a ischémie. Je významně narušena mikrocirkulace se zvýšenou permeabilitou, progredující vazodilatací, orgánovou dysfunkcí a šokem.
Vliv kauzálního agens na produkci cytokinů a role cytokinů v patogenezi sepse
Z klinické praxe víme, že klinický výsledek pacientů se sepsí se liší jednak podle místa infekce, jednak pozorujeme rozdíl také v závislosti na druhu infekčního agens [72]. Byla prokázána rozdílná produkce cytokinů v závislosti na kauzálním agens sepse [73, 74]. V této souvislosti je nutné zmínit úlohu infekčního agens na produkci cytokinů makroorganismem, respektive úlohu cytokinů v obraně proti infekci. Příkladem může být vliv mikrobů na produkci TNF-α a jeho role v obraně proti infekci. Bylo již zmíněno, že jedním z hlavních mediátorů, které se negativně uplatňují v patogenezi sepse, je TNF-α. Po vazbě TNF-α na receptory pro TNF – p55 a p75 dochází k aktivaci zánětlivé a koagulační kaskády se všemi jejími negativními důsledky. Přitom význam koncentrace TNF-α jako prognostického parametru u pacientů v sepsi nebyl spolehlivě prokázán, protože výsledky jeho monitorování byly protichůdné [75, 76]. Kritickým momentem je zřejmě časový faktor, který omezuje významným způsobem dostupnost měření TNF-α, ale také jiných cytokinů. Například u pacientů ve studii PROWESS byla detekovatelná koncentrace TNF-α naměřena u 53 % pacientů, IL-1β pouze u 9 % pacientů, IL-8 u 40 % a IL-10 u 41 % pacientů. Monitorování koncentrace TNF-α v séru jako prognostického faktoru se proto do klinické praxe neprosadilo. Naproti tomu měření produkce TNF-α ex vivo po stimulaci lipopolysacharidem se ukázalo jako užitečný prognostický marker pro konečný klinický výsledek sepse [26, 77]. Terapeutickou konsekvencí role TNF v patogenezi sepse bylo použití inhibitorů TNF-α v klinických studiích, při kterých však nebyl prokázán významný pozitivní vliv pro konečný výsledek, respektive mortalitu [78, 79]. To vedlo ke značnému pesimismu v náhledu na další budoucnost této terapie sepse. Je nutné ale zdůraznit, že v těchto studiích se neverifikovala aktuální schopnost produkce TNF-α u léčených pacientů. Je tedy otázkou, zda použití inhibitorů TNF může mít terapeutický vliv u pacientů, kteří nejsou schopni tento mediátor produkovat, nebo jej produkují ve značně sníženém množství. Kromě toho však má TNF-α rovněž zásadní pozitivní vliv na úspěšné zvládnutí infekce makroorganismem. Jedním z důkazů je práce německých autorů, kteří prokázali závislost mortality experimentálních zvířat se sepsí na množství produkovaného TNF. Po navození imunoparalýzy mechanismem ligace céka a následnou punkcí (CLP) zjištovali vnímavost zvířat k superinfekci navozenou Salmonella enterica nebo Listeria monocytogenes. U zvířat s navozenou imunoparalýzou byla prokázána snížená produkce TNF-α. Superinfekce v této fázi vedla ke zvýšené letalitě zvířat. Po aplikaci exogenního rekombinantního TNF před superinfekcí a v okamžiku jejího nástupu byla navozena ochrana vůči salmonele, ale nikoli proti listerii [80]. Význam TNF-α pro zvládnutí infekce dokládá rovněž použití inhibitorů TNF-α u interních nebo revmatologických onemocnění – revmatoidní artritidy, Crohnovy choroby a dalších. U některých pacientů bylo závažným vedlejším účinkem této terapie propuknutí latentní tuberkulózy nebo vznik sepse, kde kauzální agens je primárně komensál nebo nepatogenní pro člověka. Naproti tomu u IL-6 se nepodařilo prokázat obdobnou závislost. Němečtí autoři na obdobném experimentálním modelu prokázali závislost mortality na koncentraci IL-6, ale jeho chybění mortalitu neovlivnilo [81]. Z uvedeného vyplývá, že potřebujeme lépe verifikovat a monitorovat aktuální stav imunitního systému, protože jen tak dospějeme k racionální indikaci imunomodulační, respektive protizánětlivé terapie.
