
Genetické testování familiární hypercholesterolemie v klinické praxi: stanovisko výboru České společnosti pro aterosklerózu
Genetic testing of familial hypercholesterolemia in clinical practice: statement of Czech Society for Atherosclerosis
Although awareness about familial hypercholesterolemia (FH) is increasing, this widespread and potentially fatal, but treatable disease, remains underdiagnosed. Although FH is an inherited condition, genetic testing for this disease is still rare. The Familial Hypercholesterolemia Foundation convened an international panel of experts to evaluate the usefulness of molecular genetic testing for FH. The findings that form the basis for justification of the examination are as follows: (1) detection of a causal mutation facilitates the definitive diagnosis (2) presence of a pathogenic mutation involves a higher cardiovascular risk and potentially requires more aggressive lipid lowering (3) for genetically demonstrated FH, there is an increased likelihood of early initiation of and adherence to treatment; and (4) the knowledge of the causal mutation essentially facilitates the cascade examination of relatives at risk. The consensus of the panel of experts recommends that genetic testing for FH becomes a standard in the care of patients with verified or likely FH and their relatives at risk. The genes encoding the low-density lipoprotein receptor (LDLR), apolipoprotein B (apoB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) should be tested. Other genes can be included in the examination according to the patient’s phenotype. The benefits will be an increase in the number of diagnosed patients, a more effective cascade examination, initiation of treatment at an earlier age and more accurate risk identification.
Keywords:
cardiovascular risk – APOB gen – Familial Hypercholesterolemia Foundation – genetic testing – LDLR gen – PCSK9 gen – familiární hypercholesterolemie
Authors:
Michal Vrablík 1; Vladimír Bláha 2; Renata Cífková 3; Tomáš Freiberger 4; David Karásek 5; Pavel Kraml 6; Jan Piťha 7; Hana Rosolová 8; Vladimír Soška 9; Tomáš Štulc 1; Lukáš Tichý 10; Zuzana Urbanová Za Výbor Čsat 11; Jana Mašková 12
Authors‘ workplace:
III. interní klinika - endokrinologie a metabolismu 1. LF UK a VFN v Praze
1; III. interní gerontometabolická klinika LF UK a FN Hradec Králové
2; Centrum kardiovaskulární prevence 1. LF UK a Thomayerova nemocnice, Praha
3; Centrum kardiovaskulární a transplantační chirurgie, Brno
4; III. interní klinika – nefrologická, revmatologická a endokrinologická LF UP a FN Olomouc
5; II. interní klinika 3. LF a FN Královské Vinohrady, Praha
6; Interní klinika 2. LF UK a FN Motol a Laboratoř pro výzkum aterosklerózy IKEM, Praha
7; II. interní klinika LF UK a FN Plzeň
8; Oddělení klinické biochemie, II. interní klinika LF MU a FN u sv. Anny v Brně
9; Interní hematoonkologická klinika, Centrum molekulární biologie a genové terapie FN Brno
10; Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
11; NEOX Clinical Research, Praha
12
Published in:
AtheroRev 2020; 5(3): 147-157
Category:
Guidelines
Overview
Ačkoliv se povědomí o familiární hypercholesterolemii (FH) zvyšuje, přesto zůstává toto velmi rozšířené a současné potenciálně fatální, avšak léčitelné onemocnění nedostatečně diagnostikováno („poddiagnostikováno“). Přestože je FH dědičným onemocněním, genetické testování tohoto onemocnění se provádí stále ještě vzácně. Nadace Familial Hypercholesterolemia Foundation svolala mezinárodní panel expertů, aby zhodnotili užitečnost molekulárně genetického testování u FH. Zjištěná fakta, která tvoří základ pro odůvodnění vyšetření, jsou následující: (1) průkaz kauzální mutace usnadní stanovení definitivní diagnózy (2) přítomnost patogenní mutace znamená vyšší kardiovaskulární riziko a potenciálně vyžaduje agresivnější snižování hladiny lipidů (3) u geneticky prokázané FH je vyšší pravděpodobnost včasného zahájení léčby a adherence k léčbě a (4) znalost kauzální mutace zásadně usnadní kaskádové vyšetření příbuzných v riziku. Konsenzus panelu expertů doporučuje, aby se genetické testování u FH stalo standardem při péči o pacienty s jistou nebo pravděpodobnou FH a jejich příbuzné v riziku. Testovat je třeba geny kódující receptor pro lipoprotein s nízkou hustotou (low-density lipoprotein receptor – LDLR), apolipoprotein B (apoB) a proprotein konvertázu subtilizin-kexin typu 9 (PCSK9). Další geny lze do vyšetření zahrnout podle fenotypu pacienta. Přínosem bude zvýšení počtu diagnostikovaných pacientů, efektivnější kaskádové vyšetření, zahájení léčby v nižším věku a přesnější určení rizika.
Klíčová slova:
Familial Hypercholesterolemia Foundation – familiární hypercholesterolemie – genetické testování – gen LDLR – gen APOB – gen PCSK9 – kardiovaskulární riziko
Preambule
V srpnu 2018 byl v Journal of American College of Cardiology (JACC) publikován konsenzus expertů o genetickém testování familiární hypercholesterolemie v klinické praxi [1]. Výbor České společnosti pro aterosklerózu (ČSAT) zpracoval následující souhrn nejdůležitějších informací a postupů, které jsou vhodné i pro zdravotní péči v České republice. Tento souhrn v českém jazyce nenahrazuje publikované doporučení a zájemcům o detailní znění doporučujeme k prostudování původní kompletní text. Číslování referencí v tomto souhrnu kromě první z nich odpovídá původní publikaci.
