
Primární plicní sarkomy
Primary Pulmonary Sarcomas
Primary pulmonary sarcomas are rare diseases unlike lung carcinomas. The occurence of these sarcomas is between 0.013–0.40% of all malignant lung tumours. There are malignant mesenchymal tumours. They are flowing from the soft tissue of lung. The pulmonary sarcomas are heterogenic group with various biological behaviour. Their morfologic structure does not digger from the sarcomas of soft tissue. The primary pulmonary sarcomas occur more often in childhood and in young people unlike lung carcinomas. Radiation and some toxic substances are noted risk factors. Some gene mutations, infectious pathoghens and contraception have a possible impact on the origin of some types of the sarcomas. The current hypothesis is, that most of the sarcomas, if not all sarcomas, stem from primitive multipotent mesenchymal cell by malignant transformation in one or more lines. The diagnostic standard is biopsy from tumour with histologic and immunohistochemistry examination of a sample. The basic diagnostic problem is exclusion of a secondary origin of sarcomatic cells in the lung, because pulmonary metastasis of extrapulmonary sarcomas are more often than the primary pulmonary involvement. The optimal treatment is a resection of the tumour. The other therapeutic modalities are radiotherapy and chemotherapy, but results of these modalities are unsatisfactory. There are various chemotherapeutic regimes, monotherapy or combination regimes. The basic cytostatics are doxorubicine, iphosphamide, dacarbazine. Problems of the chemotherapy are high toxicity and relatively low curative effect about 20%. The first studies with biological treatment of the sarcomas of soft tissue have been published recently. This types of drugs could be a part of the complex management of these primary pulmonary tumours in the future. The primary pulmonary sarcomas have mostly aggresive course and often recur. Their prognosis is usually not very good. The survival median is 48 months and 5-years survival ranges between 38 and 48%. Prognostic factors are the size of tumour, histological type, grading, clinical stage and measure of a surgery major.
Key words:
primary pulmonary sarcoma – classification of diseases – disease detection – disease management
Authors:
T. Jakubcová; P. Jakubec
Authors‘ workplace:
Klinika plicních nemocí a tuberkulózy FN a LF UP, Olomouc
Published in:
Klin Onkol 2009; 22(4): 139-153
Category:
Reviews
Overview
Primární plicní sarkomy jsou na rozdíl od plicního karcinomu vzácná onemocnění. Tvoří přibližně jen 0,013–0,40% všech maligních plicních nádorů. Jedná se o maligní mezenchymální tumory, které vycházejí z měkkých tkání plic. Jsou heterogenní skupinou nádorů různého biologického chování a morfologicky se neliší od sarkomů měkkých tkání. Na rozdíl od plicního karcinomu se plicní sarkomy vyskytují více v dětském a mladším věku. Jejich etiologie není dosud jasná. Jako rizikové faktory jsou uváděny radiace, některé toxické látky, u některých sarkomů je možný vliv genetických mutací, hormonální antikoncepce a infekčních patogenů. Současné studie ukazují, že většina sarkomů, ne li všechny, vychází z primitivní multipotentní mezenchymové buňky, která prochází maligní transformací v jedné nebo více liniích. Diagnostickým standardem je biopsie tumoru s histologickým a imunohistochemickým vyšetřením vzorku. Základním diagnostickým problémem je vyloučení sekundárního původu plicního sarkomu, protože plicní metastázy mimoplicních sarkomů jsou mnohem častější než primární plicní postižení. Optimální léčbou je resekce tumoru. Dalšími léčebnými modalitami jsou radioterapie a chemoterapie, ale jejich výsledky jsou neuspokojivé. Používají se různé chemoterapeutické režimy – monoterapie nebo kombinované režimy. Základními cytostatiky jsou doxorubicin, ifosfamid a dakarbazin. Problémem chemoterapie je vysoká toxicita a poměrně nízká léčebná odpověď kolem 20%. V současnosti se objevují první studie biologické léčby sarkomů měkkých tkání a tyto léky by mohly být v budoucnosti součástí komplexní léčby primárních plicních nádorů. Primární plicní sarkomy mají většinou agresivní průběh a často recidivují. Jejich prognóza nebývá příliš dobrá. V literatuře je uváděn medián přežití 48 měsíců a pětileté přežití se pohybuje mezi 38 a 48%. K prognostickým faktorům patří velikost tumoru, histologický typ, grading, klinické stadium sarkomu a rozsah operačního výkonu.
Klíčová slova:
primární plicní sarkom – klasifikace nemocí – diagnostika a léčba nemocí
Úvod
Maligní plicní nádory patří v dnešní době dle četnosti k nejčastějším zhoubným onemocněním. Mohou vycházet z jakékoliv anatomické struktury plicní tkáně. Jednoznačně nejpočetnější je bronchogenní karcinom, který tvoří více než 90% všech maligních plicních onemocnění. Jedná se o nejčastější maligní nádor u mužů, u žen se pak řadí na třetí místo. Jeho incidence v České republice dosahovala v roce 2005 hodnoty 94,4 případů/100 000 obyvatel u mužů, u žen pak 30,8 případů/100 000 obyvatel. Poměrně častý je karcinoid, který tvoří asi 1% všech plicních nádorů [1]. Naopak mezenchymální maligní onemocnění – primární plicní sarkomy – jsou vzácná. Jejich výskyt se dle různých publikací pohybuje mezi 0,013 a 0,40% všech maligních plicních nádorů a udává se, že jeden plicní sarkom připadá na přibližně 500 případů plicního karcinomu [2]. Etiologie sarkomů není dodnes jednoznačně určena. Jako rizikové faktory jsou uváděny radiace, některé toxické látky (fenoxyherbicidy, chlorfenoly, dioxiny, polyvinylchlorid), ale u hemangioendoteliomu se uvažuje i o možném negativním vlivu hormonální antikoncepce. U Kaposiho sarkomu se předpokládá vliv infekčního agens, resp. herpetického viru. Některé sarkomy jsou spojovány i s dědičnými onemocněními, známá je např. neurofibromatóza. Sarkomy vycházejí z měkkých tkání plic, ke kterým patří pojivová tkáň, tuková tkáň, příčně pruhované svalstvo, krevní a lymfatické cévy a periferní nervový systém. Z embryonálního hlediska jsou většinou mezodermového původu, s výjimkou periferních nervů vznikajících z neuroektodermu. Plicní sarkomy jsou heterogenní skupinou nádorů různého biologického chování, které se morfologicky neliší od sarkomů měkkých tkání [3]. Původně se předpokládalo, že sarkomy vznikají maligním zvratem plně diferencovaných buněk (např. fibrosarkom z fibroblastů). Nová data ale ukazují, že většina sarkomů, ne li všechny, vychází z primitivní multipotentní mezenchymové buňky, která prochází maligní transformací v jedné nebo více liniích [4]. Na rozdíl od plicního karcinomu se plicní sarkomy vyskytují více v dětském a mladším věku. Sarkomová ložiska v plicích bývají mnohem častěji sekundárními projevy mimoplicního sarkomu než primárním plicním nádorem, protože plíce jsou místem s nejvyšším výskytem vzdálených metastáz sarkomů. Proto je vždy při nálezu sarkomových buněk v plicní tkáni nezbytné provést všechna dostupná vyšetření k vyloučení eventuálního primárního extrapulmonálního tumoru [5].