Rozpoznávací mechanismy vrozeného systému imunity
Systém vrozené imunity disponuje řadou mechanismů, které jsou schopné reagovat při kontaktu s mikroorganismem. Mezi tyto výkonné mechanismy patří imunokompetentní buňky schopné produkovat cytokiny. Imunokompetentní buňky mají na svém povrchu tzv. PRRs, které jsou schopny rozeznat bakteriální motivy jednotlivých infekčních agens (PAMPs). Předpokládá se, že imunitní systém je schopen rozpoznat 103 PAMPs. Jako příklad PAMPs můžeme uvést: lipopolysacharid, peptidoglykan, bakteriální lipoproteiny a lipopeptidy, lipoteichoovou kyselinu, bakteriální a virovou jadernou ribonukleovou kyselinu.Kromě toho PRRs mají schopnost rozeznávat signály – endogenní mediátory, které vznikají v důsledku procesů, jakými jsou ischémie, trauma nebo nekróza. Tyto mediátory nazýváme alarminy a společně s PAMPs tvoří skupinu tzv. DAMPs (s poškozením asociované molekulární vzory – damage associated molecular patterns). Úlohou alarminů je zachování homeostázy, přičemž je charakterizuje sekrece Golgiho aparátem a to, že nejsou produkovány apoptotickými buňkami [82]. Mezi alarminy patří např.: HMGB1 (High mobility group box 1), HSPs proteiny (heat shock proteins), S100 proteiny a řada dalších.
Rozeznáváme dvě skupiny PRRs, které se liší svou funkcí – endocytární rozpoznávací receptory a signální receptory rozpoznávající motivy. Endocytární receptory rozpoznávající motivy jsou na povrchu fagocytujících buněk a jejich příkladem jsou např. opsoninové receptory, scavengerové receptory nebo manózové receptory. Jejich funkcí je vazba mikrobů na fagocytující buňky s jejich následnou destrukcí. Signální receptory rozpoznávající motivy se exprimují na buněčném povrchu a po jejich vazbě s PAMPs dochází k aktivaci systému vrozené a adaptivní imunity. Jejich příkladem jsou TLR, které představují rodinu molekul přítomných na povrchu imunokompetentních buněk. V současné době známe 10 TLR se zásadním významem pro přenos informace o přítomnosti infekčního agens a následnou produkcí prozánětlivých cytokinů [83]. Poltorak et al. popsali schopnost TLR-4 rozpoznávat lipopolysacharid, který je v případě gramnegativní sepse komponentou spouštějící zánětlivou kaskádu [84]. Postupně byly objeveny další vazby mezi jednotlivými TLR a komponenty infekčních agens, takže dnes jsou TLR považovány za zásadního zprostředkovatele interakcí mezi infekčním agens a vrozeným systémem imunity [85]. TLR-2 a TLR-6 rozeznávají peptidoglykany, TLR-5 flagelin, TLR-9 bakteriální DNA. Genetický polymorfismus TLR je predisponujícím faktorem k rozvoji některých infekčních onemocnění [86]. Kromě rozpoznávání motivů infekčních agens se TLR uplatňují také ve spouštění koagulační kaskády charakteristické pro sepsi. V současné době se diskutuje o tom, zdali tyto receptory pouze „zprostředkují “ rozvoj zánětlivé reakce nebo jsou přímo odpovědné za její nedostatečnost, s důsledkem nekontrolovatelnosti zánětu. Zatímco TLR jsou na povrchu buněk, další zástupci těchto receptorů jsou přítomni v cytoplazmě. Jejich příkladem jsou NOD proteiny – cytosolové proteiny umožňujícíintracelulární rozpoznávání peptidoglykanů nebo CARD-containing proteins(caspase activating and recruitment domain) – což jsou cytoplazmatické receptory pro přítomnost virové DNA a RNA, která je produkována infikovanými buňkami. Výsledkem interakcí PRRs, PAMPs a DAMPs je spuštění zánětlivé a koagulační kaskády.