Úvod
Familiární hypercholesterolemie (FH) je genetické onemocnění, které se projevuje předčasným aterosklerotickým kardiovaskulárním onemocněním v důsledku celoživotního zvýšení hladiny cholesterolu v lipoproteinech o nízké hustotě (LDL-C). FH je nejčastější genetickou příčinou kardiovaskulárního onemocnění (KVO) s odhadovanou prevalencí přibližně 1 : 220 [7–10]. Pokud není FH již v časném věku identifikována a vhodně léčena, mají muži 50% riziko fatální nebo nefatální koronární příhody ve věku 50 let a ženy 30% riziko téhož ve věku 60 let [12,13]. Časná diagnóza FH a léčba statiny a dalšími hypolipidemiky (lipid-lowering treatment – LLT) zahájená již v dětství mohou snížit incidenci aterosklerózy až na úroveň jedinců bez FH [15–17].
FH zahrnuje celé spektrum klinických fenotypů, které částečně odráží zastoupení rozličných patogenních variant v zodpovědných genech (schéma 1).
![Schéma 1 | Spektrum fenotypů familiární hypercholesterolemie. Upraveno podle [1]](https://pl-master.mdcdn.cz/media/image_pdf/8765d9c13b0c9d35028244f7c1a6000f.png?version=1605300163)
Heterozygotní forma FH (HeFH) je obvykle způsobena jedinou patogenní mutací v 1 ze 3 hlavních genů souvisejících s FH: receptor pro lipoproteiny s nízkou hustotou (low-density lipoprotein receptor – LDLR), apolipoprotein B (APOB) a proprotein konvertázu subtilizin-kexin typu 9 (proprotein convertase subtilisin/kexin 9 – PCSK9) [18]. Nejčastější jsou patogenní mutace v genu LDLR. Homozygotní forma FH (HoFH) je způsobena přítomností patogenní mutace na obou alelách, obvykle také v LDLR [19]. Prevalence molekulárně potvrzené HoFH činí podle poslední dat přibližně 1 na 200 000 až 300 000 osob [20]. HoFH způsobuje závažné předčasné aterosklerotické KVO a, pokud není léčena, předčasné úmrtí z těchto příčin.
Hladinu LDL-C u pacientů s mutací, která způsobuje FH, ovlivňují také zděděné varianty v dalších genech. Na klinickém fenotypu FH, alespoň u některých pacientů, se proto kromě významného vlivu patogenní mutace v hlavním genu pro FH podílí i polygenní složka [21,22]. Pacienti s klinickým fenotypem FH (s vysokou hladinou LDL-C i s pozitivní rodinnou anamnézou) mohou mít negativní výsledek genetického testování všech 3 hlavních genů, a přesto mohou být přítomny alely jiných genů přispívající k vysokým hladinám LDL-C.
Významná část pacientů s FH není diagnostikována, i když mají zvýšené hladiny LDL-C nebo předčasnou ischemickou chorobu srdeční (ICHS) včetně infarktu myokardu (IM) [7,23]. Kromě „poddiagnostikování“ jsou pacienti s FH podle nedávných studií vyhodnocujících údaje z elektronických zdravotních záznamů také nedostatečně léčeni, přitom tyto studie dokládají, že přiměřeně je léčena nanejvýše jedna polovina pacientů, a až jedna třetina není léčena vůbec [7,26].
Ačkoliv má molekulárně genetické vyšetření potenciál zlepšit diagnostiku, poskytnout data o prognóze a upřesnit riziko, data z registru Cascade Screening for Awareness and Detection of FH (CASCADE FH) ukazují, že ve Spojených státech amerických není dostatečně využíváno, protože jej podstoupilo pouze 3,9 % jedinců z registru pacientů s klinickou diagnózou FH [27]. Genetické testování FH se provádí častěji a/nebo u širší populace např. v Nizozemí, Norsku, Velké Británii, Španělsku, Dánsku, Belgii, České republice, Slovensku, Islandu, Švýcarsku, Kanadě, Austrálii, Novém Zélandu a Jižní Africe [24,25,28].
Odůvodnění molekulárně genetického vyšetření u FH v klinické praxi
Genetické testování umožní s jistotou stanovit molekulání diagnózu FH
Diagnóza FH je historicky založena na několika různých sadách diagnostických kritérií jako například Dutch Lipid Clinic Network Diagnostic Criteria (DLCNC) [29], nebo Simon Broome Register Diagnostic Criteria [30], která zahrnují zvýšenou hladinu LDL-C, předčasné KVO v klinické anamnéze, hypercholesterolemii a/nebo KVO v rodinné anamnéze, nález při fyzickém vyšetření (včetně šlachových xantomů a arcus lipoides corneae) a prokázání patogenní mutace způsobující FH při vyšetření deoxyribonukleové kyseliny (DNA). Právě molekulárně genetické vyšetření je klíčovým pro diagnózu jisté FH. Identifikace patogenní mutace, nebo mutací v genech LDLR, APOB nebo PCSK9 byla popsána jako „zlatý standard“ pro diagnózu FH [3,31].
Díky nedávným studiím s rozsáhlým sekvenováním DNA bylo prokázáno, že diagnostické využití určité určené hladiny LDL-C jako hranice pro odlišení pacientů s patogenní mutací způsobující FH je nepřesné. I když je LDL-C u pacientů s FH obecně vyšší, bylo popsáno poměrně široké rozmezí hladin (schéma 1, s. 148) [7,36]. Pacienti s mutací způsobující FH nemusí dosáhnout hladiny LDL-C nad určenou mez, ale přesto mají zvýšené riziko ICHS. Odlišení pacientů s FH a pacientů se zvýšenou hladinou cholesterolu z jiných příčin komplikuje překrývání intervalů hladin LDL-C u jedinců s a bez patogenní mutace způsobující FH [10,36,41] (schéma 2). Odlišení na základě hladin LDL-C je nejpřesnější v mladém věku.

Genetické testování tedy pomáhá při diagnóze FH identifikací pacientů s patogenní mutací, kteří nesplňují diagnostická kritéria založená na hladině lipidů, klinických příznacích a/nebo rodinné anamnéze. Jedním z argumentů pro odůvodnění genetického testování u FH je tedy usnadnění diagnózy FH u jedinců, kteří by jiným způsobem diagnostikováni nebyli.