Speciální klasifikace plicních sarkomů neexistuje. V aktuální histologické klasifikaci plicních a pohrudničních nádorů Světové zdravotnické organizace a Mezinárodní asociace pro studium plicní rakoviny jsou sarkomy řazeny do skupiny 2 – nádory měkkých tkání (tab. 1). V této klasifikaci je však většina sarkomů vedena v nic neříkající podskupině 2.9 jiných nádorů měkkých tkání. Mimoto jsou některé sarkomy zařazeny i do jiných skupin plicních nádorů, např. plicní blastom do skupiny 1.3.6 (karcinomy s pleomorfními sarkomatoidními a sarkomatózními elementy), světlobuněčný tumor do skupiny 4 (různorodé tumory) a zánětlivý myofibroblastický tumor do skupiny 8 (tumorům podobné léze). Dle vyjádření našich onkologů lze však pro klasifikaci plicních sarkomů použít Klasifikace nádorů měkkých tkání Světové zdravotnické organizace z roku 2002 (tab. 2). Součástí této klasifikace je i rozdělení tumorů měkkých tkání dle jejich biologické povahy do čtyř kategorií. První kategorií jsou benigní tumory, které se obvykle nešíří do okolí, a pokud ano, tak nedestruují okolní tkáně a většinou jsou resekabilní. Dále to jsou intermediární, lokálně agresivní tumory, kterou infiltrují a destruují okolní struktury, ale netvoří metastázy. Také tyto nádory se léčí resekcí i s okolní zdravou tkání. Třetí skupinou jsou intermediární, taktéž lokálně agresivně rostoucí nádory, které ale i vzácně metastazují (riziko asi 2%), nejčastěji do lymfatických uzlin. Poslední kategorií jsou maligní tumory (sarkomy), u kterých se riziko vzniku metastáz pohybuje dle histologického typu od 20 až do téměř 100%. Velikost nádoru má prognostický význam [3]. Riziko metastazování a smrti přímo koreluje s velikostí primárního tumoru [4].
![Histologická klasifikace plicních a pleurálních nádorů Světové zdravotnické organizace a Mezinárodní asociace pro studium plicní rakoviny z roku 1999 – výňatek [64].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/de72dfa5b44cf27133e7d7a6713a54c8.png)
![Klasifikace nádorů měkkých tkání Světové zdravotnické organizace z roku 2002 [65].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/0a938de9422ac0cf599d7dc78909687f.png)
![Pokračování tab. 2. Klasifikace nádorů měkkých tkání Světové zdravotnické organizace z roku 2002 [65].](https://pl-master.mdcdn.cz/media/image/88c7d489b2b03f42a81c08ded18371ef.png?version=1537795093)
Protože histologický grading sarkomů je taktéž důležitým prognostickým faktorem, je TNM klasifikační schéma modifikováno na GTNM staging systém, do kterého je zakomponován i grading (tab. 3). Americký výbor pro rakovinu provedl dle GTNM stagingu rozdělení tumorů měkkých tkání do jednotlivých klinických stadií (tab. 4).
![GTNM staging tumorů měkkých tkání [66].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/aa297d5bcf32a7df183f9378a0cac116.png)
![AJCC GTNM klasifikace a stagingové skupiny tumorů měkkých tkání [67–68].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a65c70de081de119c2192afef23cf493.png)
V této práci jsou primární plicní sarkomy rozděleny a podrobněji popsány dle Klasifikace nádorů měkkých tkání Světové zdravotnické organizace z roku 2002. Dalším významný zdrojem informací je velmi podrobný 13. svazek Atlasu patologie nádorů, pojednávající o tumorech dolního respiračního traktu. V tomto textu se zmiňujeme pouze o těch nádorech, které jsou popisovány jako primárně plicní, i když v plicní tkáni může být nalezen jakýkoliv sarkom ve formě metastáz. V prezentované práci jsou uvedeny pouze tumory s maligním potenciálem, proto se nepopisují chondrom, hamartom, sklerozující hemangiom a kalcifikující fibrózní pseudotumor pleury. Stejně tak primární plicní světlobuněčný tumor je v dostupné literatuře uváděn jako jednoznačně benigní nádor. Lymfosarkom je uváděn spíše ve skupině lymfoproliferativních onemocnění než sarkomů. Nejsou zahrnuty ani smíšené nádory – karcinosarkomy. Difuzní plicní lymfangiomatóza je do přehledu zahrnuta i přes svůj benigní charakter, a to z důvodu svého progresivního průběhu, který často vede ke smrti na respirační selhání.
Klasifikace
1) tuková tkáň
liposarkom
2) hladké svaly
leiomyosarkom
difuzní plicní lymfangiomatóza
3) příčně pruhované svaly
rabdomyosarkom
4) skeletální systém
chondrosarkom
osteosarkom
5) fibroblastické/fibrohistiocytární sarkomy
intrapulmonální solitární fibrózní tumor
zánětlivý myofibroblastický tumor (zánětlivý pseudotumor)
maligní fibrózní histiocytom (myofibrohistiocytární tumor)
hemangiopericytom
hyalinizující vřetenobuněčný tumor
fibrosarkom
kongenitální peribronchiální myofibroblastický tumor
6) vaskulární sarkomy
Kaposiho sarkom
epitelioidní hemangioendoteliom
angiosarkom
sarkom plicní arterie a vény
7) pericytární (perivaskulární) tumory
maligní glomus tumor
8) neurogenní sarkomy
neurosarkom (maligní tumor pochvy periferních nervů, maligní schwannom)
9) sarkomy nejasného původu
pleuropulmonální blastom
plicní blastom
synoviální sarkom
primitivní neuroektodermální tumor
desmoplastický malokulatobuněčný tumor
maligní tumor ze zrnitých buněk
intimální sarkom
maligní mezenchymom
Liposarkom
Liposarkom patří k častým sarkomům. Typicky se vyskytuje na končetinách a v retroperitoneu. Intratorakálně bývá lokalizován vzácněji. V tomto případě bývá nejčastěji postiženo mediastinum, kde může růst velmi pomalu a být asymptomatický i po dobu několika let. Plicní lokalizace liposarkomu je velmi vzácná, dosud bylo popsáno jen několik případů, v naprosté většině u lidí starších 40 let věku [6]. V literatuře se spekuluje, že možným patogenetickým faktorem je maligní zvrat plicního lipomu nebo azbestózy. Klinicky se projevuje bolestmi na hrudi, progredující dušností a suchým kašlem. Na skiagramu hrudníku bývá nacházen různě veliký, nehomogenní, ale dobře ohraničený ložiskový stín. Může být přítomen pleurální výpotek. Liposarkom může metastazovat do plic, na pleuru, do regionálních lymfatických uzlin, jater, kostí, nadledvin a ledvin. Podle histologického nálezu se liposarkom dělí na dobře diferencovaný, nediferenciovaný, myxoidní, kulatobuněčný, pleomorfní, smíšený a nespecifikovaný. Můžou být nalezeny bizarní mnohojaderné buňky obsahující v cytoplazmě tukové kapénky v případě dobře diferencovaného liposarkomu, u myxoidního typu bývají přítomny lipoblasty a plexiformní kapilární síť v myxoidním stromatu, u nediferencovaného typu a pleomorfního typu převažují nediferencované vřetenovité buňky a fokální nekrózy. Histologický typ nádoru koreluje s jeho biologickým chováním a prognózou. Dobře diferencovaný liposarkom má nízký maligní potenciál, zatímco typ pleomorfní a kulatobuněčný má velkou tendenci k lokálnímu šíření a metastazování [7]. Léčba je chirurgická s resekcí nádoru a disekcí regionálních lymfatických uzlin. Adjuvantní radioterapie a chemoterapie má význam u dobře diferencovaných liposarkomů, u ostatních histologických typů liposarkomu se ale neosvědčila.