Apoptóza v procesu sepse
Apoptóza je fyziologický děj, programovaná smrt buňky. Podobně jako u zánětu je to proces primárně fyziologický, který má za úkol zachování homeostázy, remodelaci tkání, aktivní proces imunitní odpovědi na exo - a endogenní podněty. Její objev spadá do počátku 70. let, výzkum apoptózy u kriticky nemocných se rozvinul v 90. letech minulého století. Podněty pro apoptózu jsou endogenní a exogenní. Známe tři způsoby navození apoptózy – vnější (extrinsic), vnitřní (intrinsic – mitochondriální) a cestu přes endoplazmatické retikulum (stresem indukovaná). Pro vnější cestu navození apoptózy je důležitý Fas antigen, člen rodiny receptorů TNF-α (CD95), exprimovaný na imunokompetentních buňkách – monocytech, makrofázích, ale také na buňkách jednotlivých orgánů (plic, jater, srdce). Po vazbě Fas antigenu na Fas ligand (FasL) probíhá proces tvorby smrt indukujícího vazebného komplexu (DISC – death-induced signaling complex) a následně tvorby molekuly obsahující Fas associated death domain (FADD). Systémem kaspáz je signál přenesen do jádra buňky, kde navodí apoptózu. U vnitřní cesty navození apoptózy je ústředním aktivačním momentem uvolnění cytochromu C, který opět aktivuje systém kaspáz. Významnou regulační roli zde hraje systém antiapoptotických proteinů Bcl-2 rodiny, které blokují uvolnění cytochromu C. U sepse je zvýšená apoptóza imunokompetentních buněk, které ve svém důsledku působí imunosupresivně [87, 88, 89]. Zatímco u lymfocytů B a CD4 pozitivních buněk bylo prokázáno jejich dramatické snížení, sepse nesnižovala počet CD8 pozitivních T lymfocytů a NK buněk. Byla popsána rozdílná míra apoptózy v jednotlivých orgánech. Největší počet apoptotických buněk byl prokázán ve slezině a střevě [90]. Takový pokles imunokompetentních buněk má pochopitelně odezvu ve snížené schopnosti organismu reagovat na infekci. Apoptotické buňky navíc aktivně potlačují zánětlivou odpověď [91, 92]. Byla prokázána změna v produkci cytokinů – prozánětlivých (TNF-α, IL-1β a IL-12) i protizánětlivých – IL-10. Otázkou zůstává význam apoptózy pro klinický výsledek sepse. Zatímco byla prokázána závislost klinického výsledku sepse na počtu lymfocytů, kauzalita mezi zvýšenou apoptózou lymfocytů a letalitou pacientů se sepsí prokázána nebyla. Naproti tomu v experimentálních modelech byla prokázán vliv inhibitorů kaspáz na lepší přežívání. Bylo tomu u modelu pneumokokové meningitidy [93], u myšího modelu s deficientní produkcí FasL [94] nebo modelu septické peritonitidy [95]. V klinické práci řeckých autorů byla časná apoptóza monocytů u pacientů se sepsí pozitivním prediktorem pro konečný klinický výsledek [96]. V současné době se proto diskutuje o otázce možného terapeutického přístupu s využitím signálních molekul pro apoptózu jako cíl pro terapii sepse [97].
Koncepce endogenně navozeného SIRS a sepse
Z klinické praxe víme, že klinická symptomatologie charakteristická pro sepsi je přítomna také u pacientů, u kterých není přítomna infekce. Hovoříme pak o syndromu sepse, který je spojen s horečkou, tachykardií, tachypnoí a šokem, bez ohledu na vyvolávající příčinu. S možným vysvětlením patogeneze těchto stavů, založeným na funkci TLR, přišli Tang et al. [98]. TLR jsou schopny reagovat nejen na infekční agens, ale také na různé stavební součásti makroorganismu, např. heparan sulfát. Za fyziologických podmínek se TLR nacházejí na buňkách ve stavu konstituční inhibice. Podněty, jako je např. hypoxie nebo operační trauma, aktivují cestou různých mechanismů, např. komplementového systému, enzymy – proteázy, které obnažují intercelulární matrix, uvolňují TLR ze stavu konstituční suprese a aktivují je. Stejným způsobem působí infekční agens. TLR jsou schopny reagovat následně s endogenními agonisty, jako je např. heparan sulfát nebo PAMPs. To ve svém důsledku přináší aktivaci koagulačního systému, produkci prozánětlivých cytokinů. Autoři z těchto poznatků dovozují mechanismy odpovědné za vznik syndromu systémové zánětlivé odpovědi neinfekční etiologie i sepse.
Aplikace poznatků imunopatogeneze sepse do terapie sepse
Ambicí poznání patogeneze onemocnění je přenést tyto poznatky do terapie. Pomineme-li základní strukturu kauzální terapie sepse – chirurgická sanace ložiska, použití korektní antibiotické terapie, další úsilí se zaměřuje na ovlivnění intenzity zánětu. Strategie dosavadní tzv. biologické léčby vycházela z konceptu hyperzánětlivé odpovědi, nebo naopak imunoparalýzy. Příkladem léčby postihující hyperzánětlivou odpověď je použití inhibitorů různých mediátorů uplatňujících se při rozvinutí zánětlivé kaskády. Znamená to např. použití intravenózních imunoglobulinů, inhibitorů C5a složky komplementu a C1 inhibitoru, inhibitorů TNF-α, systémových kortikosteroidů a celé řady dalších [99, 100, 101]. Zvláštní zmínku si zaslouží použití monoklonálních protilátek proti jednotlivým molekulám zúčastněným v procesu zánětu (adhezivní molekuly, antagonisté TLR) [102]. Je však nutné zdůraznit problematičnost těchto zásahů a opatrnost při jejich aplikaci z experimentálního modelu na člověka. Jako příklad můžeme uvést použití anti-CD28 monoklonální protilátky v první fázi klinické studie u terapie sepse a spuštění cytokinové bouře s katastrofálním důsledkem pro dobrovolníky, kterým byla tato léčebná látka aplikována [103]. Na opačné straně stojí terapie založená na uplatnění imunostimulačního účinku, tedy zesílení zánětlivé aktivity makroorganismu. Sem patří použití interferonu-γ nebo GM-CSF [104, 105]. Bohužel, ani jeden z obou přístupů dosud nevedl k významnému efektu v klinických studiích. Rekombinantní protein C(Xigris) je dosud jediným lékem schváleným FDA používaným v terapii sepse. Součástí jeho účinku je protizánětlivý efekt. Účinnost léčby je ale v současné době podrobována významné kritice [106].