Molekulárně genetické vyšetření poskytuje informaci pro stanovení prognózy a stratifikaci rizika
Genetické testování FH umožňuje získat informaci o prognóze a upřesnit stratifikaci rizika. Ve skupině případů a kontrol z populace hodnocené skupinou Myocardial Infarction Genetics Consortium bylo riziko ICHS vyšší u nositelů patogenní mutace způsobující FH v porovnání s jedinci bez patogenní mutace při jakékoliv hodnotě LDL-C (graf) [36]. V porovnání s referenční skupinou s hladinami LDL-C < 3,6 mmol/l a bez patogenní mutace, měly subjekty s hladinami LDL-C ≥ 4,9 mmol/l a bez patogenní mutace 6krát vyšší riziko ICHS, zatímco subjekty s hladinami LDL-C ≥ 4,9 mmol/l a zároveň patogenní mutací způsobující FH měli riziko vyšší 22krát. Přítomnost patogenní mutace způsobující FH zvyšuje riziko ICHS při stejné hladině LDL-C 3násobně zřejmě z důvodu celoživotní expozice zvýšené hladiny LDL-C.
![Molekulárně genetické vyšetření FH poskytuje informaci o prognóze a umožňuje přesnější stratifikaci
rizika. Vliv potvrzené mutace způsobující FH na riziko ICHS podle hladiny LDL-C. Upraveno podle [36]](https://pl-master.mdcdn.cz/media/image_pdf/76c5ba2422b496d809afb0a5e0791d19.png?version=1605300483)
Konkrétní typ patogenní mutace a její závažnost (např. LDLR-defektní versus nulová mutace) má souvislost se stupněm hypercholesterolemie a rizikem ICHS, přičemž nejzávažnější je nulová mutace LDLR [7,36] a ostatní mutace LDLR, mutace APOB a PCSK9 mají obvykle mírnější fenotyp [7]. Typ patogenní mutace je také nezávislým prediktorem dosažení cílové hladiny LDL-C při léčbě [34].
Detekce patogenní mutace indikuje vyšší kardiovaskulární riziko a potřebu agresivnějšího snižování hladiny LDL-C. Pozitivní výsledek vyšetření zvyšuje pravděpodobnost zahájení léčby a adherenci k této léčbě.
Genetické testování usnadňuje kaskádové vyšetření FH v rodině
Identifikace patogenní mutace nebo mutací u probanda umožňuje cílené molekulárně genetické testování příbuzných v riziku s velmi vysokou senzitivitou a specificitou. Takový přístup jednoznačně určí příbuzné s a bez FH.
Genetické testování zvyšuje přesnost genetického poradenství
Molekulárně genetické testování umožňuje rozlišit na molekulární úrovni mezi HeFH, složeným heterozygotem, dvojitě heterozygotní FH, HoFH, autosomálně recesivní FH a pacienty bez identifikovatelné patogenní mutace, ale s fenotypem FH. Riziko postižení pro příbuzné a postup při plánování rodičovství se u vyjmenovaných možností liší.
Genetické testování má dopad na terapii FH
Patogenní mutaci/e ovšem nelze identifikovat u všech pacientů s fenotypem FH, a proto nesmí být léčba odepřena pacientům s klinickou diagnózou FH, u kterých při vyšetření patogenní mutace detekována nebyla.
Výsledky molekulárně genetického vyšetření mají jednoznačně pozitivní vliv na rozhodnutí o zahájení LLT i na adherenci pacientů k léčbě, a tím ve výsledku i na snížení hladiny LDL-C [35,44]. Dokonce i u pacientů s FH, kterým již byla terapie LLT předepsána, byla po potvrzení diagnózy genetickou analýzou pozorována vyšší adherence k léčbě, a v důsledku toho i příznivý vliv na snížení hladiny LDL-C [49]. Například v Norsku se četnost užívání LLT zvýšila v době genetického testování na 89 % z 53 % před testováním, a to se snížením hladiny celkového cholesterolu o 21 % za 6 měsíců po vyšetření [50].
Genetické testování je důležité pro pediatrické pacienty s FH
Děti s FH, které zahájí léčbu statiny, mají statisticky nižší četnost příhod než jejich postižení rodiče [16]. Podle věku zahájení terapie statiny je možné kumulativní zátěž LDL-C snížit až na úroveň srovnatelnou s úrovní u běžné populace [73]. Časným identifikováním dítěte s FH je možné zavést změny životního stylu, které ovlivní nejen zvýšenou hladinu LDL-C, ale i další rizikové faktory jako například kouření [40].
Osobní přínos genetického testování a psychosociální dopady
Podle dostupných dat má diagnóza FH potvrzená vyšetřením DNA minimální nežádoucí psychologický dopad [76,77] a molekulárně genetické vyšetření na FH nevyvolává u pacientů úzkost [78]. Naopak byla pozorována vyšší adherence k léčbě, a v důsledku toho i příznivý vliv na snížení hladiny LDL-C [49].
Metodika genetického testování
Ze 3 hlavních genů, jejichž patogenní mutace způsobují FH (LDLR, APOB a PCSK9), jsou nejčastější mutace v LDLR: > 90 % identifikovaných mutací způsobujících FH jsou v genu LDLR, dalších 5–10 % v genu pro apoB a < 1 % v genu PCSK9 [82–84]. V souvislosti s FH již bylo identifikováno více než 2 000 unikátních mutací, z nichž byla u přibližně 1 000 dostatečně prokázána jejich patogenita nebo pravděpodobná patogenita [85]. Patogenní mutace LDLR zahrnují předčasné ukončení translace (nonsense), mutace se změnou smyslu/aminokyseliny (missense), mutace v promotoru a konzervativních sekvencích místa sestřihu, malé inzerce a delece i rozsáhlá přeskupení DNA [86,87]. Právě rozsáhlá přeskupení DNA představují asi 10 % patogenních mutací, proto by strukturní analýza velkých přestaveb genu LDLR měla být rutinní součástí vyšetření [88–90].