Leiomyosarkom
Jde o jeden z nejčastějších primárních plicních sarkomů. Průměrný věk nemocných je 50 let. Muži bývají postiženi častěji než ženy (v poměru 2,5 : 1). Plicní leiomyosarkom se vyskytuje ve třech různých lokalizacích – endobronchiálně či peribronchiálně, v plicní arterii a periferní plicní tkáni [8]. Klinicky se projevuje bolestmi na hrudi, kašlem, dušností, hemoptýzou. Doprovodný pleurální výpotek je vzácný. Na skiagramu hrudníku bývá popisováno laločnaté, ostře ohraničené ložisko různé velikosti, v případě endobronchiální lokalizace obraz obstrukční pneumonie [9]. V histologickém nálezu jsou vřetenovité buňky uspořádané do proplétajících se svazků. Buňky mají protáhlá, doutníkovitá jádra a eozinofilní cytoplazmu. Mohou být přítomny i obrovské nádorové buňky. Imunohistochemicky je pozitivní desmin a vimentin. Léčba je chirurgická. U inoperabilních nádorů se zkouší radioterapie, u metastazujících chemoterapie. Obě modality ale mají jen nevelký efekt.
Difuzní plicní lymfangiomatóza (DPL)
DPL je vzácné onemocnění, které někteří autoři řadí k nádorům hladkého svalstva, jiní k vaskulárním tumorům. Část odborníků se dokonce přiklání k lymfoproliferativní etiologii nemoci. Dochází u ní k difuzní proliferaci abnormálních lymfatických cév v plicích podél normálních lymfatických cév. Obvyklý je chylotorax a často dochází k infiltraci měkkých tkání mediastina [10]. Na CT hrudníku je popisováno ztluštění interlobulárních sept a peribronchiálně, pleurální ztluštění a hydrotorax [11]. Imunohistochemicky jsou pozitivní markery hladkých svalů (aktin, desmin) a markery vaskulárních buněk. Postihuje hlavně děti a mladé dospělé. Klinicky se projevuje progredující dušností, suchým kašlem a hemoptýzou. Často vede ke smrti z důvodu respiračního selhání. Neexistuje žádná specifická léčba DPL. Paliativní postupy zahrnují léčbu interferonem - α, drenáž výpotku, event. pleurodézu.
Rabdomyosarkom
Ačkoliv je rabdomyosarkom jedním z nejčastějších sarkomů, primární plicní postižení je vzácné. Dospělí bývají postiženi lehce více než děti, mírně převažují muži a obvyklý věk nemocných se pohybuje mezi 40 a 60 lety. U dětí tvoří plicní rabdomyosarkom jen 0,5% všech rabdomyosarkomů a dle některých autorů 4,4% primárních plicních malignit [12]. Často vznikají v preexistujících cystických útvarech a bývá popisována souvislost s kongenitální cystickou adenoidní malformací. Nádor má solidní nebo multicystický charakter a může prorůstat do bronchovaskulárních struktur. Z klinických symptomů jsou přítomny kašel, dušnost, teploty a tumor může být příčinou spontánního pneumotoraxu. V histologickém obrazu dominují velké vřetenovité rabdomyoblasty uspořádané do paralelních vrstev. Obsahují eozinofilní cytoplazmu a časté jsou atypické mitózy. Mezi rabdomyoblasty se nacházejí shluky nízce diferencovaných myoblastů a hvězdicovitých primitivních mezenchymálních buněk [13]. Imunohistochemicky je prokazována pozitivita desminu. Léčbou je resekce tumoru s eventuální adjuvantní radioterapií a chemoterapií.
Chondrosarkom
Primární plicní chondrosarkom je velmi vzácné onemocnění. Průměrný věk nemocných je 55 let a není rozdíl v četnosti nemoci mezi pohlavími. Nádor může postihovat tracheobronchiální strom nebo periferní plicní tkáň, přičemž obě lokalizace jsou přibližně rovnoměrně zastoupeny. Předpokládá se ale, že chondrosarkomy tracheobronchiálního stromu mají lepší prognózu. V počáteční fázi tumor roste pomalu a průběh bývá dlouho asymptomatický. Následná symptomatická fáze pak probíhá obvykle rychle. Nádor může obturovat bronchy, invadovat do cévních struktur nebo prorůstat do mediastina, pleury nebo perikardu. Může metastazovat do krčních uzlin, kostí, kůže a ledvin [14]. Způsobuje kašel, bolesti na hrudi, dušnost, hemoptýzu. Na skiagramu hrudníku se popisuje solidní, dobře ohraničené ložisko různé velikosti. Tumor se obvykle skládá z chondromatózní, myxoidní a fibrózní komponenty [15]. Podle diferenciace mohou být přítomny chondroblasty či chondrocyty. Při imunohistochemickém vyšetření se zjišťuje pozitivita S-100 proteinu a vimentinu. Léčba je chirurgická a může být doplněna adjuvantní nebo neoadjuvantní radioterapií a chemoterapií. Existují práce o dobrém efektu radioterapie a parciálních remisích po chemoterapii u inoperabilních případů.
Osteosarkom
Plicní osteosarkom je výjimečně se vyskytující nádor. Postihuje dospělé, ženy a muže ve stejném poměru. Vytváří velké, solitární, heterogenní, kalcifikované masy v plicní tkáni a rychle progreduje. Metastazuje do lymfatických uzlin, kostí, jater a kůže [16]. Většina pacientů má výrazné potíže, dominují kašel, dušnost, bolesti na hrudi, hemoptýzy a recidivující pneumonie. V histologickém nálezu jsou přítomny vřetenovité buňky, anaplastické mnohojaderné buňky, buňky podobné osteoklastům. Imunohistochemicky jsou pozitivní vimentin, osteonektin, osteocalcin a někdy S-100 protein. Resekce nádoru bývá doplněna chemoterapií a radioterapií. Onemocnění má agresivní povahu a prognóza pacientů je špatná, zvláště v případě tumorů větších než 5cm. Velká část nemocných umírá během jednoho roku od zjištění diagnózy [17].