Jsme přesvědčeni o tom, že nasazení terapie ovlivňující imunologickou reaktivitu pacienta musí vycházet z posouzení aktuálního stavu imunitního systému pacienta. Proč používat inbitory TNF v případě, že pacient tento mediátor neprodukuje? Rovněž načasování terapie je extrémně důležité, jak ukazují experimentální studie. Použití terapie postihující několik úrovní zánětlivé odpovědi se zdá být nevyhnutelné, ale také racionální.
Závěr
Sepse představuje dynamickou a komplexní interakci makro - a mikroorganismu. Je charakterizována dysfunkcí imunitního systému, která může vyústit až do stavu imunitního selhání. Detailní poznání patogeneze sepse, robustní diagnostika a komplexní přístup v terapii ovlivňující makro - i mikroorganismus povede k lepší prognóze v léčbě tohoto onemocnění.
Došlo 14. 5. 2008.
Přijato 20. 8. 2008.
Adresa pro korespondenci:
MUDr. Miroslav Průcha, Ph.D.
OKBH, Nemocnice Na Homolce
Roentgenova 2
150 30 Praha 5
e-mail: miroslav.prucha@homolka.cz
Sources
1. Martin, G. S., Mannino, D. M., Eaton, S., Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. England J. M., 2003, 348, p. 1546–1554.
2. Černý, V., Novák, I., Šrámek, V. Prevalence těžké sepse v České republice – prospektivní multicentrická jednodenní studie. Anest. intenziv. Med., 2003, 14, p. 218–222.
3. Angus, D. C., Linde-Zwirble, W. T., Lidicker, J. et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med., 2001, 29, p. 1303–1310.
4. Dombrovskiy, V. Y., Martin, A. A., Sunderram, J. et al. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: A trend analysis from 1993 to 2003. Crit. Care Med., 2007, 35, p. 1244–1250.
5. Dellinger, R. P. Cardiovascular management of septic shock. Crit. Care Med., 2003, 31, 3, p. 946–955.
6. Esper, A., Martin, G. S. Is severe sepsis increasing in incidence AND severity? Crit. Care Med., 2007, 35, 5, p. 1414–1415.
7. Bone, R. C., Balk, R. A.,Cerra, F. B. et al. American College of Chest Physicians/Society of Critical Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of Innovative therapies in sepsis. Chest, 1992, 101, p. 1644–1655.
8. Levy, M. M., Fink, M. P., Marshall, J. C. et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med., 2003, 29, p. 530–538.
9. Calandra, T., Baumgartner, J. D., Grau, G. et al. Prognostic value of tumor necrosis factor/cachectin, interleukin-1, interferon-α, and interferon-γ in the serum of patients with septic shock. J. Infect. Dis., 1990, 161, p. 982–987.
10. Cassatella, M. A., Meda, L., Bonora, S. et al. Interleukin-10 (IL-10) inhibits the release of proinflammatory cytokines from human polymorphonuclear leukocytes. Evidence for an autocrine role of tumor necrosis factor and IL-1β in mediating the production of IL-8 triggered by lipopolysaccharide. J. Exp. Med., 1993, 178, p. 2207–2211.
11. Damas, P., Reuter, A., Gysen, P. et al. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit. Care Med., 1989, 17, p. 975–978.
12. Emery, P., Salmon, M. The immune response: systemic mediators of inflammation. Br. J. Hosp. Med., 1991, 45, 1, p. 64–168.
13. Abbas, A. K., Murphy, K. M., Sher, A. Functional diversity of helper T lymphocytes. Nature, 1996, 383, p. 787–793.
14. Borowikova, L. V., Ivanova, S., Zhang, H. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature, 2000, 405, p. 458–462.
15. Woiciechowsky, C., Asadullah, K., Nestler, D. et al. Sympathetic activation triggers systemic IL-10 release in immunodepression induced by brain injury. Nat. Med., 1998, 4, p. 808–813.
16. Heidecke, C. D., Hensler, T., Weighradt, H. et al. Selective defects of T lymphocytes function in patiens with lethal intraabdominal infection. Am. J. Surg., 1999, 178, p. 288–292.
