
Diagnostika a stratifikace systémové AL amyloidózy ve světle „Doporučení České myelomové skupiny 2013“
Diagnosis and stratification of systemic AL amyloidosis in the light of the 2013 Czech Myeloma Group recommendation
The paper is concerned with classification of amyloidosis and presenting clinical manifestations, examination methods and an effective diagnostic algorithm related to systemic AL amyloidosis. Greater attention is paid to current investigation techniques and their role in the differential diagnosis and stratification of systemic AL amyloidosis as a starting point for prognostic prediction and individual selection of therapy. The review has been prepared in the light of the 2013 Czech Myeloma Group recommendations “Diagnosis and Treatment of Systemic AL Amyloidosis”.
Key words:
classification of amyloidosis, systemic AL amyloidosis, diagnostic algorithm, differential diagnosis, prognostic stratification.
:
V. Ščudla 1; T. Pika 1; P. Látalová 2; P. Flodr 2; V. Maisnar 3; R. Hájek 4; Česká Myelomová Skupina
:
III. interní klinika-revmatologická, nefrologická a endokrinologická LF UP a FN, Olomouc
1; Ústav klinické a molekulární patologie LF UP a FN, Olomouc
2; IV. Interní hematologická klinika LF UK a FN, Hradec Králové
3; Klinika hematoonkologie LF OU a FN, Ostrava
4
:
Klin. Biochem. Metab., 22 (43), 2014, No. 2, p. 49-60
Náplní předloženého sdělení je klasifikace amyloidóz, představení klinických projevů, přehled vyšetřovacích metod a účelný diagnostický algoritmus systémové AL amyloidózy. Hlubší pozornost je věnována recentním vyšetřovacím technikám a jejich postavení v diferenciální diagnostice a stratifikaci systémové AL amyloidózy jako východiska prognostické predikce a individuálního výběru léčby. Přehledové sdělení je zpracováno ve světle Doporučení České myelomové skupiny 2013 „Diagnostika a léčba systémové AL amyloidózy“.
Klíčová slova:
klasifikace amyloidóz, systémová AL amyloidóza, diagnostický algoritmus, diferenciální diagnostika, prognostická stratifikace.
Úvod
Amyloidóza je neobyčejně heterogenní onemocnění postihující většinu orgánů a tkání organismu. Podstatou onemocnění je agregace a depozice bílkovinných substancí ve formě nerozpustných amyloidových fibril v antiparalelním β-uspořádání, uložených extracelulárně v podobě homogenních amorfních hmot, tj. amyloidu. Tkáňová depozita amyloidu mají při barvení konžskou červení cihlově červenou barvu, při použití polarizovaného světla dochází k typické metachromázii, tj. výskytu žlutozeleného zbarvení. Každý typ amyloidu má svůj specifický fibrilární prekurzorový protein, od něhož se odvíjí i jeho název, viz. zjednodušená, klinicky orientovaná klasifikace nejčastějších amyloidóz (Tabulka 1) [1]. Vzhledem k heterogenitě v současnosti známých amyloidóz byla v roce 2012 vypracována „Nomenclature Committee of the International Society of Amyloidosis“ recentní klasifikace, zahrnující > 30 amyloidóz [2]. Amyloidóza se vyskytuje ve formě lokalizované, s pouze ložiskovou depozicí amyloidu, mnohem častější je ale její forma systémová, vyznačující se postižením mnoha orgánů a tkání. Nejčastější je systémová AL amyloidóza (ALA) (70 %), méně častá její ložisková forma (19 %), zatímco ostatní typy se vyskytují mnohem vzácněji, např. senilní a hereditární v 4 % a AA reaktivní amyloidóza v 3 % [1,3]. Historické názvosloví, rozpoznávající amyloidózu primární (AL typ) a amyloidózu sekundární (AA typ) se v současnosti již prakticky nepoužívá. Vzhledem k řídkému výskytu a zejména nízké informovanosti lékařů uniká i v současnosti amyloidóza časnému podchycení a většinou je rozpoznána až pokročilá forma nemoci se závažným, případně nezvratným poškozením životně důležitých orgánů. Amyloidóza se vyznačuje sklonem k trvalé progresi s nepříznivou prognózou, neboť všechny typy amyloidóz jsou léčebně jen omezeně ovlivnitelné [4,5].
![Simplified classification of the most frequent types of amyloidosis [1]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/72dbcb2f60fe0792bb4361147a8fb82a.png)
Charakteristika, epidemiologie a patogeneze systémové AL amyloidózy
Systémová ALA je onemocnění, vyznačující se pozvolnou monoklonální proliferací plazmocytů v kostní dřeni („plazmocelulární dyskrázie“), provázené lineární tkáňovou depozicí nerozpustného bílkovinného materiálu, tj. amyloidu, jehož prekurzorem jsou lehké řetězce (l. ř.) monoklonálního imunoglobulinu (MIg), 3x častěji λ nežli ĸ, případně jejich fragmenty prokazatelné v séru a/ nebo v moči. Název AL amyloidóza odpovídá charakteru prekurzorového proteinu, tj. A – amyloid, L – l. ř. MIg [6, 7]. Progresívní extracelulární akumulace AL amyloidu v cévní a parenchymové složce životně důležitých orgánů vede k tkáňové dezorganizaci, úbytku normálních elementů a k progresívní poruše funkce zejména srdce, ledvin, jater, zažívacího traktu, periferního i autonomního nervstva, nikoliv ale CNS. Mezi tíží amyloidových depozit a závažností orgánové dysfunkce není přímá závislost, neboť za narušení, případně selhání funkce orgánů, zejména srdce končící záhy letálně, je odpovědná vysoká tkáňová toxicita amyloidogenních l. ř. [8].
Incidence ALA, nejčastějšího typu systémové amyloidózy je v USA odhadována na 5,1 – 12,8, v Evropě na 8-10 nemocných/1 milion obyvatel/rok [3,9]. Postihuje především jedince vyššího věku, v 66 % jde o nemocné v rozmezí 50-70 let, pouze 4 % jsou mladší 40 let, poněkud častěji jsou postiženi muži [1, 6]. Nejčastější je typ IgG (35 %), méně často IgA (10 %), IgM (5 %), IgD (1 %), ve zbývajících případech je přítomen pouze l. ř. λ nebo ĸ.