Intrapulmonální solitární fibrózní tumor
Solitární fibrózní tumor je poměrně častý nádor pleury, ale může postihovat i další orgány a tkáně, jako jsou plíce, mediastinum, perikard, peritoneum, játra, meningy, štítnici, paranazální dutiny, dutinu ústní a nosní. Primární plicní lokalizace je vzácná. Současná hypotéza předpokládá, že nádor vychází ze subpleurálních mezenchymálních buněk, jež někteří autoři označují jako „dendritické intersticiální buňky“. Obvykle postihuje dospělé. Většinou probíhá asymptomaticky [18]. Tvoří okrouhlá, dobře ohraničená homogenní ložiska. Nádor obsahuje svazky vřetenovitých fibroblast like buněk, které mají protáhlá jádra. Tyto svazky nádorových buněk jsou obklopeny pásy kolagenních vláken, je přítomna hypervaskularizace i fokální nekrózy. Imunohistochemicky je pozitivní vimentin a CD34 [19]. Velká většina případů je benigních, ale někdy má nádor maligní potenciál. Indikována je kompletní resekce tumoru. Adjuvantní chemoterapie a radioterapie jsou obecně považovány za málo efektivní, ale byly popsány případy pozitivního účinku adjuvantní terapie u inkompletně resekovaných tumorů.
Zánětlivý pseudotumor (zánětlivý myofibroblastický tumor)
Je to vzácné onemocnění, o kterém dlouho probíhaly diskuze, zda se jedná o tumor či zánětlivou entitu. Poslední výzkumy ale svědčí pro primárně nádorovou povahu této nemoci [20]. Postihuje všechny věkové skupiny, ale 60% nemocných je starších 40 let věku. Ženy a muži jsou postiženi ve stejném poměru. V naprosté většině případů roste v plicním parenchymu, endobronchiální umístění je zřídkavé. Nádor může infiltrovat okolní struktury, jako jsou bronchy, plicní cévy, pleura, srdce, hrudní stěna, bránice a páteř. Může tvořit vzdálené metastázy v mozku nebo kostech, a dokonce je popsána dermatomyositida jako paraneoplastický syndrom tohoto onemocnění. V literatuře se udává, že 30–70% všech případů je asymptomatických. Obvykle popisované potíže jsou kašel, dušnost, bolesti na hrudi, hemoptýzy, teploty. Laboratorně bývá zjištěna anémie, trombocytóza, zvýšená sedimentace a polyklonální hypergamaglobulinemie. Nejčastějším radiologickým nálezem je solitární uzel nebo masa, mnohočetné nodulace jsou popsány jen asi u 5% případů. V histologickém obrazu dominují vřetenovité myofibroblasty s oblými jádry a eozinofilní cytoplazmou, které tvoří proplétající se svazky. Tumor je infiltrován velkým množstvím zánětlivých buněk zahrnujícím plazmocyty, lymfocyty a histiocyty. Imunohistochemickým vyšetřením je zjištěna pozitivita vimentinu, aktinu hladkých svalů, CD68. Nejefektivnější léčbou je kompletní resekce tumoru. Výsledky systémové kortikoterapie jsou velmi variabilní a efekt radioterapie a chemoterapie je kontroverzní. Prognóza nemoci je dobrá v případech kompletní resekce tumoru, ale obecně je pětiletá doba přežití jen 74% [21].
Maligní fibrózní histiocytom (myofibrohistiocytární tumor)
Maligní fibrózní histiocytom (MFH) je primitivní sarkom. Histologicky se jedná o vysoce buněčný nádor, složený z vřetenovitých a histiocytárních buněk, které jsou uspořádány do svazků nebo fasetovitě. Mimo tyto dvě entity bývají v tumoru přítomny i bizarní, obrovské, mnohojaderné buňky. Dalšími nálezy jsou zvýšená mitotická aktivita, atypické mitózy a nekrózy. Imunohistochemicky bývá pozitivní vimentin a faktor XII, zatímco cytokeratin je negativní. Ačkoliv se jedná o jeden z nejčastějších sarkomů měkkých tkání dospělých (tvoří kolem 10% všech sarkomů), primární plicní původ je velmi neobvyklý [22]. Etiologie MFH je neznámá. Onemocnění postihuje hlavně dospělé, průměrný věk je 52 let a lehce více bývají postiženy ženy. Ke klinickým příznakům patří bolesti na hrudi, kašel a hemoptýza, ale častý je i asymptomatický průběh, zvláště v časných stadiích. Typický je rychlý růst tumoru a poměrně časté je intrabronchiální prorůstání a intravaskulární invaze nádoru. Metastazování do regionálních lymfatických uzlin je popisováno asi u 20% případů. Na RTG hrudníku bývá většinou nalezen solitární ložiskový stín různé velikosti. Optimální léčbou je resekce tumoru a kompletní disekce regionálních lymfatických uzlin [23]. Často jsou popisovány lokální recidivy či vzdálené metastazování. Role chemoterapie či radioterapie v léčbě MFH není zatím jasně definována. Prognosticky nepříznivými znameními jsou vzdálené metastázy a invaze do mediastina nebo hrudní stěny, pětileté přežití je u operovaných 43%, u neoperovaných 0%.
Hemangiopericytom
Hemangiopericytom se může vyskytnout v jakémkoliv věku a postihnout jakoukoliv část těla, ale plicní postižení je zřídkavé [24]. Vychází z mezenchymálních buněk s pericytární diferenciací. Jeho přesný charakter není dosud definován a neexistuje konsenzus ohledně adekvátního histologického zařazení tumoru. Dle poslední klasifikace nádorů měkkých tkání je zahrnut do skupiny myofibroblastických tumorů. Většina tumorů je v plicích uložena centrálně a má tendenci růst periferním směrem a utlačovat okolní plicní tkáň. Za normálních okolností je nádor opouzdřen tenkou fibrózní tkání. V určitých případech ale tumor penetruje pouzdrem a prorůstá do okolních nitrohrudních struktur nebo intrabronchiálně. Obvyklými příznaky jsou hemoptýza a bolesti na hrudi, méně častá je dušnost a kašel. Nezřídka ale onemocnění probíhá asymptomaticky. Radiologicky je prokazováno okrouhlé nebo lehce laločnaté, homogenní, dobře ohraničené ložisko. Variabilním nálezem je doprovodný pleurální výpotek. Metastázy jsou nalézány nejčastěji v plicích a kostech. Histologicky je nádor tvořen rozvětvenými primitivními cévami, které jsou obklopeny radiální sítí jemných retikulárních vláken. Mezi nimi jsou uloženy kulaté nebo vřetenovité nádorové buňky s velkými vezikulárními jádry a světlou cytoplazmou. Dosud neexistuje žádný specifický imunohistochemický marker hemangiopericytomu. Léčba je chirurgická. Na rozdíl od většiny ostatních sarkomů je u tohoto nádoru popisován dobrý efekt radioterapie i chemoterapie [25].
Hyalinizující vřetenobuněčný tumor
Jde o velmi neobvyklý mezenchymální tumor, který má mnohé charakteristiky shodné s low-grade fibromyxoidním sarkomem. Jako primární sarkom plic se vyskytuje zcela výjimečně, do roku 2007 byly v literatuře popsány dva případy tohoto onemocnění. Skládá se ze dvou histologických komponent. První jsou okrsky vřetenovitých buněk. Druhou částí jsou dobře ohraničené, téměř nebuněčné hyalinizované oblasti palisádovitě obklopené oválnými buňkami. Tyto útvary jsou popisovány jako tzv. rozety [26]. Imunohistochemicky je v celém tumoru pozitivní vimentin, zatímco CD57, neuron specifická enoláza a protein S100 jsou pozitivní jen v buňkách rozet [27]. Na druhé straně vřetenovité buňky jsou pozitivní na kolagen typ IV a faktor XIII, což svědčí pro histiocytární původ buněk. Tento tumor tvoří velké solitární ložisko v plicích, klinicky může být asymptomatický nebo se může projevovat nespecifickými příznaky, jako jsou bolesti na hrudi, kašel, hemoptýzy. Léčba je chirurgická.