17. Pellegrini, J. D., De Ak, J., Kodys, K. et al. Relationships between T lymphocytes apoptosis and anergy following trauma. J. Surg. Res., 2000, 88, p. 200–206.
18. Hotchkiss, R. S., Tinsley, K. W., Swanson, P. E. et al. Sepsis-induced apoptosis cause progressive profound depletion of B and CD4+ T lymphocytes in humans. J. Immunol., 2001, 166, p. 6952–6963.
19. Hotchkiss, R. S., Tinsley, K. W., Swanson, P. E. et al. Depletion of dendritic cells, but not macrophages, in patiens with sepsis. J. Immunol., 2002, 168, p. 2493–2500.
20. Fukuzuka, K., Edwards, C. K., Clare-Walter, M. et al. Glucocorticoid-induced, caspase – dependent organ apoptosis early after burn injury. Am. J. Physiol. Regul. Integr. Comp. Physiol., 2000, 278, p. R1005–1018.
21. Greene, D. R., Beere, H. M. Apoptosis:gone but not forgotten. Nature, 2000, 405, p. 28–29.
22. Fadok, V. A., Bratton, D. L., Rose, D. M. et al. A receptor for phosphatidylserine-specific clearence of apoptotic cells. Nature, 2000, 405, p. 85.
23. Eichacker, P. Q., Parent, C., Kalil, A. et al. Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am. J. Respir. Crit. Care Med., 2002, 166, p. 1197–205.
24. Macias, W. L., Nelson, D. R., Wiliams, M. et al. Lack of evidence for qualitative treatment by disease severity interactions in clinical studies of severe sepsis. Crit. Care, 2005, 9, R607–622.
25. Marshall, J. C. The staging of sepsis: understanding heterogeneity in treatment efficacy. Crit. Care, 2005, 9, p. 626–628.
26. Průcha, M., Herold, I., Zazula, R., Dubská, L., Kavka, B. Monocytární deaktivace a produkce tumor nekrotizujícího faktoru-α ex vivo – prognostické parametry u pacientů jednotek intenzivní péče. Anest. intenziv. Med., 2003, 5, p. 223–228.
27. Calandra, T., Echtenacher, B., Le Roy, D. et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nature Medicine, 2000, 6, p. 164–170.
28. Wang, H., Zhang, M. et al. HMG-I as a late mediator of endotoxin letality in mice. Science, 1999, 285, p. 248–251.
29. Tschaikowsky, K., Geissing, M. H., Schiele, A. et al. Coincidence of pro - and anti-inflammatory responses in the early phase of severe sepsis: Longitudinal study of mononuclear histocompatibility leukocyte antigen – DR expression, procalcitonin, C-reactive protein, and changes in T-cell subsets in septic and postoperative patients. Crit. Care Med., 2002, 30, p. 1015–1023.
30. Oberholzer, A., Sousa, S. M., Tschoeke, S. K. et al. Plasma cytokine measurements augment prognostic scores as indicators of outcome in patients with severe sepsis. Shock, 2005, 23, p. 488–493.
31. Groeneveld, A. B. J., Bossink, A. W. J., Mierlo, G. J., Hack, C. E. Circulating inflammatory mediators in patients with fever: predicting bloodstream infection. Clin. Diagn. Lab. Immunology, 2001, 8, p. 1189–1195.
32. D’Andrea, A., Aste-Amezaga, M., Valiante, N. M. et al. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma production by supressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med., 1993, 178, p. 1041–1048.
33. De Vries, J. E., de Waal Malefyt K. Immunosuppressive and anti-inflammatory effects of human interleukin-10. In Faits, E., Baue, A. E., Schildberg, F. W. (Eds.) The immune consequences of trauma, shock and sepsis. Mechanisms and therapeutic approaches 1996. Pabst Science Publishers, p. 303–307.
34. Oberholzer, A., Oberholzer, C., Moldawer, L. L. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med., 2002, 30, S58–S63.
35. Janeway, C. A., Bottomly, K., Babich, J. Quantitative variation in Ia antigen expression plays a central role in immune regulation. Immunol. Today, 1984, 5, p. 99–105.
36. Docke, W. D., Syrbe, U., Meinecke, A. et al. Improvement of monocyte function – a new therapeutic approach? Update to Intensive Care and Emergency Medicine, 1994, 18, p. 473–500.
37. Rothe, G., Oser, A., Valet, G. Dihydrorhodamin 123: a new flow cytometric indicator for respiratory burst activity in neutrophil granulocytes. Naturwissenschaften, 1988, 73, p. 354–356.
38. Platzer, C., Richter, G., Uberla, K. et al. Analysis of cytokine mRNA levels in interleukin-4 transgenic mice by quantitative polymerase chain reaction. Eur. J. Immunol., 1992, 22, p. 1179–1184.