Patogeneze AL amyloidózy je složitá a není doposud zcela objasněna. Základní příčinou je porucha funkce zvolna proliferujících CD38+ a CD138+ monoklonálních plazmocytů produkujících monoklonální „amyloidogenní“ l. ř., tj. „plazmocelulární dyskrázie“ [10]. Dochází k tvorbě makromolekulárních agregátů bílkovinného fibrilárního materiálu s kodepozicí kofaktorů nefibrilárního charakteru. Toxicita amyloidogenních l. ř. je vázána na jejich biochemické charakteristiky a interakce s povrchovým buněčným receptorem RAGE („receptor for advanced glycation end-product“). Umístění tkáňových depozit závisí na „tkáňovém tropizmu“, např. podtyp IgVlĸVI má vztah k rozvoji amyloidové neuropatie zatímco IgVlλII/nebo III k amyloidové kardiomyopatii [11], důležitý je i genový expresní profil („amyloid associated Vλ germline“ gen). Amyloidová depozita obsahují glykoprotein SAP (amyloidový sérový protein), který je vysoce odolný proti proteolýze, který se podílí na rezistenci fibril amyloidu vůči degradaci. Plazmocelulární dyskrazie u ALA je blízká monoklonální gamapatii nejistého významu (MGUS), což dokládá výskyt cytogenetických změn typu del 13 a translokace 14q [12].
Klinický obraz systémové AL amyloidózy
Nejčastější klinické a orgánové projevy, které jsou indikací k dalšímu vyšetření
Klinické projevy systémové ALA jsou pestré, individuálně značně rozdílné a závisí na typu i tíži orgánového postihu [13]. Vyskytují se jednak celkové, nespecifické projevy, tj. slabost, malátnost, únavnost, snížení chuti k jídlu a úbytek hmotnosti a současně i pestré projevy vyplývající z postižení jednotlivých orgánů a tkání [6, 7, 14]. Zahrnují příznaky subjektivní, objektivní a klinické syndromy, jejichž řešení může vést ke kauzální diagnóze (Tabulka 2) [4, 6, 7, 15]. V souboru 868 nemocných byl zaznamenán výskyt slabosti v 68 %, otoky v 62 %, pokles hmotnosti v 43 %, dušnost v 40 %, ortostatická hypotenze v 27 %, dysestézie a parestézie v 23 %, dysgeusie v 18 %, makroglosie v 14 %, kožní purpura v 11 % a průjem v 9 %, ostatní příznaky a projevy jsou méně časté [9]. Postižení jednoho orgánu bývá v 25 %, dvou v 36 %, tří a více orgánů u 39 % nemocných, amyloidová nefropatie se vyskytuje v 72 %, kardiomyopatie v 63 %, postižení jater v 27 %, periferní neuropatie v 19 %, postižení autonomního nervstva v 16 %, postižení měkkých tkání v 12 % a amyloidová kožní purpura v 11 % [9]. Jedinci s IgM typem MIg se vyznačují častějším postižením měkkých tkání, lymfatických uzlin a dýchacího systému, méně častým postižením srdce a v 73 % infiltrací kostní dřeně (KD) lymfoplazmocytárními elementy [13].
Postižení orgánů u systémové AL amyloidózy
Amyloidová nefropatie. Postižení funkce ledvin, zejména glomerulární funkce je nejčastějším projevem a není vázáno pouze na přítomnost depozit amyloidu, ale i na výskyt intersticiální fibrózy a tubulární atrofie. Klíčovým projevem amyloidové nefropatie je výrazná proteinurie, obvykle ale plně vyjádřený nefrotický syndrom (NS) s eventuálním selháním ledvin a se sonografickým průkazem jejich zvětšení [4,16]. Součástí klinického obrazu bývá negativní dusíková bilance s atrofií svalstva, hypoimunoglobulinémie provázená zvýšeným výskytem infekcí, tromboembolická nemoc, projevy akcelerované aterosklerózy v rámci hyperlipoproteinémie a refrakterní anémie navozená mj. ztrátami transferinu močí.
Amyloidová kardiomyopatie. Postižení srdce u systémové ALA se vyskytuje u 2/3 nemocných, izolované postižení myokardu je vzácné (~ 5 %). Myokardiální depozita amyloidu vedou k hypertrofické restriktivní kardiomyopatii, infiltrace chlopní a závěsného aparátu k mitrální a trikuspidální insuficienci, vzácně lze pozorovat perikarditidu. U 1/4 nemocných je hlavním příznakem pravostranné srdeční selhání, ale i hypotenze v důsledku sníženého srdečního výdeje a autonomní neuropatie projevující se ortostatickou i chronickou hypotenzí se synkopálními stavy. Diastolická, posléze systolická dysfunkce vyúsťuje v globální, refrakterní srdeční selhání s dušností, otoky, případně až anasarkou. Častým projevem jsou palpitace a dysrytmie, např. supraventrikulární arytmie a A-V blokády. Postižení srdce amyloidem je nejvýznamnějším prognostickým faktorem a nejčastější příčinou smrti v důsledku elektromechanické disociace nebo maligní komorové arytmie [13,17,18].
Periferní a autonomní neuropatie. Periferní neuropatie (PN) se projevuje pestrou škálou příznaků, zprvu obvykle v oblasti DKK. Má charakter axonální, distální, smíšené a symetrické PN projevující se dysestézií, parestézií s progresívní ztrátou citlivosti, rozvojem svalové slabosti a nezřídka i „syndromem neklidných nohou“ [13]. Senzitivní neuropatie je symetrická, někdy značně bolestivá, motorická neuropatie je podstatně vzácnější, častý syndrom karpálního tunelu je způsoben depozity amyloidu v oblasti vazů [1]. Autonomní neuropatie se podílí na rozvoji posturální hypotenze, dysgeusie a poruše motility GIT, erektilní dysfunkce a anhidrózy [19].
Amyloidóza GIT a jater. Gastrointestinální projevy provázejí depozita amyloidu ve stěně GIT a přítomnost vegetativní dysfunkce. Projevují se pocitem předčasné sytosti, nauzeou a zvracením, postižením distálních oblastí bolestmi břicha, obstipací (pseudoobstrukce), postprandiálním průjmem, malabsorpcí a enteroragií. Příznačným projevem amyloidózy je makroglosie vedoucí k poruše polykání, případně i zúžení dýchacích cest. Zvětšení jater s navýšením zejména jaterní frakce alkalické fosfatázy a GMT bývá nezřídka spojeno s portální hypertenzí, ascitem a splenomegalií [6].
Postižení ostatních orgánů a tkání. Infiltrace dýchacích cest se projevuje kašlem, dušností a hemoptýzou, postižení laryngu způsobuje inspirační dušnost a dysfonii, vzácným projevem je nodulární postižení plic (amyloidom) a/nebo amyloidová infiltrace tracheobronchiálního stromu, postižení pleury vede k torpidnímu pleurálnímu výpotku. U 1/4 nemocných se vyskytují poruchy hemostázy, nejčastěji v podobě vaskulární kožní purpury patrné periorbitálně a/nebo v oblasti hřbetu rukou, splenomegalie může vést k trombocytopenii. Významné snížení hladiny f. X může vést v případě jaterní a renální biopsie k život ohrožujícímu krvácení [6]. Postižení skeletu bývá asymptomatické, infiltrace endokrinních orgánů se projevuje hypotyreózou nebo Addisonovou chorobou, lymfatických uzlin lymfadenomegálií, imbibice příčně pruhovaného svalstva svalovou slabostí a „pseudohypertrofií“, synoviální depozita amyloidu obrazem neerozivní symetrické artropatie, zatímco postižení kůže jejím ztluštěním nebo přítomností papul, v případě nehtů obrazem onychodystrofie [14].