Fibrosarkom
Fibrosarkom tvoří asi 12% všech primárních plicních sarkomů [28]. Může se vyskytnout v jakémkoliv věku a postihuje přibližně stejně obě pohlaví. Roste ve dvou lokalizacích – buď přímo v plicním parenchymu, nebo endobronchiálně, kde tvoří polypoidní masy. Endobronchiální typ se nachází hlavně u dětí a mladých dospělých. Klinicky bývá často asymptomatický, případně se projevuje bolestmi na hrudi, kašlem, hemoptýzou, teplotami, celkovou únavou. Tumor je velmi buněčný, obsahuje vřetenovité buňky, které tvoří proplétající se svazky, někdy až připomínající obraz rybí kosti. Buňky obsahují štíhlá, zašpičatělá jádra a jen skromné množství cytoplazmy. Obvyklé jsou i fokální nekrózy. Imunohistochemicky je pozitivní vimentin. Léčebnou první volbou je chirurgické odstranění tumoru. Radioterapie a chemoterapie je používána jako adjuvantní léčba nebo je vyhrazena pro inoperabilní případy. U čistě endobronchiálně rostoucích fibrosarkomů je možné se pokusit o radikální intervenční bronchologický výkon [29]. Endobronchiální typ tumoru má obvykle nízký maligní potenciál s relativně dobrou prognózou a mortalitou dosahující 32%.
Kongenitální peribronchiální myofibroblastický tumor
Jde o velmi vzácný nádor novorozenců, prozatím bylo v literatuře popsáno jen přibližně 10 případů tohoto onemocnění [30]. Vyvíjí se z buněk mezenchymu, které obklopují respirační dukty, z nichž se formují velké bronchy. Může být příčinou fetálního hydropsu [31]. Jedná se o solidní, heterogenní tumor. Klinicky se projevuje respiračním nebo kardiálním selháním. Nádor se skládá z proplétajících se svazků krátkých vřetenovitých buněk, které obsahují velká, prodloužená jádra a jen menší množství cytoplazmy. Typická je hypervaskularizace tumoru. Nádorové buňky se pozitivně barví na calponin, pozitivita desminu může svědčit pro dobře diferencovaný typ nádoru. Léčba je chirurgická.
Kaposiho sarkom
Před vypuknutím AIDS epidemie a rozvojem orgánových transplantací byl Kaposiho sarkom (KS) v evropské populaci vzácným nádorem s výskytem přibližně 20 případů na 100 000 obyvatel dle recentních údajů. Častěji postihoval starší muže v populaci aškenázských židů a v oblasti Středomoří. Naopak v rovníkové Africe se jedná o častý nádor, který může tvořit téměř jednu desetinu všech malignit. V dnešní době jsou nejčastěji postižené HIV pozitivní osoby, a to zvláště homosexuální muži. Plicní KS je popsán u 10% nemocných s AIDS a udává se, že 25% HIV pozitivních s mukokutánním KS má současně i plicní formu tohoto nádoru [32]. Samotný plicní KS při absenci mukokutánního postižení je vzácný. Rizikový faktor vzniku plicního KS je snížení absolutního počtu CD4+ lymfocytů pod 150/mm3. Etiopatogeneze KS není ještě zcela vyjasněna, ale předpokládá se vliv lidského herpesviru 8, tzv. KS Associated Herpes Virus (KSHV), který je nalézán ve všech formách KS. Jeho DNA je přítomna v lymfatickém systému, mononukleárech, slinách a spermatu nemocných s KS. Některé sekvence DNA KSHV mají onkogenní potenciál, jako např. bcl-2 homolog, který ovlivňuje apoptózu. Další sekvence DNA pak kódují např. G-protein vazebný receptor, který indukuje angiogenezi nebo jiné proteiny, jež imitují některé cytokiny a chemokiny [33]. V histologickém nálezu zjišťujeme neovaskularizaci se štěrbinovitými cévami, přítomnost typických vřetenovitých nádorových buněk, proliferujících endoteliálních buněk, fibroblastů a zánětlivou infiltraci s leukocyty, makrofágy a dendritickými buňkami.
Klinický obraz KS je nespecifický a může imitovat pneumonii. Nejčastější příznaky jsou dušnost a kašel. Mohou být ale přítomny i horečky, bolesti na hrudi, hemoptýza, noční pocení. Na skiagramu hrudníku je nejčastěji retikulonodulární kresba splývající ve větší tumorózní ložiska. Z dalších nálezů můžeme nalézt difuzní intersticiální infiltráty, pruhovité infiltráty, konzolidaci, pleurální výpotek či hilovou nebo mediastinální lymfadenopatii. Na CT skenech bývá přítomno ztluštění bronchiální stěny, hvězdicovité léze nebo špatně ohraničené nodulace, které se paprskovitě šíří od plicních hilů podél bronchovaskulárních struktur do interlobulárních sept. KS se může vyskytovat i endobronchiálně. Bronchoskopicky se jeví jako červené nebo fialové makuly nebo papuly, často lokalizované na bronchiálních karinách.
Diagnostika KS je samozřejmě morfologická. Výtěžnost endoskopických metod (forceps biopsie a transbronchiální plicní biopsie) se pohybuje mezi 26 a 60%. Otevřená plicní biopsie je úspěšná přibližně v 50% případů. Nejvýtěžnější metodou je torakoskopie. V diferenciální diagnostice musíme myslet na kapilární hemangiomatózu, epitelioidní hemangioendoteliom a angiosarkom.
Základní léčbou KS je chemoterapie. Jsou používány kombinace různých látek. K podávaným cytostatikům patří adriamycin, bleomycin, vinkristin, vindesin, etoposid, daunorubicin, doxorubicin, z novějších pak paclitaxel. Léčebná odpověď se dostaví u 30–50% nemocných. Medián přežití je dle různých údajů u nonresponderů 6–7 měsíců, u responderů kolem 12 měsíců. KS je velmi radiosenzitivní nádor a radioterapie vede k potlačení symptomu plicního KS, bohužel medián přežití není lepší než u chemoterapie. Novější práce ukazují možný pozitivní vliv vysoce aktivní antiretrovirální léčby (High Active Antiretroviral Aherapy – HAART) na vznik a průběh plicního KS, pravděpodobně svým působením na KSHV.