39. Munoz, C., Carlet, J., Fitting, C. et al. Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Invest., 1991, 88, p. 1747–1754.
40. Abraham, E. Physiologic stress and cellular ischemia: Relationship immunosuppression and susceptibility to infection. Crit. Care Med., 1991, 19, p. 613–618.
41. Dinarello, C. A. The proinflammatory cytokine interleukin-1 and tumor necrosis factor and treatment of the septic shock syndrome. J. Infect. Dis., 1991, 163, p. 1177–1184.
42. Doughty, L., Patrene, K., Boggs, S. et al. Supression of native IL-10 production by constitutive expression of viral IL-10 (vIL-10) or soluble TNF receptor p75 (sTNFR) in mice. Abstr. Crit. Care Med., 1996, 24 (Suppl): A32.
43. Hamilton, G., Hofbauer, S., Hamilton, B. Endotoxin, TNF-α, interleukin-6 and parameters of the cellular immune system in patients with intraabdominal sepsis. Scand. J. Infect. Dis., 1992, 24, p. 361–368.
44. Cheadle, W. G., Hershman, M. J., Wellhausen, S. R. et al. Role of monocytic HLA-DR expression following trauma in predicting clinical outcome. In Faist, E., Ninnemann, J., Green, D. (Eds) Immune Consequences of Trauma, Shock and Sepsis. Springer-Verlag : Berlin, Heidelberg, New York 1989, p. 199–222.
45. De Waal Malefyt, K., Haanen, J., Spits, H. et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen – specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med., 1991, 174, p. 915–924.
46. Durez, P., Abramowicz, D., Gerard, C. et al. In vivo induction of interleukin 10 by anti - CD3 monoclonal antibody or bacterial lipopolysaccharide: differential modulation by cyclosporin. J. Exp. Med., 1993, 177, p. 551–555.
47. D’Andrea, A., Aste-Amezaga, M., Valiante, N. M. et al. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma production by supressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med., 1993, 178, p. 1041–1048.
48. De Vries, J. E., de Waal Malefyt, K. Immunosuppressive and anti-inflammatory effects of human interleukin-10. In Faits, E., Baue, A. E., Schildberg, F. W. (Eds.) The immune consequences of trauma, shock and sepsis. Mechanisms and therapeutic approaches 1996. Pabst Science Publishers, p. 303–307.
49. Chandry, D., Turner, M., Abney, A. et al. Modulation of cytokine production by transforming growth factor β. J. Immunol., 1989, 140, p. 4217–4222.
50. Espenik, T., Figari, I. S., Shalaby, M. R. et al. Inhibition of cytokine production by cyclosporin A and transforming growth factor β. J. Exp. Med., 1987, 166, p. 571–576.
51. Czarniecki, C. W., Chiu, H. H., Wong, G. H.W. et al. Transforming growth factor β modulates the expression of class II histocompatibility antigens on human cells. J. Immunol., 1988, 140, p. 4217–4222.
52. Oczenski, W., Krenn, H., Jilch, R. et al. HLA-DR as a marker for increased risk for systemic inflammation and septic complications after cardiac surgery. Intensive Care Med., 2003, dx.doi.org/10.1007/s00134-003-1826-8.
53. Perry, S. E., Modrala, S. M., Wenstone, R. et al. Is low monocyte HLA-DR expression helpful to predict outcome in severe sepsis? Intensive Care Med., 2003, dx.doi.org/10.1007/s00134-003-1686-2.
54. Braude, A. E., Jones, J. L., Douglas, H. The behavior of Escherichia coli endotoxin (somatic antigen) during infectious arthritis. J. Immunol., 1963, 90, p. 297 – 301.
55. Andriole, V. T. Urinary tract infections in the 90s: Pathogenesis and management. Infection, 1992, 20 (suppl. 4), S251–S256,
56. Feltis, B. A., Jechorek, R. P., Erlanden, S. L. et al. Bacterial translocation and lipopolysaccharide – induced mortality in genetically macrophage-deficient op/op mice. Shock, 1994, 2, p. 29–33.
57. Go, L. L., Healey, P. J., Watkins, S. C. et al. The effect of endotoxin on intestinal mucosal permeability to bacteria in vitro. Arch. Surg., 1995, 130, p. 53–58.
58. Sedman, P. C., MacFie, J., Sagar, P. et al. The prevalence of gut translocation in humans. Gastroenterology, 1994, 107, p. 643–649.
59. Cabie, A., Farkas, J. C., Fitting, C. et al. High levels of portal TNF-α during abdominal aortic surgery in man. Cytokine, 1993, 5, p. 448–453.
60. Owens, W. E., Berg, R. D. Bacterial translocation from the gastrointestinal tract of thymectomized mice. Curr. Microbiol., 1982, 7, p. 169–174.