Nejčastější patogenní mutace APOB způsobující FH jsou missense mutace v oblasti, v níž se apolipoprotein B100 váže na LDLR, jejichž důsledkem je defektní apolipoprotein, který není schopen se navázat na LDLR; tato porucha se někdy označuje jako familiární defekt apolipoproteinu B [91]. V Evropě se v APOB dominantně vyskytuje mutace způsobující záměnu aminokyseliny v pozici 3 527 peptidové sekvence (p.Arg3527Gln) [92], ale v této oblasti se může objevit i jiná mutace [93]. Mutace, která naopak zvyšuje funkci proteinu, je popsána u PCSK9, při níž zvýšená schopnost PCSK9-proteinu degradovat LDLR vede ke snížení počtu receptorů na povrchu buněk.
Výtěžnost molekulárně genetického vyšetření na FH závisí na pravděpodobnosti FH podle klinických diagnostických kritérií a dalších klinických faktorech, jako jsou předčasná ICHS a/nebo extrémní hypercholesterolemie bez známých sekundárních příčin. Patogenní mutace v 1 ze 3 genů, o kterých se ví, že souvisí s FH, bývá identifikována u přibližně 60 až 80 % pacientů s „jistou“ FH podle klinických diagnostických kritérií a u přibližně 21 % až 44 % pacientů s „pravděpodobnou“ FH [83,101–103]. U pediatrických pacientů se silným podezřením na FH je výsledek genetického testování pozitivní u přibližně 60 % až 95 % [39,104]. Negativní výsledek molekulárně genetického vyšetření u pacienta s fenotypem FH, jak je definován klinickými kritérii, tedy nevylučuje diagnózu FH. Výsledek může být negativní pro technické problémy a/nebo mutaci v dosud neobjevených genech. Diagnóza FH by měla být stanovena klinicky (jistá/pravděpodobná/možná) i při negativním výsledku molekulárně genetického vyšetření (tito pacienti mají fenotyp pozitivní a genotyp negativní), pokud má pacient závažnou hypercholesterolemii a v rodinné anamnéze hypercholesterolemii a/nebo předčasnou ICHS, protože kardiovaskulární riziko odpovídající fenotypu FH zůstává vysoké [105].
Minimálně by tedy u pacientů se suspektní diagnózou FH měla v rámci molekulárně genetického vyšetření proběhnout analýza LDLR, regionu v APOB, kterým se váže na LDLR, a PCSK9. Důležité je, že diagnózu FH nelze vyloučit, ani pokud při molekulárně genetickém vyšetření není žádná patogenní mutace nalezena, protože fenotyp FH může způsobovat neodhalená patogenní mutace v uvedených genech, mutace v jiném genu a/nebo mutace v dosud neobjeveném genu [3].
Známá je také autosomálně recesivní forma hypercholesterolemie způsobená bialelickou patogenní mutací genu pro LDLR-adaptorový protein (LDLRAP1) [110]. Tuto možnost je třeba zvážit u pacientů se závažnou hypercholesterolemií, u kterých nebyla detekována žádná mutace v genech LDLR, APOB nebo PCSK9 a v rodině by přenos mezi generacemi mohl odpovídat autozomálně recesivní dědičnosti.
Mezi další příčiny fenotypu FH patří patogenní mutace v genu pro apolipoprotein E, která vykazuje autosomálně dominantní dědičnost a projevuje se předčasnými infarkty myokardu, šlachovými xantomy, xantelazmaty a zvýšenou hladinou LDL-C [111].
Uvažovat lze i o dalších poruchách lipidového metabolizmu s Mendelovským typem dědičnosti, které mohou fenotypem odpovídat FH [112,113]. Sitosterolemie (způsobená autosomálně recesivní patogenní mutací v genech ABCG5 nebo ABCG8) [114] se může projevovat xantomy a hypercholesterolemií. Deficit kyselé lyzosomální lipázy způsobený autosomálně recesivní patogenní mutací v genu LIPA se může také projevovat zvýšenou hladinou LDL-C a je často doprovázen jaterní steatózou [113,115]. Genetické testování na tyto mutace může u suspektních pacientů přímo ovlivnit volbu terapie.
Rovněž polygenní etiologie může vysvětlit klinický fenotyp FH u velké části pacientů s negativním výsledkem vyšetření 3 hlavních genů, kteří mohou být nositeli vyššího počtu jednonukleotidových polymorfizmů (single-nucleotide polymorphism – SNP) zvyšujících hladinu LDL-C [22,107,116]. U pacientů s fenotypem FH bez detekované patogenní mutace bývá rovněž často zvýšená hladina lipoproteinu (a) [117].
Genetické testování u FH je dostupné v mnoha laboratořích ve Spojených státech i ve světě [82]. Cena vyšetření se neustále snižuje mimo jiné díky technologii sekvenování nové generace (Next Generation Sequencing – NGS) [32,82]. Nejčastěji laboratoře nabízejí panely NGS včetně sekvenování celého genu LDLR, APOB a PCSK9 současně s analýzou delecí/duplikací LDLR. Cena genetického testování u prvního pacienta v rodině se může dostat pod částku 500 USD [32]. Vyšetření známé specifické mutace u dalších příbuzných v dané rodině (kaskádové genetické testování) je poté ještě výrazně levnější. K dispozici jsou také panely NGS pro poruchy lipidového metabolizmu, které nabízejí vyšetření nejen hlavních genů u FH, ale také dalších genů s možným vlivem na fenotyp podobný FH.
Doporučení pro genetické testování u FH
Tab. 1 shrnuje naše doporučení a rozvahu u genetického testování u FH. Tento přehled zahrnuje doporučení pro probandy i kaskádové vyšetření příbuzných v riziku. Molekulárně genetické vyšetření by mělo minimálně zahrnovat analýzu LDLR, APOB a PCSK9. Konkrétně se jedná o sekvenování všech exonů a hraničních oblastí exon/intron u genů LDLR a PCSK9, včetně analýzy delecí/duplikací, a exonů APOB v oblasti, která se váže na LDLR.