Epitelioidní hemangioendoteliom
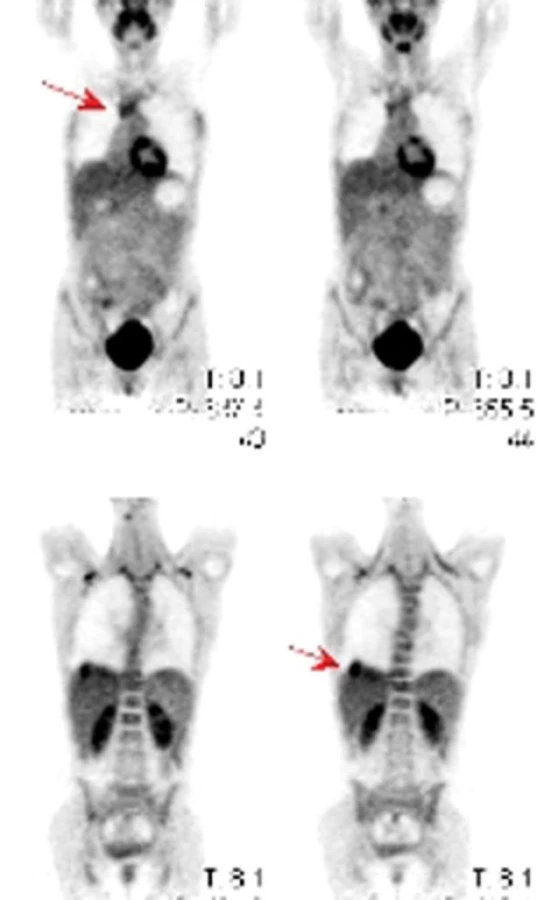
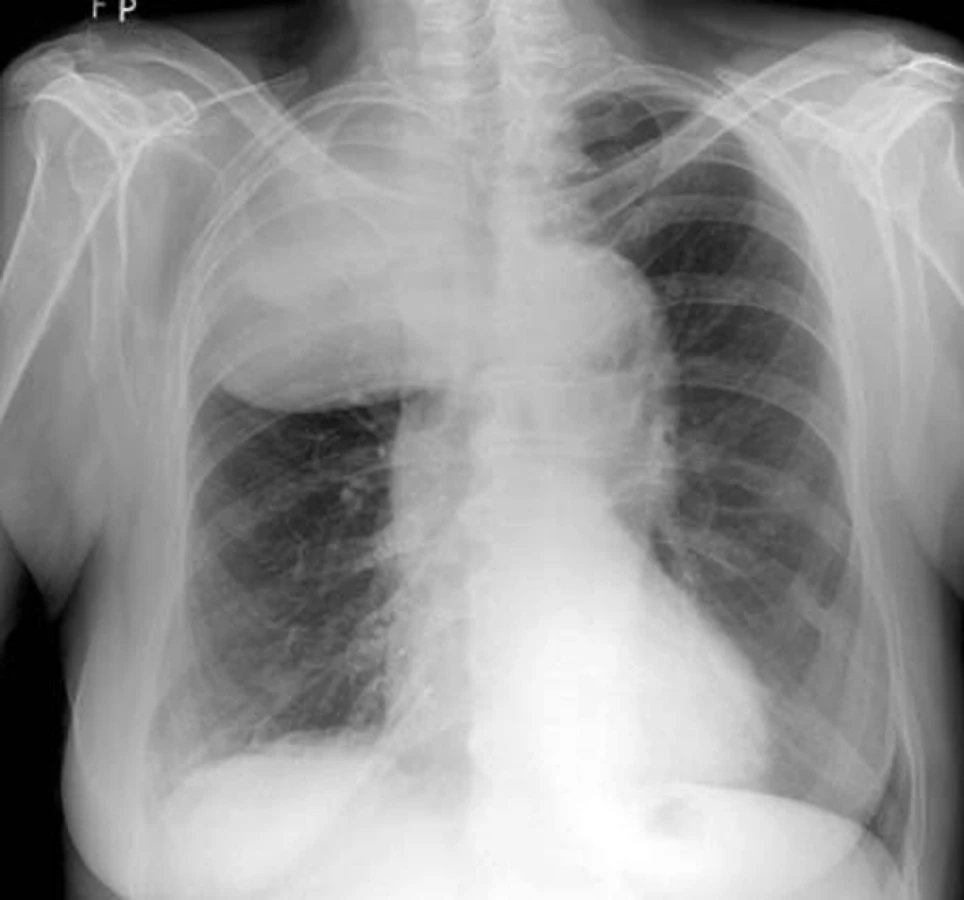
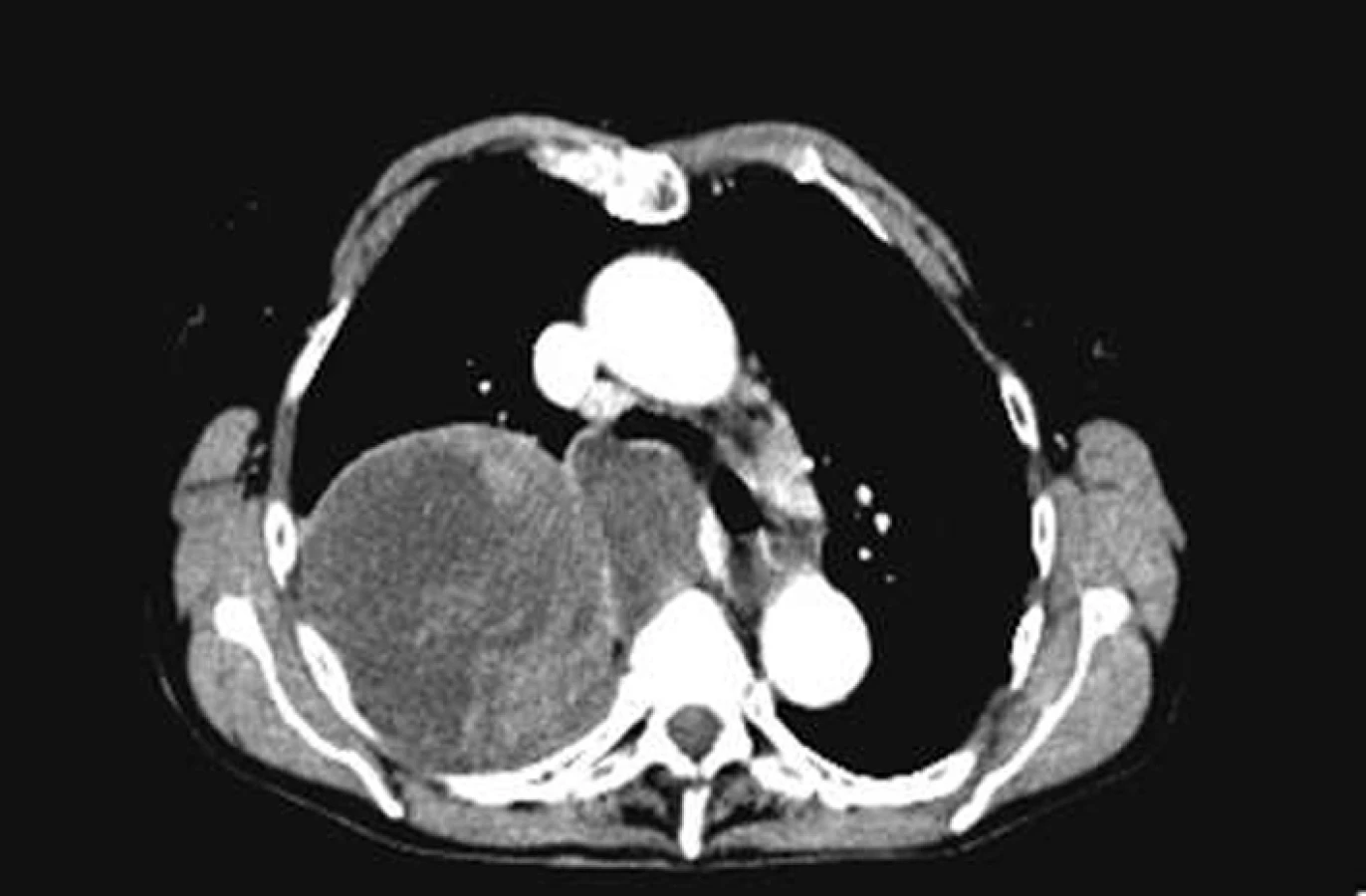
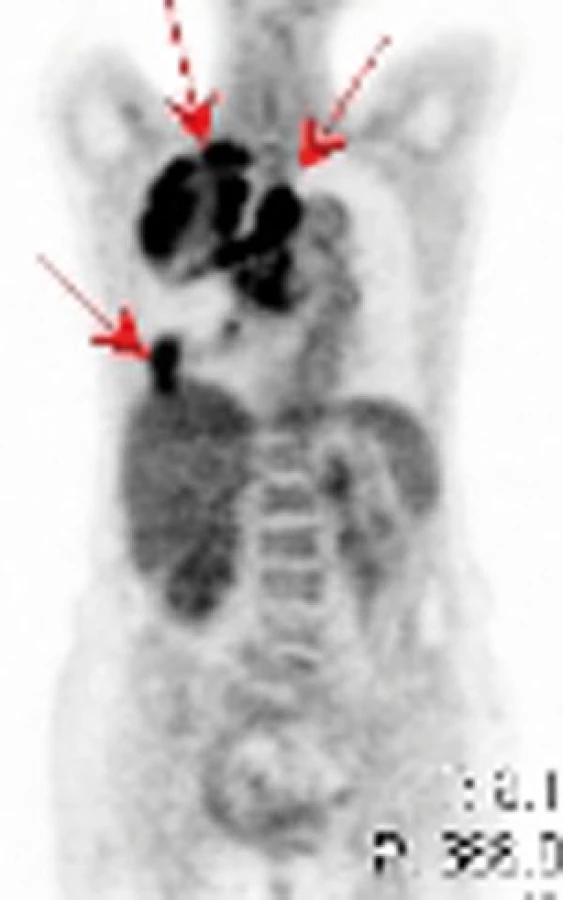
Plicní epitelioidní hemangioendoteliom (PEHE) je vzácný nízce až středně maligní primární sarkom plic a jeho biologická aktivita ho řadí mezi hemangiom a angiosarkom. Poprvé byl popsán v roce 1975 Dailem a Liebowem pod názvem intravaskulární bronchoalveolární tumor (Intravascular Bronchoalveolar Tumour – IVBAT), protože byl považován za agresivní formu bronchoalveolárního karcinomu prorůstající do přilehlých krevních cév. Následně byl ale dalšími výzkumy prokázán endoteliální původ nádorových buněk. Současný název epitelioidní hemangioendoteliom byl poprvé použit Weissem et al v roce 1986. PEHE má multicentrický původ a může postihovat mimo plíce i játra, měkké tkáně hlavy, krku, končetin, břicha, anogenitální oblasti, kosti, lymfatické uzliny, mediastinum, peritoneum, prsní žlázy, kostní dřeň, mozek, mozkové pleny, srdce, oblast gastrointestinálního traktu a kůži. V současné době neexistuje žádný diagnostický klinický ani laboratorní marker PEHE. Incidence tumoru ani jeho etiologie nejsou známy, ale v různých pracích je popisován možný vliv hormonální antikoncepce a vinylchloridu. Onemocnění postihuje častěji ženy než muže (poměr 3,8 : 1,0). Věkový průměr je 37 let a přes 40% nemocných je mladších 30 let. Výskyt nemoci má dva vrcholy, a to ve třetí a páté dekádě života. Přibližně polovina případů je asymptomatických a onemocnění se zjistí náhodně na skiagramu hrudníku. Druhá polovina nemocných má nespecifické příznaky jako dušnost, suchý kašel, pleurální bolest, váhový úbytek a celkovou slabost. Vzácně se nemoc může projevovat alveolárním krvácením nebo rychle progredující plicní hypertenzí. Nádor může postihovat regionální lymfatické uzliny a pleuru a pleurální postižení může mít podobu suché pleuritidy nebo pleurálního výpotku. Vzdálené metastázy se objevují asi u 25% případů PEHE. Na skiagramu hrudníku jsou typickým nálezem mnohočetné nodulace velikosti do 2cm postihující obě plíce. Asi v 10% případů je na skiagramu hrudníku nalezeno solitární ložisko velikosti až 5cm. Na HRCT plic mají tato ložiska perivaskulární distribuci a jsou často kalcifikována. Vzácnějším nálezem při HRCT vyšetření jsou opacity typu mléčného skla a nepravidelná intersticiální ztluštění. Konzistence nádoru je pryžovitá až chrupavčitá. Na řezu je neprůsvitný nebo poloprůsvitný, šedobílé až žlutohnědé barvy. Histologicky se noduly skládají z hypocelulárního až acelulárního centra myxoidního vzhledu, které obsahuje velké množství polysacharidů. V centrech často dochází ke koagulační nekróze, hyalinizaci, kalcifikaci až osifikaci. Centra jsou obklopena zónou nádorových buněk. Tyto nádorové buňky jsou popisovány jako epitelioidní nebo histiocytoidní. Jsou