61. O’Boyle, C. J., Mac Fie, J., Mitchell, C. J. et al. Microbiology of bacterial translocation in humans. Gut, 1998, 42, p. 29–35.
62. Morin, M., Schindler, R., Wakabayashi, G. et al. Picogram concentrations of endotoxin stimulate synthesis of IL-1b and TNF-α by human peripheral blood mononuclear cells exposed to recombinant human C5a. Eur. Cytokine Netw., 1991, 2, p. 27–30.
63. Quereshi, S. T., Lariviere, G., Leveque, S. et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4. J. Exp. Med., 1999, 189, p. 615–625.
64. Cleveland, M. G., Gorham, J. D., Murphy, T. L. et al. Lipoteichoic acid preparations of Gram-positive bacteria induce interleukin-12 through a CD14 – dependent pathway. Infect. Immun., 1996, 64, p. 1906–1912.
65. Nwariaku, F., Sikes, P., Lightfoot, E. et al. Role of CD14 in hemorrhagic shock-induced alterations of the monocyte tumor necrosis factor response to endotoxin. J. Trauma, 1996, 40, p. 564–567.
66. Medzhitov, R., Janeway, C. Innate immunity: the virtues of a nonclonal system of recognition. Cell, 1997, 91, p. 295–298.
67. Hemmi, H. A Toll-like receptor recognizes bacterial DNA. Nature, 2000, 408, p. 740–745.
68. Ozinsky, A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. USA, 2000, 97, p. 13766–13771.
69. Wiel, E., Lebuffe, G., Vallet, B. Bacterial CpG DNA in septic shock. In Vincent, J. L. (Ed.) Yearbook of Intensive Care and Emergency Medicine. 2002, p. 388–398.
70. Schouten, M., Wiersinga, W. J., Levi, M., van der Poll Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol., 2008, 83, p. 536–545.
71. Stief, T. W., Ijagha, O., Weiste, B. et al. Analysis of hemostasis alterations in sepsis. Blood Coagul. Fibrinolysis, 2007, 18, p. 179–186.
72. Cohen, J., Cristofaro, P., Carlet, J., Opal, S. New method of classifying infections in critically ill patients. Crit. Care Med., 2004, 32, p. 1510–1526.
73. Warris, A., Verweij, P. E., Gaustad, P. et al. Various Filamentous fungi digger in thein ability to induce TNF-α and IL-6 release in human monocyte culture. Agents Chemother., 2000, 40, p. 373–375.
74. Opal, S. M., Gerber, G. E., La Rosa, S. P. et al. Systemic host response in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated). Clin. Infect. Diseases, 2003, 37, p. 50–58.
75. Damas, P., Reuter, A., Gysen, P. et al. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit. Care Med., 1989, 17, p. 975–978.
76. Borrelli, E. et al. Plasma concentrations of cytokines, their soluble receptors, and antioxidant vitamins can predict the development of multiple organ failure in patients at risk. Crit. Care Med., 1996, 24, p. 392–397.
77. Dadák, L., Šťouračová, M., Štětka, P. et al. Rozšířený imunologický profil v prvních dnech pobytu a prognóza nemocných dlouhodobě hospitalizovaných na JIP. Anest. intenziv. Med., 2007, 3, s. 164-170.
78. Abraham, E., Laterre, P. F., Garbino, J. et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double blind, placebo-controlled multicenter phase III trial with 1 342 patients. Crit. Care Med., 2001, 29, p. 503–510.
79. Fisher, C. J. Jr et al. Treatment of septic shock with the tumor necrosis factor receptor-fusion protein. N. Engl. J. Med., 1996, 334, p. 1697–1702.
80. Echtenacher, B., Urbaschek, R., Weigl, K. et al. Treatment of experimental sepsis-induced immunoparalysis with TNF. Immunobiology, 2003, 208, p. 381–389.
81. Remick, D. G., Bolgos, G., Copeland, S., Siddiqui, J. Role of interleukin 6 in mortality from a physiological response to sepsis. Infection and Immunity, 2005, 73, p. 2751–2757.
82. Bianchi, M. E. DAMPS, PAMPs and alarmins: all we need to know about danger. J. Leuko. Biology, 2007, 81, p. 1–5.
83. Wiersinga, W. J., van der Poll, T. The Role of Toll-like Receptors in Sepsis. In Yearbook of Intensive and emergency medicine. Springer : Berlin, Heidelberg 2006, p. 3–13.
84. Poltorak, A., He, X., Smirnova, I. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScR mice: Mutation in TLR4 gene. Science, 1998, 282, p. 2085–2088.
85. Beutler, B. Science review: key inflammatory and stress pathways in critical illness – the central role of the Toll-like receptors. Crit. Care, 2003, 7, p. 39–46.