Vyšetřovací techniky u systémové AL amyloidózy
Klíčovým předpokladem rozpoznání systémové ALA je požadavek, aby lékaři v klinické praxi na toto onemocnění mysleli a byli obdařeni žádoucí dávkou diagnostické invence. K podezření by měla vést zejména přítomnost NS s případnou renální insuficiencí, nedilatační kardiomyopatie s restriktivním plněním, nejasná periferní a/nebo autonomní neuropatie a další mnohotvárné subjektivní příznaky a objektivní projevy (Tabulka 2). Za těchto okolností je povinností lékaře pátrat po přítomnosti MIg standardní a imunofixační elektroforézou (IFE) séra a zejména po zvýšených hladinách volných lehkých řetězců (VLŘ) včetně jejich patologického poměru ĸ/λ (FreeliteTM). V případě pozitivity uvedených vyšetření je nutné odeslání nemocného k dovyšetření na hematologické oddělení [6, 7, 14, 19, 20]. Přehled základních i výběrově indikovaných vyšetření při podezření na systémovou ALA je uveden v Tabulce 3.
![Clinical manifestations of systemic amyloidosis [4, 6, 15]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/edd44dafaa2a9d066c48916b5eea3215.png)
![Overview of examinations necessary for diagnosis and staging of AL amyloidosis [35]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/09905e6be92c528a336012079c5c84b8.png)
Standardní techniky v diagnostice systémové AL amyloidózy
Histologické vyšetření v základním a speciálním barvení. Depozita amyloidu lze ve většině případů rozpoznat již v základním barvení histologických preparátů (hematoxylin-eosin), kde se znázorňuje jako amorfní, cihlově červený materiál ve stěnách cév, v intersticiu i v oblasti bazálních membrán. Pro specifické znázornění amyloidu je nejčastěji používáno barvení konžskou červení, kdy depozita amyloidu vykazují oranžové až červené zbarvení. Při vyšetření polarizačním mikroskopem vzniká při použití konžské červeně ve vazbě na amyloid tzv. dvojlom („birefrigerence“) a dichroismus, vyznačující se „apple-green“ zbarvením [21] při použití klasického zpracování histologického materiálu (fixace formalinem a zalití do parafinu), kvalitnější výsledek ovšem poskytují zmrazené řezy nefixované tkáně. Při histologickém vyšetření vzorku KD nutno odlišit plazmocelulární dyskrázii u AL amyloidózy od zmnožení plazmocytů u MM (koincidence 12-20 %) a od hereditární amyloidózy provázené ve 3-10 % přítomnosti MIg [13,19].
Imunohistochemické vyšetření. V současné diagnostice ALA je standardem typizace amyloidu metodou nepřímé imunohistochemie (IHC) s pomocí protilátek proti ĸ a λ l. ř. MIg, transthyretinu a amyloidu A, přičemž spektrum komerčně dostupných protilátek proti hereditárním formám amyloidózy se stále rozšiřuje [22]. Vzhledem k proměnlivé senzitivitě IHC (maskování antigenu, variabilita domén l. ř. aj.) je některými patology preferováno imunofluorescenční vyšetření vzorků ze zmrazené tkáně. Vzhledem k odlišnému rozpoznávání epitopů jednotlivými protilátkami se ke zvýšení senzitivity a specificity vyšetření používá více polyklonálních a monoklonálních protilátek proti epitopům ĸ a λ l. ř. [23].
Fluorescenční vyšetření. Přímá fluorescence, používající řezy ze zmrazené nefixované tkáně prokazuje přímou vazbu fluorochromu (konžské červeně) na amyloid. Ve srovnání s použitím polarizačního mikroskopu se vyznačuje, a to zejména v případě málo objemných depozit amyloidu, vyšší senzitivitou [24]. Zcela stěžejní a dnes již standardní metodou je tzv. nepřímá fluorescence, využívající protilátky proti l. ř. MIg označených fluorochromem.
Elektronová mikroskopie umožňuje průkaz nevětvených splývajících mikrofibril šíře 7,5 – 10 nm, vytvářejících nepravidelnou síť amyloidu. Vysoce specializovaná pracoviště mají k dispozici imunoelektronovou mikroskopii, spojující výhody vysokého rozlišení elektronového mikroskopu se specificitou reakce antigenprotilátka, umožňující ověření koincidence imunohistochemické reakce v místě amyloidových fibril [25].
Podrobnější informaci lze nalézt v připravovaných „guidelines“ („Doporučený postup pro histopatologickou diagnostiku amyloidu/amyloidózy“) Společnosti českých patologů ČLS JEP.
Průkaz MIg a VLŘ v séru a/nebo v moči
Průkaz přítomnosti MIg a patologické hladiny VLŘ včetně abnormálního poměru ĸ/λ je základním stavebním kamenem v diagnostice systémové ALA. Standardní elektroforéza bílkovin séra je stále základní, ovšem málo citlivou metodou, odhalující přítomnost MIg pouze u ~ 50 % ALA, přičemž hodnota M-proteinu nebývá vysoká (u ~ 70 % jedinců je < 20 g/l) [20,26]. Základním vyšetřením je proto IFE, odhalující přítomnost MIg nebo l. ř. v 71-84 % se schopností citlivé detekce všech imunochemických typů MIg. Nevýhodou IFE je nemožnost monitorace léčby, neboť neposkytuje kvantitativní výsledky [20, 26]. Většina nemocných s ALA se vyznačuje nízkou hladinou, případně chyběním intaktního MIg, avšak charakteristické je zvýšení hladiny VLŘ λ nebo ĸ [13, 27]. V současnosti je proto zcela standardním postupem kvantitativní stanovení sérových hladin VLŘ, včetně indexu ĸ/λ s pomocí nefelometrické nebo turbidimetrické techniky (FreeliteTM), dosahující při kombinaci s IFE až 99 % senzitivity [20, 27, 28]. Zvýšená hladina VLŘ v séru spolu s patologií indexu ĸ/λ se vyskytuje u systémové ALA v 86-98 % a vydává vlastně zprávu o výši prekurzorového proteinu a koreluje s náloží amyloidu. Spolu s imunohistochemickým vyšetřením KD (index ĸ/λ CD138+ plazmocytů) umožňuje analýza VLŘ odhalení monoklonality plazmocelulárního procesu. Sledování pohybu sérových hladin VLŘ u ALA je metodou volby při monitorování léčby a hodnocení hloubky léčebné odezvy. Výše hladiny VLŘ při diagnóze je velmi důležitým prognostickým faktorem a jedním ze 3 kritérií revidovaného stratifikačního systému systémové ALA [29, 30]. Z výsledků recentních studií vyplývá nadějný přínos vyšetřování párů těžkých/lehkých řetězců („heavy /light chain“, metoda HevyliteTM) v diagnostice a monitorování průběhu systémové ALA [20, 27, 31].