kulaté nebo oválné s bohatou růžovou cytoplazmou, která obsahuje velké množství vakuol. Jádra buněk mají oválný nebo kulatý tvar, jsou excentricky uložená s periferní kondenzací chromatinu a prominujícími jadérky. Mitózy jsou vzácné (< 1 na 10 zorných polí). Vzácně mohou mít nádorové buňky vzhled buněk dendritických s hyperchromatickým vřetenovitým jádrem. Nádor může prorůstat do malých plicních arterií, žil, lymfatik a respiračních bronchiolů. Imunohistochemickým potvrzením histologického nálezu PEHE je pozitivita faktoru VIII podobného antigenu (F VIII Related Antigen), vimentinu, lektinu vázajícího se na endoteliální buňky (Ulex europeus-1), CD31 (Platelet Endothelial Cell Adhesion Molecule – PECAM 1) a CD34 (antigen na povrchu hemopoetických a endoteliálních kmenových buněk) [34]. Elektronovou mikroskopií jsou nalézány typická Weibel-Paladeho tělíska, která jsou specifická pro endoteliální buňky. Jsou to protáhlé, multitubulární struktury dlouhé 1,0–2,5 μm a široké 0,1 μm. V nádorových buňkách byly také nalezeny četné chromozomální abnormality, např. ztráta Y chromozomu, monosomie chromozomu 11, translokace páru chromozomu 14 nebo translokace mezi chromozomy 7 a 22. Strategie léčby PEHE není jednoznačná a existují různá doporučení stran optimálního léčebného postupu. Někteří autoři dokonce doporučují asymptomatické formy neléčit. Terapeutickou volbou u solitárních nebo několika izolovaných lézí je resekce. Radioterapie se opakovaně ukázala v léčbě PEHE neúčinnou a používá se jen jako analgetická paliace u kostních metastáz. U mnohočetných forem se zkoušela léčba různými cytostatiky v monoterapii nebo kombinaci. Použity byly cyklofosfamid, etoposid, gemcitabin, docetaxel, doxorubicin, 5-fluorouracil, mitomycin C, vincristin, tegafur a oba platinové preparáty. Bohužel remise po chemoterapii jsou sledovány jen výjimečně a žádný chemoterapeutický režim není v léčbě PEHE standardně doporučován. V posledních letech jsou vkládána velká očekávání do biologické terapie PEHE pomocí interferonu, interleukinu 2 a retinoidů, ale dosavadní úspěšnost této léčby není příliš vysoká. Prognóza nemoci a její průběh jsou ve většině případů nepředvídatelné. Doba přežití se dle různých údajů v literatuře pohybuje od několika měsíců do 28 let. Většina pacientů umírá na respirační selhání. U malé skupiny nemocných je příčinou smrti mimoplicní šíření nádoru. Bylo ale zaznamenáno i několik spontánních remisí. K nepříznivým prognostickým faktorům PEHE plic se řadí přítomnost klinických potíží, endobronchiální nebo intravaskulární šíření tumoru, přítomnost pleurálního výpotku, mediastinální nebo periferní lymfadenopatie, současné postižení jater a/nebo jiných orgánů. Nález opacit typu mléčného skla a/nebo iregulárních intersticiálních ztluštění při HRCT vyšetření plic je známkou agresivního chování nemoci a je také negativním prognostickým faktorem.