86. Smirnova, I., Mann, N., Dols, A. et al. Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc. Natl. Acad. Sci. USA, 2003, 100, p. 6075–680.
87. Ayala, A., Lomas, J. L., Grutkoski, P. S., Chung, C. S. Pathological aspects of apoptosis in severe sepsis and shock? Int. J. Biochem. & Cell Biol., 2003, 35, p. 715–720.
88. Chung, C. S., Chaudry, I. H., Ayala, A. The apoptotic response of the lymphoid immune system to trauma, shock and sepsis. In Vincent, J. L. (editor) Yearbook of Intensive Care and Emergency Medicine. Springer-Verlag : Berlin 2000. p. 27–40.
89. Hotchkiss, R. S., Swanson, P. E., Freeman, B. D., Tinsley, K. W., Cobb, J. P., Matuschak, G. M. et al. Apoptotic cell death in patients with sepsis, shock and multiple organ dysfunction. Crit. Care Med., 1999, 27, p. 1230–1251.
90. Hotchkiss, R. S., Swanson, P. E., Knudson, C. M., Chang, M. et al. Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J. Immunol., 1999, 162, p. 4148–4156.
91. Vol, R. E., Hermann, M., Roth, E. A. et al. Immunosuppressive effects of apoptotic cells. Nature, 1997, 390, p. 350.
92. Fadok, V. A., Bratton, D. L., Rose, D. M. et al. A receptor for phosphatidy serine-specific clearence of apoptotic cells. Nature, 2000, 405, p. 485–490.
93. Braun, J. S., Novak, R., Herzog, K. H. et al. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat. Med., 1999, 5, p. 298.
94. Chung, C. H. S., Ying, X. X., Wang, W., Ayala, A. Is Fas ligand or endotoxin responsible for mucosal lymphocyte apoptosis in sepsis? Arch. Surg., 1998, 133, p. 1213–11215.
95. Hotchkiss, R. S., Swanson, P. E., Freeman, B. D. et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc. Natl. Acad. Sci. USA, 1999, 96, p. 14541–14546.
96. Giamarellos-Bourboulis, E. J., Routsi, Ch., Plachouras, D. et al. Early apoptosis of blood monocytes in the septic host: is it a mechanism of protection in the event of septic shock? Critical Care, 2006, 10R76 (doi 10.1186/cc4921).
97. Wesche-Soldato, D. E., Stan, R. Z., Chung, Ch. S., Ayala, A. The apoptotic pathway as a therapeutic target in sepsis. Curr. Drug Targets, 2007, 8, p. 493–500.
98. Tang, A. H., Brunn, G. J., Cascalho, M., Platt, J. L. Pivotal advance: endogenous pathway to SIRS, sepsis, and related conditions. J. Leukoc. Biol., 2007, 82, p. 282–285.
99. Liu, D., Lu, F., Qin. G. et al. C1 inhibitor – mediated protection from sepsis. J. Immunol., 2007, 179, p. 3966–3972.
100. Guo, R. F., Ward, P. A. C5a, a therapeutic target in sepsis. Recent Patents Anti-Infect. Drug Disc., 2006, 1, p. 57–65.
101. Werdan, K. Immunoglobulin treatment in sepsis – is the answer „no“? Crit. Care Med., 2006, 34, p. 1542–1544.
102. Ishii, K. J., Uematsu, S., Akira, S. Toll Gates for future immunotherapy. Curr. Pharm. Des., 2006, 120, p. 4135–4142.
103. Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D., Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med., 2006, 355, 10, p. 1018–1028.
104. Nierhaus, A., Montag, B., Timmler, N. et al. Reversal of immunoparalysis by recombinant human granulocyte – macrophage colony stimulating factor in patients with severe sepsis. Intensive Care Med., 2003, (on-line, Springer-Verlag 2003, 10.1007/s00134-003-1666-6).
105. Flohe, S. B., Agrawal, H., Flohe, S. et al. Diversity of interferon gamma and granulocyte-macrophage colony-stimulating factor in restoring immune dysfunction of dendritic cells and macrophages during polymicrobial sepsis. Mol. Med., 2008, Feb 24 [Epub ahead of print].
106. Eichacker, P. Q., Natanson, C. Increasing evidence that the risks of rhAPC may outweigh its benefits. Intensive Care Med., 2007; 33 p. 396–399.
Labels
Anaesthesiology, Resuscitation and Inten Intensive Care MedicineArticle was published in
Anaesthesiology and Intensive Care Medicine

2008 Issue 5
Most read in this issue
- Celková anestezie s bdělou fází u neurochirurgických výkonů
- Úskalia uplatňovania odporúčaní pre diagnostiku a liečbu ťažkej sepsy
- Imunopatogeneze sepse
- Čtvrtstoletí selektivní dekontaminace trávicího traktu – řada otázek zůstává nezodpovězena