Důkazy, na jejichž základě je doporučení pro genetické testování vytvořeno, jsou popsány v odstavcích výše. Ve stručnosti je doporučení genetického testování probanda založeno na následujícím: (1) zvýšené riziko u jedinců s patogenní mutací související s FH (2) dostupnost účinné léčby pro snížení LDL-C (3) efektivnější kaskádové testování a (4) pravděpodobné zlepšení adherence k léčbě při molekulárně geneticky potvrzené diagnóze. U pacientů, u kterých je genetické testování doporučeno nebo je zvažováno na základě uvedených kritérií, je dostatečně velká pravděpodobnost identifikace patogenní mutace. Provádění kaskádového vyšetření bylo podpořeno extenzivními epidemiologickými daty a ekonomickými analýzami nákladů, které zohlednily přínos časnějšího rozpoznání FH a také efekt terapie statiny jasně demonstrovaný v randomizovaných studiích.
K dispozici jsou také panely NGS pro poruchy lipidového metabolizmu, které nabízejí vyšetření nejen hlavních genů pro FH, ale také dalších genů s možným vlivem na fenotyp podobný FH. Tento rozšířený panel by měl být zvážen u pacientů s fenokopií FH, která vyžaduje specifickou léčbu, a měl by zahrnovat následující geny: LDLR, APOB, PCSK9, LDLRAP1, LIPA, ABCG5, ABCG8 a APOE.
Genetické testování u FH je vhodné doplnit důkladným informováním pacientů o genetické povaze onemocnění a významu genetického testování před vlastním testováním, jakož i podrobným vysvětlením získaných výsledků po testování, aby byl pacient seznámen s jeho přínosy, omezeními, možnými riziky a důsledky vyplývajícími pro celou rodinu (tab. 2). Postupy genetické konzultace u pacientů s FH, včetně specifických doporučení před a po vlastním testování, vlivu na rodinu, ochrany osobních dat, možných důsledků týkajících se diskriminace a zákonů, které se týkají této problematiky [127], již byly publikovány [2,18,128,129].
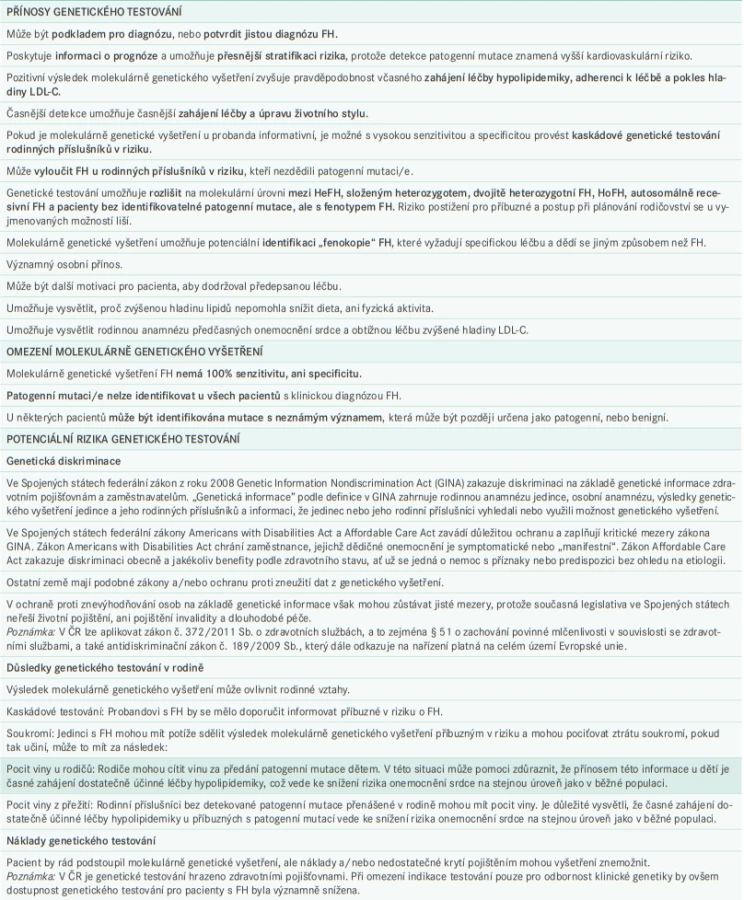
Doporučený postup při předání informací o genetické povaze onemocnění a genetickém testování shrnuje tab. 3 a schéma 3 níže, v nichž je uveden nejen postup pro probanda a jeho rodinu, ale zahrnuje i postup pro pacienty, kteří souhlasí, nebo nesouhlasí s genetickým testováním, a také návod, jak použít pozitivní, negativní, nebo nejistý výsledek genetického testování. U kaskádového genetického vyšetření, pokud důležitý příbuzný v rodové linii buď nesouhlasí s genetickým testováním, nebo již zemřel, a tedy nemůže vyšetření podstoupit, by měla kaskáda pokračovat příbuznými 1. stupně tohoto člena rodiny, aby bylo možné určit, zda mají, nebo nemají riziko onemocnění FH.


Závěr a mezery v důkazech
Molekulárně genetické vyšetření pacientů se suspektní diagnózou FH se dostatečně nevyužívá, a to přestože cena sekvenování DNA neustále klesá, a vyšetření se tím stává dostupnější. Poslední výzkum ukázal, že zavedení genetického testování u FH jako součásti standardní péče o pacienty a jejich příbuzné, u nichž je jistá, pravděpodobná nebo možná diagnóza FH by zlepšilo diagnostikování FH, protože by bylo možné snáze dohledat příbuzné každého pacienta s FH, kteří jsou také v riziku, a doporučená léčba LLT by tak mohla být zahájena v nižším věku, což by dlouhodobě snížilo následky onemocnění. Pacienti díky vyšetření také obdrží přesnější informace o prognóze a riziku.