Zobrazovací techniky u systémové AL amyloidózy
Použití zobrazovacích metod u systémové ALA sleduje dva hlavní aspekty, tj. morfologické a funkční zhodnocení orgánového postižení amyloidem.
Radiografické vyšetření (RTG) nemá v diagnostickém algoritmu samotné ALA větší přínos, RTG skeletu slouží k pátrání po osteolytických lézích, a tím i vyloučení asociace ALA s MM. RTG hrudníku může odhalit rozšíření srdečního stínu s městnáním v malém oběhu, pleurální výpotek a vzácně i postižení plic v rámci ALA.
Počítačová tomografie (CT) může odhalit ložiskové léze v oblasti orbity, nasofaryngu a laryngu, především je ale vhodná k detekci depozit amyloidu v oblasti tracheobronchiálního stromu, plicního parenchymu a rozpoznání hepatomegalie (šíře > 15 cm).
Ultrazvukové vyšetření. Echokardiografie (ECHO) je stěžejní metodou pro diagnostiku a monitorování amyloidové kardiomyopatie a spolu s vyšetřením srdečních biomarkerů klíčovou metodou k hodnocení léčebné odezvy nebo progrese nemoci. Slouží k detekci koncentrické hypertrofie myokardu levé komory (LK), měření šíře interventrikulárního septa (> 12 mm), rozpoznání diastolické dysfunkce s restriktivním plněním LK a se ztrátou longitudinální kontraktility, snížení ejekční frakce a dilatace levé síně [19]. Typický je obraz jemně žíhaného myokardu („granular sparkling“) [18]. Vysoce specializovaná pracoviště používají k detailní diagnostice tkáňovou dopplerometrii („strain, specle cracking“) [32].
Magnetická rezonance (MRI) srdce umožňuje morfologické a funkční zobrazení. Kontrastní vyšetření gadoliniem slouží stejně jako fenomén pozdního sycení subendokardiálně či transmurálně („delayed enhancement“) k průkazu imbibice myokardu amyloidem [32]. MRI je rovněž užitečná v odhalení ložiskové amyloidózy v oblasti laryngu, nasofaryngu, míchy a urogenitálního traktu.
Radioscintigrafie s využitím značeného 123I-SAP je neinvazivní, kvantitativní a velmi citlivá metoda pro celotělovou detekci amyloidu, vhodná k monitorování výsledku léčby [29]. Vzhledem k potenciálnímu riziku přenosu infekce není v ČR zavedena.
Diagnostická kritéria a postupy u systémové AL amyloidózy
Diagnostická kritéria. Vzhledem k neexistenci samostatného laboratorního ukazatele nebo zobrazovací techniky, které by samy o sobě umožnily rozpoznání systémové ALA, opírá se diagnóza o splnění souboru kritérií, sestavených v roce 2011 IMWG (International Myeloma Working Group, Tabulka 4) [5,13,19]. Spolehlivá diagnóza systémové ALA vyžaduje splnění všech čtyř základních kritérií nemoci:
- Jednoznačný průkaz, že k postižení některého z orgánů, tj. ledvin, srdce, GIT včetně jater, nervové tkáně a vzácněji svalů, kůže aj. došlo v důsledku amyloidózy.
- V klinické praxi se k detekci amyloidu používá standardně současná aspirační biopsie podkožního tuku (pozitivita 60-80 %) a kostní dřeně (50-65 %), při jejich negativitě i slinné žlázy (15 % pozitivita při negativitě amyloidu v podkožním tuku), případně, a to i v souladu s klinickým obrazem, (např. makroglosie) jazyka, nebo rekta a gingivy. Při negativitě necíleného odběru se volí cílená biopsie dominantně postiženého orgánu, tj. ledviny, myokardu (endomyokardiální biopsie), jater, GIT, ojediněle i n. suralis, kosterního svalstva, uzliny aj., vyznačující se 80-100 % výtěžností [5].
- Vzhledem k určité nespolehlivosti imunohistochemické identifikace AL amyloidu (v 50 % ztráta antigenních epitopů v průběhu fibrilogenezy) se stále významněji uplatňuje hmotnostní spektrometrie, případně imunoelektronová mikroskopie.
- Monoklonální charakter plazmocytů lze prokázat detekcí intaktní molekuly MIg a/nebo klíčového znaku ALA, tj. zvýšené hladiny VLŘ MIg s patologickým indexem ĸ/λ [5,20]. Samotný průkaz MIg k průkazu ALA nepostačuje, k rozpoznání monoklonality je nutno využít imunochemického vyšetření se stanovením poměru exprese ĸ/λ plazmocytů získaných při trepanobiopsii KD, nebo analýzou aspirátu KD pomocí multiparametrické průtokové cytometrie (MFC) [33]. Nutno mít na paměti, že zastoupení plazmatických buněk je při ALA nízké (medián 7 %) a morfologie bez větších atypií [13].
![The IMWG diagnostic criteria for systemic AL amyloidosis [13]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d5d28802180ab4840175dd5e51d8e487.png)
Pozorované chromozomální aberace, např. monozomie chromozomu 18 (72 %), trisomie chromozomů 9, 7, 11, 15 a 18 nebo t(11;14), del (13q14) a zisk 1q21 nejsou z hlediska systémové ALA specifické [12,13].
Od přímého průkazu amyloidu v tkáni lze ustoupit v případě typického nálezu pro ALA (vysoká hladiny VLŘ s patologií indexu ĸ/λ, průkaz monoklonality plazmocytů s pomocí imunohistochemie a/nebo M-FC a nálezu amyloidu v podkožním tuku) a typického poškození orgánové funkce bez zřejmé jiné příčinné nemoci. Pro potřeby klinické praxe byla v rámci „10th International symposium on amyloid and amyloidosis, Tours 2004“ dohodnuta kritéria, přesně definující klinicko-laboratorní známky orgánového postižení (Tabulka 5) [19].
![Evidence of organ involvement in systemic AL amyloidosis [19]
(A consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, 2004)](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/621d31559ca3624155eec436a1526d7f.png)
Diagnostická kritéria postižení jednotlivých orgánových systémů u systémové ALA
Diagnostika amyloidové kardiomyopatie.
Laboratorní vyšetření. Nedílnou součástí diagnostického algoritmu ALA je vyšetření sérové hladiny srdečních biomarkerů troponinu T (TnT) a NT-pro „brain“ natriuretického peptidu (NT-proBNP), ojediněle i troponinu I (TnI) laboratorně standardizovanými metodami [17]. Hladina TnT, citlivého ukazatele myokardiálního postižení má při vyšetření v období diagnózy úzký vztah k prognóze (> 0.035 μg/l), přetrvávající zvýšení signalizuje pokračující ztrátu kardiomyocytů [29, 34]. Hladina NTproBNP má rovněž prognostický a stratifikační potenciál (> 332 ng/l), jeho normální hladina prakticky vylučuje amyloidovou kardiomyopatii [34]. Vzhledem k tomu, že při účinné léčbě dochází k poklesu hladin obou srdečních biomarkerů, jsou používány jako kritéria kardiologické remise. Vzhledem k přetrvávání elevace TnT při léčbě lenalidomidem a NT-proBNP při renální nedostatečnosti jeví se v těchto situacích výhodnější stanovení samotného BNP („brain natriuretic peptide“) [35].