Při uplatnění genetického testování existují i určité mezery ve znalostech a důkazech. Zejména je třeba pokračovat v odlišení definice FH podle fenotypu a genotypu a uznat variabilitu klinických projevů. HeFH způsobená mutací v LDLR se může projevovat v rozmezí od normální až po extrémně vysokou hladinu LDL-C a také u pacientů s molekulárně definovanou HoFH (2 mutace) je rozmezí podobné, jak bylo dříve definováno pro HeFH [18]. Naopak pacienti ve vysokém riziku mohou splnit kritéria pro FH (vysoká hladina LDL-C a v rodinné anamnéze hypercholesterolemie a předčasná ICHS), ale mohou mít negativní výsledek genetického testování z důvodu jiné dosud neznámé genetické příčiny vysoké hladiny LDL-C. Budoucí výzkum tedy musí dále upřesnit a rozšířit možnosti genetického testování pacientů s hypercholesterolemií, včetně polygenních rizikových faktorů a dalších metabolických drah.
Genetické testování je příležitostí, jak pro danou hladinu LDL-C identifikovat osoby s významně vyšším rizikem ICHS oproti běžné populaci. Screening příbuzných podle výsledků genetického vyšetření umožňuje identifikovat a léčit osoby s nerozpoznanou FH. Právě časná diagnóza FH a léčba podle platných doporučení vede ke změně přirozeného průběhu tohoto vysoce závažného dědičného onemocnění.
prof. MUDr. Michal Vrablík, Ph.D. | michal.vrablik@athero.cz | www.athero.cz
Doručeno do redakce | Doručené do redakcie | Received 18. 9. 2020
Doručené do redakcie | Doručeno do redakce | Received 18. 9. 2020
Sources
Seznam citované literatury je číslován podle originálního anglického textu [1]. Položky [4–6], [11], [14], [33], [37,38], [42,43], [45–48], [51–72], [74,75], [79–81], [94–100], [106,108,109], [118–126] ve výše uvedeném textu stanoviska citovány nejsou. Úplný seznam citované literatury je dostupný z DOI: <http://dx.doi.org/10.1016/j.jacc.2018.05.044>.
1. Sturm AC, Knowles JW, Gidding SS et al. [Convened by the Familial Hypercholesterolemia Foundation]. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel J Am Coll Cardiol 2018; 72(6): 662–680. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2018.05.044>.
2. Watts GF, Sullivan DR, Poplawski N et al. Familial hypercholesterolaemia: a model of care for Australasia. Atherosclerosis 2011; 12(2): 221–263. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosissup.2011.06.001>.
3. Watts GF, Gidding S, Wierzbicki AS et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol 2014; 171(3): 309–325. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ijcard.2013.11.025>.
7. Abul-Husn NS, Manickam K, Jones LK et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016; 354(6319): aaf7000. Dostupné z DOI: <http://dx.doi.org/10.1126/science.aaf7000>.
8. Benn M, Watts G, Tybjærg-Hansen A et al. Corrigendum to “Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary Artery Disease, and Cholesterol-Lowering Medication”. J Clin Endocrinol Metab 2014; 99(11): 4758–4759. Dostupné z DOI: <https://doi.org/10.1210/jc.2014–3926>.
9. Benn M, Watts GF, Tybjaerg-Hansen A et al. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012; 97(11): 3956–3964. Dostupné z DOI: <https://doi.org/10.1210/jc.2012–1563>.
10. Wald DS, Bestwick JP, Morris JK et al. Child-parent familial hypercholesterolemia screening in primary care. N Engl J Med 2016; 375(17): 1628–1637. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1602777>.
12. Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet 1969; 2(7635): 1380–1382. Dostupné z DOI: <http://dx.doi.org/10.1016/s0140–6736(69)90930–1>.
13. Stone NJ, Levy RI, Fredrickson DS et al. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation 1974; 49(3): 476–488. Dostupné z DOI: <http://dx.doi.org/10.1161/01.cir.49.3.476>.
15. Versmissen J, Oosterveer DM, Yazdanpanah M et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008; 337: a2423. Dostupné z DOI: <http://dx.doi.org/10.1136/bmj.a2423>.
16. Braamskamp MJ, Kastelein JJ, Kusters DM et al. Statin initiation during childhood in patients with familial hypercholesterolemia: consequences for cardiovascular risk. J Am Coll Cardiol 2016; 67(4): 455–456. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2015.11.021>.
17. Neil A, Cooper J, Betteridge J et al. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: a prospective registry study. Eur Heart J 2008; 29(21):2625–2633. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehn422>.
18. Gidding SS, Champagne MA, de Ferranti SD et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation 2015; 132(22): 2167–2192. Dostupné z DOI: <http://dx.doi.org/10.1161/CIR.0000000000000297>.
19. Cuchel M, Bruckert E, Ginsberg HN et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014; 35(32): 2146–2157. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehu274>.
20. Sjouke B, Hovingh GK, Kastelein JJ et al. Homozygous autosomal dominant hypercholesterolaemia: prevalence, diagnosis, and current and future treatment perspectives. Curr Opin Lipidol 2015; 26(3):200–209. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000179>.
21. Futema M, Shah S, Cooper JA et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem 2015; 61(1): 231–238. Dostupné z DOI: <http://dx.doi.org/10.1373/clinchem.2014.231365>.
22. Talmud PJ, Shah S, Whittall R et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 2013; 381(9874): 1293–1301. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(12)62127–8>.
23. Braenne I, Kleinecke M, Reiz B. et al. Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet 2016; 24(2): 191–197. Dostupné z DOI: <http://dx.doi.org/10.1038/ejhg.2015.100>.
24. Nordestgaard BG, Benn M. Genetic testing for familial hypercholesterolaemia is essential in individuals with high LDL cholesterol: who does it in the world? Eur Heart J 2017; 38(20): 1580–1583. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehx136>.