Paraklinická vyšetření. EKG vyšetření se v případě pokročilé amyloidové kardiomyopatie vyznačuje snížením voltáže v končetinových svodech (< 5 mm), obrazem QS ve svodech V1-3 (obraz „pseudoinfarktu“) a záchytem arytmií či A-V blokád [18]. Jak bylo uvedeno, stěžejním vyšetřením ALA je ECHO a MRI. V případě negativity necílené biopsie a přítomnosti rozšíření interventrikulárního septa nebo jiných známek vyvolávajících podezření na ALA je plně indikována endomyokardiální biopsie. Závažnost postižení vegetativního nervového systému lze posoudit provedením testu variability srdeční frekvence (Ewingův test).
Diagnostika amyloidové nefropatie.
Postižení ledvin, nejčastěji postiženého orgánu u systémové ALA (50-80 %), se projevuje neselektivní proteinurií dosahující intenzity NS (2,5-30 g/24 hod.), případně i vzestupem sérového kreatininu a mikroskopickou hematurií. Vedle standardního vyšetření NS jsou nezbytnou součástí analýzy nejen standardní elektroforéza, ale i IFE zaměřené na detekci MIg a vyšetření hladin VLŘ v séru a/nebo v moči (tj. Bence-Jonesova bílkovina). Minimální proteinurie se vyskytuje pouze v případech, kdy depozita amyloidu jsou omezena pouze na tubuly, intersticium a cévní složku ledvinové tkáně [4].
Glomerulární filtrace (GF). U amyloidové nefropatie dochází k rychlému poklesu GF s častým vývinem ESRD („end-stage renal disease“). U většiny nemocných se vyvíjí v závislosti na masivitě amyloidových deposit individuálně různě rychlý pokles GF, o renální prognóze ovšem rozhoduje zejména tíže depozit v tubulo-intersticiální oblasti. Porucha orgánové funkce ovšem záleží spíše nežli na rozsahu amyloidových depozit na toxicitě prekurzorových proteinů, tj. amyloidogenních l. ř. a jejich oligomerů [4]. Při hodnocení GF nutno zohlednit rovněž pokles prokrvení ledvin v důsledku snížení srdečního výkonu a krevního tlaku, závažnosti vegetativní dysfunkce i předchozí aplikaci kontrastní látky. K náhlému, obvykle přechodnému poklesu GF dochází občas po vysokodávkované chemoterapii s podporou autologní transplantace krvetvorných buněk a po léčbě lenalidomidem. Při hodnocení GF nutno zohlednit, že pokles amyloidogenních l. ř. po léčbě vede obvykle k pouhé stabilizaci GF a v případě ztráty svalové tkáně mohou hodnoty kreatininu nadhodnocovat stupeň skutečného snížení. Reálná hodnota GF může být při použití výpočtových metod oproti GF odhadnuté pomocí vyšetření cystatinu či clearence inulinu podhodnocena.
Tubulární funkce, sonografie a biopsie ledviny. Vyšetření tubulárních funkcí je k relativně častému výskytu funkčních tubulárních poruch plně indikováno, avšak úplný obraz postižení proximálního (Fanconiho syndrom) a distálního tubulu (nefrogenní diabetes mellitus) je spíše výrazem koincidence s MM [4]. U většiny amyloidových nefropatií se setkáváme se symetrickým zvětšením a změnou echogenity v ultrazvukovém obraze [16]. V případě absence průkazu amyloidu v necílené biopsii je rozhodnutím volby cílená biopsie ledviny, vyznačující se 90-95 % záchytností a umožňující přesné zhodnocení nejen amyloidových depozit, ale i poškození ledvinové tkáně [4].
Kritéria postižení ostatních orgánů u systémové AL amyloidózy
Nervový systém. Rozpoznání distální smíšené senzomotorické PN DKK je založeno na rozboru klinického obrazu a neurologickém vyšetření včetně elektromyografie (EMG). Jen velmi individuálně se využívá histologie n. suralis. Rozpoznání autonomní neuropatie je založeno na rozboru klinického obrazu doplněného o testy vegetativní dysfunkce.
Gastrointestinální trakt. Vzhledem k tomu, že klinické projevy jsou často vázány na přímou infiltraci stěny zažívací trubice, je indikováno endoskopické vyšetření GIT s odběrem materiálu na histologické vyšetření. Biopsie jater je indikována při zvětšení šíře jater > 15 cm (sonografie a CT), přítomnost portální hypertenze se splenomegalií a > 1,5 násobkem zvýšení jaterní frakce alkalické fosfatázy (ALP).
Postižení plic. Průkaz je založen na provedení konvenční radiografie hrudníku, zejména ale vysoce citlivého HR - CT („high-resolution CT“), umožňující odhalení depozice amyloidu v oblasti plic a pleury. V případě zmnožené retikulonodulární kresby (difuzní intersticiální amyloidóza), „coin lesion“ (amyloidom) a pleurálního výpotku může správné diagnóze napomoci transbronchiální nebo video-asistovaná thorakoskopická biopsie, v případě depozit v bronchiálním stromu bronchoskopie s odběrem materiálu na histologii a spirometrické vyšetření včetně difuzní plicní kapacity.
Postižení pohybového aparátu. Pro odhalení postižení příčně pruhovaného svalstva lze využít vyšetření kreatinkinázy a myoglobinu v séru, MRI a biopsie svalu. V případě amyloidové artropatie je přínosná biopsie synovie, či histologické vyšetření tkáně získané při operaci karpálního tunelu.
Postižení krvetvorného a koagulačního systému. Vyšetření krevního obrazu může odhalit anémii chronických chorob a zvýšení počtu krevních destiček. Koagulační vyšetření může zaznamenat zvýšení fragility kožních cév, prodlouženou krvácivost a vzestup aPTT a občas i snížení hladiny f. X (důsledek vazby na amyloid). NS bývá provázen snížením hladiny AT III a vysokou koncentrací fibrinogenu [4].
Postižení ostatních orgánů a tkání. Klinický obraz je nutno doplnit odběrem tkáně k histologickému vyšetření, např. excize jazyka při makroglosii, biopsie změn přítomných na kůži, histologie lymfatické uzliny při lymfadenomegalii.