25. Nordestgaard BG, Chapman MJ, Humphries SE et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34(45): 3478–3490a. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/eht273>.
26. Knickelbine T, Lui M, Garberich R et al. Familial hypercholesterolemia in a large ambulatory population: statin use, optimal treatment, and identification for advanced medical therapies. J Clin Lipidol 2016; 10(5): 1182–1187. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2016.05.007>.
27. Ahmad ZS, Andersen RL, Andersen LH et al. US physician practices for diagnosing familial hypercholesterolemia: data from the CASCADE-FH registry. J Clin Lipidol 2016; 10(5): 1223–1229. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2016.07.011>.
28. Vrablik M, Vaclova M, Tichy L et al. Familial hypercholesterolemia in the Czech Republic: more than 17 years of systematic screening within the MedPed project. Physiol Res 2017; 66(Suppl 1): S1–S9. Dostupné z DOI: <http://dx.doi.org/10.33549/physiolres.933600>.
29. Defesche JC, Lansberg PJ, Umans-Eckenhausen MA et al. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med 2004; 4(1): 59–65. Dostupné z DOI: <http://dx.doi.org/10.1055/s-2004–822987>.
30. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991; 303(6807): 893–896. Dostupné z DOI: <http://dx.doi.org/10.1136/bmj.303.6807.893>.
31. Humphries SE, Norbury G, Leigh S et al. What is the clinical utility of DNA testing in patients with familial hypercholesterolaemia? Curr Opin Lipidol 2008; 19(4):362–368. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0b013e32830636e5>.
32. Kindt I, Mata P, Knowles JW. The role of registries and genetic databases in familial hypercholesterolemia. Curr Opin Lipidol 2017; 28(2):152–160. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000398>.
34. Perez de Isla L, Alonso R, Watts GF et al. Attainment of LDL-cholesterol treatment goals in patients with familial hypercholesterolemia: 5-year SAFEHEART registry follow-up. J Am Coll Cardiol 2016; 67(11): 1278–1285. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2016.01.008>.
35. Leren TP. Cascade genetic screening for familial hypercholesterolemia. Clin Genet 2004; 66(6): 483–487. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1399–0004.2004.00320.x>.
36. Khera AV, Won HH, Peloso GM et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol 2016; 67(22): 2578–2589. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2016.03.520>.
39. Klancar G, Groselj U, Kovac J et al. Universal screening for familial hypercholesterolemia in children. J Am Coll Cardiol 2015; 66(11): 1250–1257. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2015.07.017>.
40. Wiegman A, Gidding SS, Watts GF et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J 2015; 36(36): 2425–2437. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehv157>.
41. Huijgen R., Hutten B.A., Kindt I et al. Discriminative ability of LDL-cholesterol to identify patients with familial hypercholesterolemia: a cross-sectional study in 26,406 individuals tested for genetic FH. Circ Cardiovasc Genet 2012; 5(3): 354–359. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCGENETICS.111.962456>.
44. Umans-Eckenhausen MA, Defesche JC et al. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001; 357(9251):165–168. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(00)03587-X>.
49. Umans-Eckenhausen MA, Defesche JC, van Dam MJ et al. Long-term compliance with lipid-lowering medication after genetic screening for familial hypercholesterolemia. Arch Intern Med 2003; 163(1): 65–68. Dostupné z DOI: <http://dx.doi.org/10.1001/archinte.163.1.65>.
50. Leren TP, Manshaus T, Skovholt U et al. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway: results from a family-based screening program. Semin Vasc Med 3004; 4(1): 75–85. Dostupné z DOI: <http://dx.doi.org/10.1055/s-2004–822989>.
73. Vuorio A, Docherty KF, Humphries SE et al. Statin treatment of children with familial hypercholesterolemia – trying to balance incomplete evidence of long-term safety and clinical accountability: are we approaching a consensus? Atherosclerosis 2013; 226(2): 315–320. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2012.10.03>.
76. Marteau T, Senior V, Humphries SE et al. Psychological impact of genetic testing for familial hypercholesterolemia within a previously aware population: a randomized controlled trial. Am J Med Genet Part A 2004; 128A(3): 285–293. Dostupné z DOI: <http://dx.doi.org/10.1002/ajmg.a.30102>.
77. Jenkins N, Lawton J, Douglas M et al. How do index patients participating in genetic screening programmes for familial hypercholesterolemia (FH) interpret their DNA results? A UK-based qualitative interview study. Patient Educ Counseling 2013; 90(3): 372–377. Dostupné z DOI: <http://dx.doi.org/10.1016/j.pec.2011.09.002>.
78. Hallowell N, Jenkins N, Douglas M et al. A qualitative study of patients’ perceptions of the value of molecular diagnosis for familial hypercholesterolemia (FH). J Commun Genet 2017; 8(1):45–52. Dostupné z DOI: <http://dx.doi.org/10.1007/s12687–016–0286–0>.
82. Iacocca MA, Hegele RA. Recent advances in genetic testing for familial hypercholesterolemia. Expert Rev Mol Diagn 2017; 17(7): 641–651. Dostupné z DOI: <http://dx.doi.org/10.1080/14737159.2017.1332997>.
83. Taylor A, Wang D, Patel K et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot CASCADE project. Clin Genet 2010; 77(6): 572–580. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1399–0004.2009.01356.x>.
84. Humphries SE, Whittall RA, Hubbart CS et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006; 43(12): 943–949. Dostupné z DOI: <http://dx.doi.org/10.1136/jmg.2006.038356>.
85. Chora JR, Medeiros AM, Alves AC et al. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet Med 2018; 20(6):591–598. Dostupné z DOI: <http://dx.doi.org/10.1038/gim.2017.151>.
86. Dedoussis GV, Schmidt H, Genschel J. LDL-receptor mutations in Europe. Hum Mutat 2004; 24(6): 443–459. Dostupné z DOI: <http://dx.doi.org/10.1002/humu.20105>.
87. Leigh S, Futema M. Whittall R et al. The UCL low-density lipoprotein receptor gene variant database: pathogenicity update. J Med Genet 2017; 54(4): 217–223. Dostupné z DOI: <http://dx.doi.org/10.1136/jmedgenet-2016–104054.
88. Taylor A, Martin B, Wang D et al. Multiplex ligation-dependent probe amplification analysis to screen for deletions and duplications of the LDLR gene in patients with familial hypercholesterolaemia. Clin Genet 2009; 76(1): 69–75. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1399–0004.2009.01168.x>.
89. Bertolini S, Pisciotta L, Rabacchi C et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 2013; 227(2): 342–348. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2013.01.007>.
90. Goldmann R, Tichy L, Freiberger T et al. Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med Genet 2010; 11 : 115. Dostupné z DOI: <http://dx.doi.org/10.1186/1471–2350–11–115>.
91. Myant NB. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis 1993; 104(1–2): 1–18. Dostupné z DOI: <http://dx.doi.org/10.1016/0021–9150(93)90171-p>.
92. Myant NB, Forbes SA, Day IN et al. Estimation of the age of the ancestral arginine3500-->glutamine mutation in human apoB-100. Genomics 1997; 45(1): 78–87. Dostupné z DOI: <http://dx.doi.org/10.1006/geno.1997.4898>.
93. Andersen LH, Miserez AR, Ahmad Z et al. Familial defective apolipoprotein B-100: a review. J Clin Lipidol 2016; 10(6): 1297–1302. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2016.09.009>.
101. Amor-Salamanca A, Castillo S, Gonzalez-Vioque E et al. Genetically confirmed familial hypercholesterolemia in patients with acute coronary syndrome. J Am Coll Cardiol 2017; 70(14): 1732–1740. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2017.08.009>.
102. Silva P, Jannes CE, Oliveira TG et al. Evaluation of clinical and laboratory parameters used in the identification of index cases for genetic screening of familial hypercholesterolemia in Brazil. Atherosclerosis 2017; 263 : 257–262. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2017.06.917>.
103. Haralambos K, Whatley SD, Edwards R et al. Clinical experience of scoring criteria for familial hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis 2015; 240(1): 190–196. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2015.03.003>.
104. van der Graaf A, Avis HJ, Kusters DM et al. Molecular basis of autosomal dominant hypercholesterolemia assessment in a large cohort of hypercholesterolemic children. Circulation 2011; 123(11): 1167–1173. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.110.979450>.
105. Perak AM, Ning H, de Ferranti SD et al. Long-term risk of atherosclerotic cardiovascular disease in US adults with the familial hypercholesterolemia phenotype. Circulation 2016; 134(1): 9–19. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCULATIONAHA.116.022335>.
107. Wang J, Dron JS, Ban MR et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol 2016; 36(12): 2439–2445. Dostupné z DOI: <http://dx.doi.org/10.1161/ATVBAHA.116.308027>.
110. D’Erasmo L, Minicocci I, Nicolucci A et al. Autosomal recessive hypercholesterolemia: long-term cardiovascular outcomes. J Am Coll Cardiol 2018; 71(3): 279–288.Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacc.2017.11.028>.
111. Awan Z, Choi HY, Stitziel N et al. APOE p.Leu167del mutation in familial hypercholesterolemia. Atherosclerosis 2013; 231(2): 218–222. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2013.09.007>.
112. Hegele RA, Ban MR, Cao H et al. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol 2015; 26(2): 103–113. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000163>.
113. Chora JR, Alves AC, Medeiros AM et al. Lysosomal acid lipase deficiency: a hidden disease among cohorts of familial hypercholesterolemia? J Clin Lipidol 2017; 11(2): 477–484.e2. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2016.11.002>.
114. Berge KE, Tian H, Graf GA et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000; 290(5497): 1771–1775. Dostupné z DOI: <http://dx.doi.org/10.1126/science.290.5497.1771>.
115. Stitziel NO, Fouchier SW, Sjouke B et al. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol 2013; 33(12): 2909–2914. Dostupné z DOI: <http://dx.doi.org/10.1161/ATVBAHA.113.302426>.
116. Teslovich TM, Musunuru K, Smith AV et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466(7307): 707–713. Dostupné z DOI: <http://dx.doi.org/10.1038/nature09270>.
117. Ellis KL, Pang J, Chan DC et al. Familial combined hyperlipidemia and hyperlipoprotein(a) as phenotypic mimics of familial hypercholesterolemia: frequencies, associations and predictions. J Clin Lipidol 2016; 10(6):1329–1337.e3. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jacl.2016.08.011>.
127. Clayton EW. Why the Americans with Disabilities Act Matters for genetics. JAMA 2015; 313(22): 2225–2226. Dostupné z DOI: <http://dx.doi.org/10.1001/jama.2015.3419>.
128. Hopkins PN, Lane SR. Genotype-guided diagnosis in familial hypercholesterolemia: clinical management and concerns. Curr Opin Lipidol 2017; 28(2): 144–151. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0000000000000397>.
129. Sturm AC. The role of genetic counselors for patients with familial hypercholesterolemia. Curr Genet Med Rep 2014; 2(2): 68–74. Dostupné z DOI: <http://dx.doi.org/10.1007/s40142–014–0036–8>.
Labels
Angiology Diabetology Internal medicine Cardiology General practitioner for adultsArticle was published in
Athero Review

2020 Issue 3
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Metamizole vs. Tramadol in Postoperative Analgesia
Most read in this issue
- Hypertriglyceridemie – současnost a budoucnost
- Genetické testování familiární hypercholesterolemie v klinické praxi: stanovisko výboru České společnosti pro aterosklerózu
- Agonisty GLP1-receptorov: antidiabetiká s antiaterogénnym účinkom
- Mendelovské randomizační studie: princip a příklady využití v oblasti kardiovaskulární medicíny