Diagnostický algoritmus systémové AL amyloidózy
V klinické praxi je vhodné přistupovat k nemocnému s podezřením na systémovou ALA podle „diagnostického algoritmu“ (Tabulka 6). Vždy je ale nutné splnit 4 základní diagnostické kroky, tj.: 1) na možnost systémové ALA pomyslet; 2) potvrdit přítomnost amyloidózy s pomocí tkáňové biopsie; 3) určit typ prekurzorového proteinu; 4) posoudit rozsah orgánového postižení [35]. Nejdůležitější klinické a laboratorní „varovné“ příznaky, usnadňující časné rozpoznání systémové ALA jsou shrnuty v Tabulce 7 [36]. K rozpoznání systémové ALA, včetně jejího odlišení od ložiskové formy a od ostatních typů amyloidóz (ATTR a Aβ2 - M amyloidózy) lze použít diagnostický algoritmus, založený na systému postupných diagnostických kroků (obr. 1) [5].
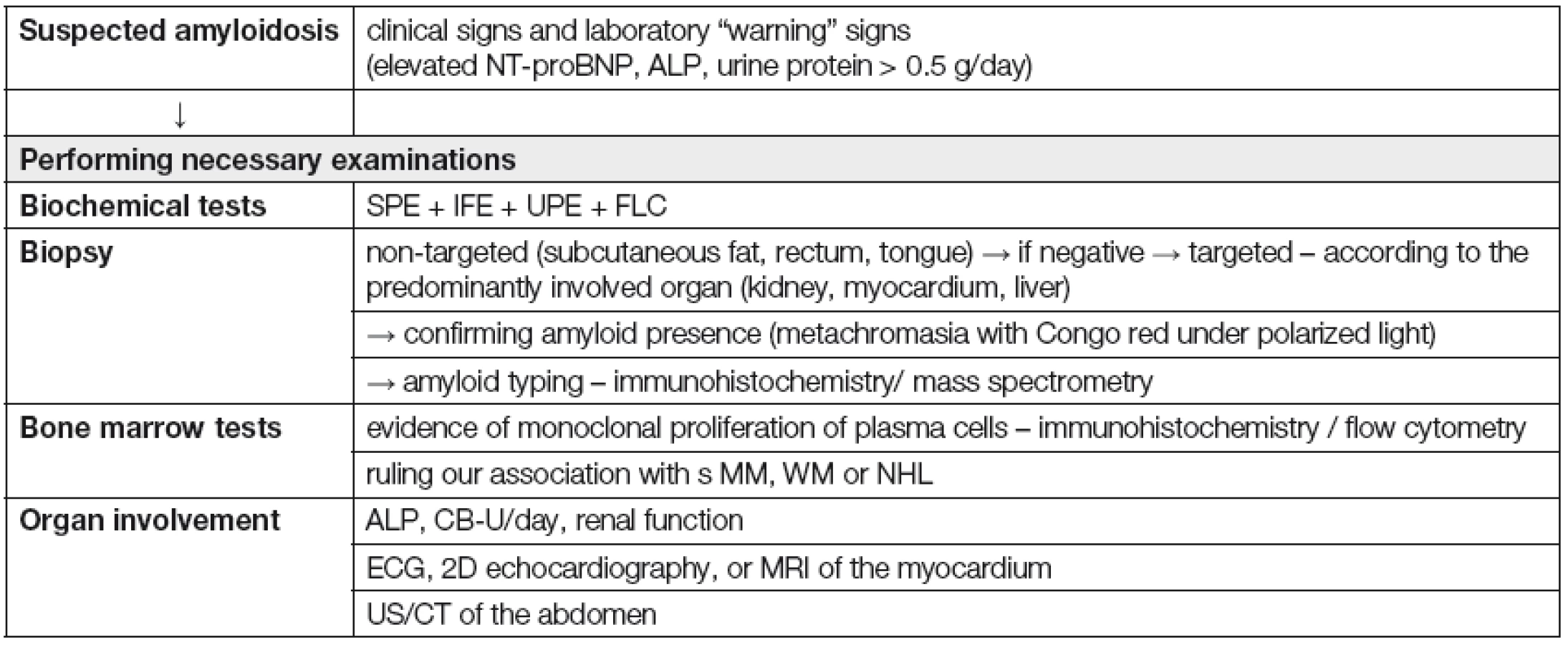
![Warning signs suggestive of systemic AL amyloidosis [36]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/351bfd222eb4e3d41fc1417e3b0b0cf2.png)
![Fig. 1. Algorithm for evaluating patients with suspected amyloidosis [5]
XRT – X-ray therapy](https://pl-master.mdcdn.cz/media/image/32ae1647c4fdfa86d254292f9aff41e9.jpg?version=1711878761)
Diferenciální diagnóza systémové AL amyloidózy
V případě histologického průkazu amyloidózy je nutné především odlišení systémové ALA od její ložiskové formy a systémové ALA provázející MM nebo jiný B-lymfoproliferativní stav. Ložisková forma AL amyloidózy se vyznačuje typickou predilekční lokalizací, nepřítomností MIg, normální hodnotou VLŘ v séru a nepřítomností monoklonálních plazmocytů v KD [33, 37]. Reaktivní AA amyloidóza navazuje na předchozí dlouhodobý průběh základního vyvolávajícího onemocnění (nejčastěji revmatoidní artritida), jsou zvýšeny hladiny reaktantů akutní fáze, zejména sérového amyloidového proteinu A (SAA) a SAP, pozitivita imunohistochemického vyšetření s použitím specifické monoklonální protilátky. Vzácný hereditární ATTR typ amyloidózy lze rozpoznat s pomocí DNA analýzy, hmotnostní spektrometrie a/ nebo imuno-elektronové mikroskopie, přičemž nutno mít na paměti možnou 5-10 % koincidenci s MGUS [6, 25, 36]. Některá z uvedených speciálních metod by měla být provedena vždy u amyloidózy s příznaky typickými pro ATTR amyloidózu, tj. postižení periferního a autonomního nervového systému a postižení srdce. U nemocných v dlouholeté hemodialyzační léčbě se ojediněle vyskytuje hemodialyzační (Aβ2-M) amyloidóza. U starších mužů s dominantním postižením srdce a s projevy městnavé srdeční slabosti a/nebo poruchami rytmu nutno pátrat po amyloidóze senilní. Na možnost systémové ALA je nutno pomýšlet u všech jedinců s MGUS a před stanovením definitivní diagnózy provést pečlivé vyšetření podle diagnostického algoritmu (Tabulka 6) [5, 36, 38]. Nutno odlišit rovněž MGRS („monoclonal gammopathy of renal significance“) provázející ojediněle MGUS s přítomností vysoce nefrotoxických l. ř. [39] a onemocnění z depozice lehkých řetězců („light chain deposition disease“, LCDD) [40], nebo z depozice těžkých řetězců imunoglobulinu („heavy chain deposition disease“, HCDD) [6]. Depozita v rámci LCDD i HCDD se vyznačují negativitou při barvení s použitím konžské červeně. Precizní zařazení rozpoznané amyloidózy je nezbytné nejen z hlediska odlišných klinických projevů a prognózy, ale především z hlediska účelné, pro jednotlivé typy amyloidóz Table 7: Warning signs suggestive of systemic AL amyloidosis [36] Involved organ or syndrome Frequency (%) Early “red flags” Sensitivity (%) Heart 70 NT-proBNP > 332ng/L BNP > 73ng/L 100 89 Kidneys 70 urine albumin > 0.5g/24hrs eGFR < 50mL/min per 1.73m2 Liver 22 Elevated ALP and/or GMT if other causes are ruled out Peripheral and autonomic neuropathy 14 Neuropathic pain and loss of sensitivity to temperature Erectile dysfunction Soft tissue Cutaneous ecchymosis Macroglossia Muscle pseudohypertrophy 13 Carpal tunnel syndrome NT-proBNP – N-terminal pro - brain natriuretic peptide, BNP – brain natriuretic peptide, eGFR – glomerular filtration rate, ALP – alkaline phosphatase, GGT – gamma-glutamyl transpeptidase specifické léčby [7]. Velmi důležitou součástí diferenciální diagnostiky systémové ALA je její odlišení od MM a rovněž podchycení současné koincidence obou chorobných stavů [1]. Současná systémová ALA a MM se pozoruje u 12-20 % jedinců, přičemž diagnóza MM obvykle předchází. V období diagnózy MM lze současnou ALA rozpoznat u 1 % nemocných [1]. Rozpoznání MM je v této situaci založeno na přítomnosti anémie, > 30 % plazmocytů v KD, hyperkalcémii a bolestech skeletu s průkazem osteolytických lézí, případně dalších nálezů včetně genetických změn (např. zvýšená exprese p53, ras, c-myc aj.), nacházených obvykle při maligní transformaci MGUS v MM. Na vývin systémové ALA v průběhu MM nutno pomýšlet v případě výskytu projevů, nepatřících do obvyklého klinického obrazu MM, např. makroglosie, NS, restriktivní kardiomyopatie, autonomní neuropatie aj. [41].
Prognostická stratifikace systémové AL amyloidózy
Prognóza nemocných se systémovou ALA je značně variabilní a v případě nemocných s amyloidovou kardiomyopatií, manifestující se městnavou srdeční slabostí a/nebo synkopálními stavy se vyznačují vysokým rizikem náhlé smrti a krátkým mediánem celkového přežití (4-6 měsíců) [1, 35]. Zatímco v historických studiích byl medián přežití 15-18 měsíců, v posledním desetiletí dosahuje dvojnásobné délky [1, 35]. V průběhu posledních let byl shromážděn úctyhodný soubor ukazatelů, majících různě významný vztah k prognóze, např.: zastoupení plazmocytů v KD, jejich charakteristiky (proliferační potenciál, zastoupení aberantního klonu vyhodnoceného s pomocí M-FC), hladina dominantního VLŘ v séru a indexu ĸ/λ, výše β2-M, závažnost a počet postižených orgánů, hloubka léčebné odezvy aj. Excelentními prediktory prognózy systémové ALA jsou sérové hladiny solubilních srdečních biomarkerů (TnT, TnI, NT-proBNP a BNP), které se staly východiskem pilotního prognostického systému, rozdělujícího nemocné se systémovou ALA do tří stádií [34], neboť závažnost postižení srdce je i u asymptomatických jedinců stěžejním prognostickým faktorem [29].
V současnosti je v klinické praxi používán tzv. „revidovaný prognostický stážovací systém“ vypracovaný na Mayo klinice, založený na hladinách cTnT, NT-proBNP a VLŘ v séru (Tabulka 9, obr. 2) [30]. Spojením parametrů srdečního postižení (srdeční biomarkery) a produktů plazmocelulárního klonu (VLŘ séra) byl sestaven komplexní stážovací systém s vysokým prognostickým potenciálem, a to i z hlediska předpovědi dlouhodobé prognózy, vytvářející dobrý základ pro „risk - adapted“ strategii léčby i v případech s „viscerálním amyloidovým syndromem“ [5, 30]. Prognostický význam revidovaného „Mayo clinic“ stážovacího systému dokládají nejen významně rozdílné délky mediánů celkového přežití, ale i zcela diferentní průběhové charakteristiky křivek přežití nemocných se systémovou ALA rozvrstvených do čtyř stádií (obr. 1). V případě potřeby lze nalézt podrobnější informace o problematice diagnostiky, diferenciální diagnostiky a léčby amyloidózy v „Doporučení České myelomové skupiny 2013“ [42].


Podpora IGA MZ ČR NT-12451-5 a IGA MZ ČR NT-14400.
Do redakce došlo 16. 1. 2014
Adresa pro korespondenci:
Prof. MUDr. Vlastimil Ščudla, CSc.,
3.IK-NRE, LF UP a FN v Olomouci
I. P. Pavlova 6 775 20 Olomouc
e-mail:vlastimil.scudla@fnol.cz
Sources
1. Kyle, R. A., Gertz, M. A. Primary systemic amyloidosis: clinical features in 474 and laboratory cases. Semin. Hematol. 1995, 32, p. 45-59.
2. Sipe, J. D., Benson, M. D., Buxbaum, J. N. et al. Amyloid fibril protein nomenclature: 2012 recommendation from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2012, 19, p. 167-70.
3. Kyle, R. A., Linos, A., Beard, C. M. et al. Incidence and natural history of primary systemic amyloidosis in Olmsted county, Minessota,1950 through 1989. Blood 1992, 79, p. 1817-22.
4. Ryšavá, R. Systémové amyloidózy a jejich léčba. Maxdorf Jessenius/Sv 61, Praha 2013, p. 124.
5. Gertz, M. A. Immunoglobulin light chain amyloidosis: 2011 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2011, 86, p. 181-86.
6. Bird, J. Guidelines on the diagnosis and management of AL amyloidosis. Br. J. Haematol. 2004, 125, p. 681-700. www.blackwellpublishing.com/bjh; www.bloodmed.com;7. Ščudla, V., Pika, T. Současné možnosti diagnostiky a léčby systémové AL-amyloidózy. Vnitř. Lék. 2009, 55 (Suppl 1), p. 77-87.
8. Rajkumar, S. V., Glassock, R. J., Kyle, R. A. et al. Pathogenesis and clinical features of primary (AL) amyloidosis and light and heavy chain deposition diseases. www.uptodate.com
9. Merlini, G. AL amyloidosis: diagnosis and prognosis. Haematologica 2007, 92(Suppl 2), p. 58-59.
10. Lavatelli, F., Palladini, G., Merlini, G. Pathogenesis of systemic amyloidosis. In: Gertz MA, Rajkumar SV. Amyloidosis: diagnosis and treatment, 1st ed., Springer Science, New York 2010, p. 49-64.
11. Comenzo, R. L., Wally, J., Kica, G. et al. Clonal immunoglobulin light chain variable region germline gene use in AL amyloidosis: association with dominant amyloidrelated organ involvement and survival after stem cell transplantation. Br. J. Haematol. 1999, 106, p. 744-51.
12. Harrison, C. J., Mazzullo, H., Ross, F. M. et al. Translocations of 14q32 and deletions of 13q14 are common chromosomal abnormalities in systemic amyloidosis. Br. J. Haematol. 2002, 117, p. 427-35.
13. Rajkumar, S. V., Glassock, R., Kyle, R. A. Clinical presentation, laboratory manifestations, and diagnosis of immunoglobulin light chain (AL) amyloidosis (primary amyloidosis). Available from: www.uptodate.com, 2013 UpToDate
14. Adam, Z., Ščudla, V. Klinické projevy a diagnostika AL amyloidózy a některých dalších typů amyloidóz. Vnitř. Lék. 2001, 47, p. 36-45.
15. Gertz, M. A., Hayman, S. R. Immunoglobulin light chain amyloidosis. In: Rajkumar SV, Kyle RA. Treatment of multiple myeloma and related disorders. Cambridge: Cambridge University Press, 2009, p. 112-128.
16. Krejčí, K., Zadražil, J., Tichý, T. Nefropatie v ultrazvukovém a histologickém obraze. 1. vyd., Praha 2011, Maxdorf. Jessenius, p. 186.
17. Pika, T., Vymětal, J., Metelka, R. et al. Postižení srdce při AL amyloidóze. Interní Med. 2008, 10, p. 466-69.
18. Fikrle, M., Paleček, T., Kuchyňka, P. et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa 2012, http://dx.doi.org/10.1016/j.crvasa.2012.11.018.
19. Gertz, M. A., Comenzo, R., Falk, R. H. et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International symposium on amyloid and amyloidosis. Am. J. Hematol. 2005, 79, p. 319 - 28.
20. Bradwell, A. R. Serum free light chain analysis (plus Hevylite) 6th ed., Birmingham: The Binding Site Ltd, 2010, p. 350.
21. Howie, A. J., Brewer, D. B., Howell, D. et al. Physical basis of colors seen in Congo red-stained amyloid in polarized light. Lab. Invest. 2008, 88, p. 232-42.
22. Picken, M. M., Westermark, P. Amyloid detection and typing: summary of current practice and recommendations of the consensus group. Amyloid. 2011, 18 (Suppl. 1), p. 48-50. doi: 10.3109/13506129.2011.574354017.
23. Schönland, S. O., Hegenbart, U., Bochtler, T. et al. Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. Blood 2012, 119, p. 488-93.
24. Marcus, A., Sadimin, E., Richardson, M., et al. Fluorescence microscopy is superior to polarized microscopy for detecting amyloid deposits in Congo red-stained trephine bone marrow biopsy specimens. Am. J. Clin. Pathol. 2012, 138, p. 590-93.
25. Arbustini, E., Verga, L., Concardi, M. et al. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid 2002, 9, p. 108-14.
26. Maisnar, V., Tichý, M. a kol. Monoklonální imunoglobuliny – výskyt, význam a možnosti jejich průkazu. 1.vyd., Nucleus, Hradec Králové 2012, p. 125.
27. Vávrová, J., Tichý, M., Maisnar, V. et al. Stanovení volných lehkých řetězců a Hevylite u monoklonálních gamapatií. In: Maisnar V, Tichý M, a kol. Monoklonální imunoglobuliny – výskyt, význam a možnosti jejich průkazu. 1. vyd., Nucleus, Hradec Králové 2012, p. 55 - 60.
28. Katzmann, J. A., Abraham, R. S., Dispenzieri, A. et al. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin. Chem. 2005, 51, p. 878-81.
29. Palladini, G., Perlini, S., Merlini, G. Imaging of systemic amyloidosis. In: Gertz MA, Rajkumar SV: Amyloidosis: diagnosis and treatment. Springer Science and Business Media, LLC 2010, p. 15 - 32.
30. Kumar, S., Dispenzieri, A., Lacy, M. Q. et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol. 2012, 30, p. 989 - 95.
31. Wechalekar, A., Faint, J., Bradwell, S. et al. Significance of abnormal serum immunoglobulin heavy/light chain ratios (Hevylite) in 294 patients with systemic AL amyloidosis. Haematologica 2011, 96, p. 392-98.
32. Piper, C., Butz, T., Farr, M. et al. How to diagnose cardiac amyloidosis early: impact of ECG, tissue doppler echocardiography, and myocardial biopsy. Amyloid 2010, 17, p. 1-9.
33. Kovářová, L., Varmužová, T., Žarbochová, P. et al. Flow cytometry in monoclonal gammopathies. Klin. Onkol. 2011, 24, p.24-29.
34. Dispenzieri, A., Gertz, M. A., Kyle, R. A. et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: A staging system for primary systemic amyloidosis. J. Clin. Oncol. 2004, 22, p. 3751-57.
35. Dispenzieri, A. Assessing response and prognosis in AL amyloidosis. In. Gertz MA, Rajkumar SV. Amyloidosis: diagnosis and treatment, 1st ed. Springer Science, New York 2010, p. 77-93.
36. Merlini, G., Wechalekar, D., Palladini, G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013, 121, p. 5124-5130.
37. Paiva, B., Vídriales, M. B., Pérez, J. J. et al. The clinical utility and prognostic value of multiparameter flow cytometry immunophenotyping in light-chain amyloidosis. Blood 2011, 117, p. 3613-16.
38. Wechalekar, A.D., Offer, M., Gillmore, J. D. et al. Cardiac amyloidosis, a monoclonal gammopathy and a potentially misleading mutation. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, p. 128-33.
39. Leung, N., Bridoux, F., Hutchison, C. A. et al. Monoclonal gammopathy of renal significance (MGRS): when MGUS is no longer undetermined or insignificant. Doi: 10.1182/blood-2012-07-445304;
40. Ščudla, V., Minařík, J., Pika, T. Nemoc z ukládání lehkých řetězců imunoglobulinu (light chain deposition disease). Vnitř. Lék. 2012, 58, p. 38-43.
41. Dinner, S., Witteles, W., Witteles, R. et al. The prognostic value of diagnosing concurrent multiple myeloma in immunoglobulin light chain amyloidosis. Br. J. Haematol. 2013, 161, p. 367-72.42. Ščudla, V., Adam, Z., Hájek, R. et al.: Diagnostika a léčba systémové AL amyloidózy. Doporučení České myelomové skupiny 2013. Transfuze Hematol. dnes 2013, 19, p. 3-52.
Labels
Clinical biochemistry Nuclear medicine Nutritive therapistArticle was published in
Clinical Biochemistry and Metabolism

2014 Issue 2
Most read in this issue
- Clinical Problems of IgM Monoclonal Gammapathies
- Risk factors of Asymptomatic (Smoldering) Multiple Myeloma
- The Standard of Analytical Quality of Glucometers
- Diagnosis and stratification of systemic AL amyloidosis in the light of the 2013 Czech Myeloma Group recommendation