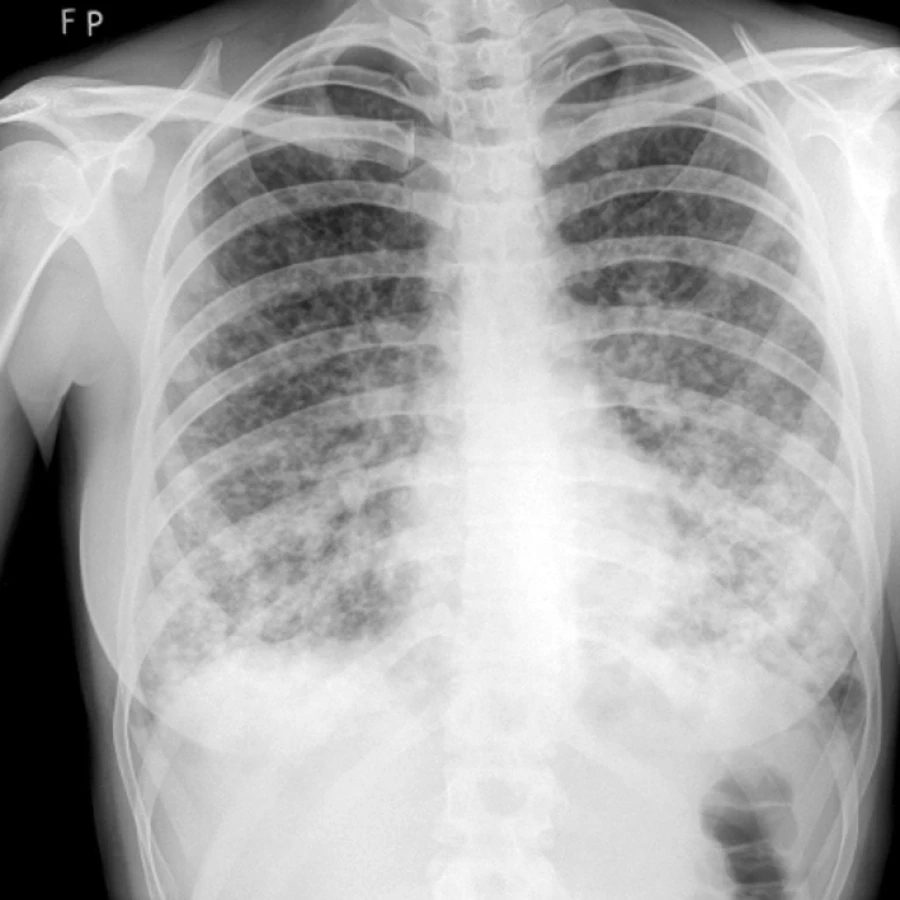
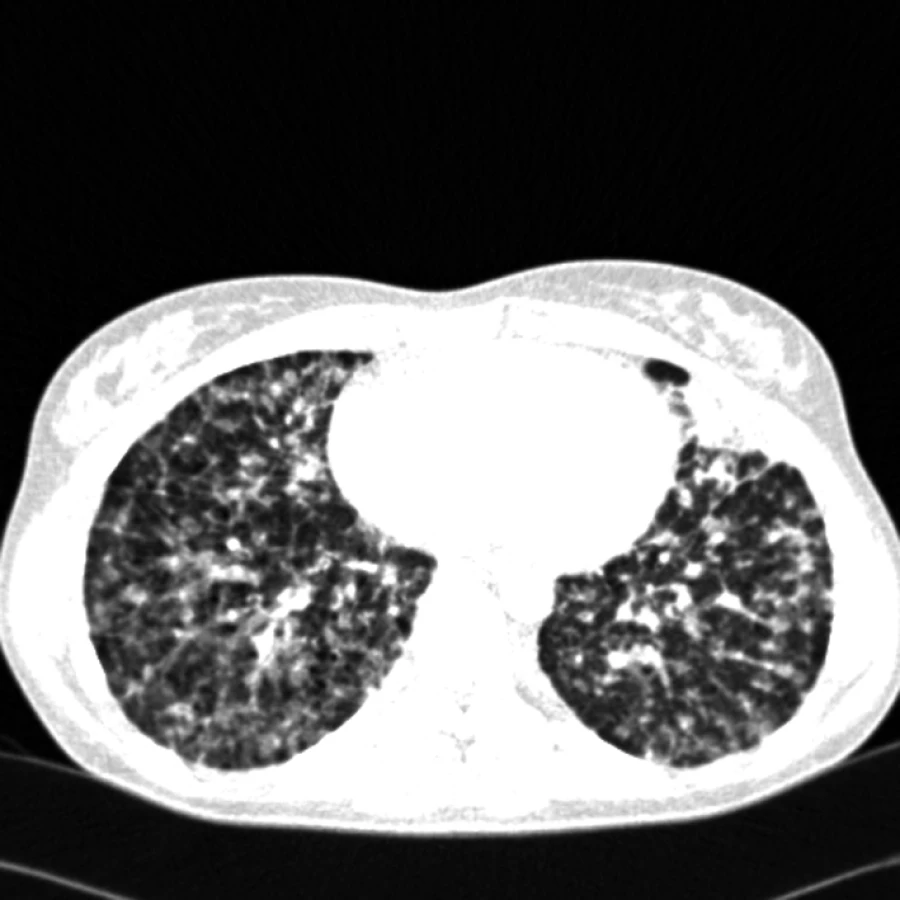
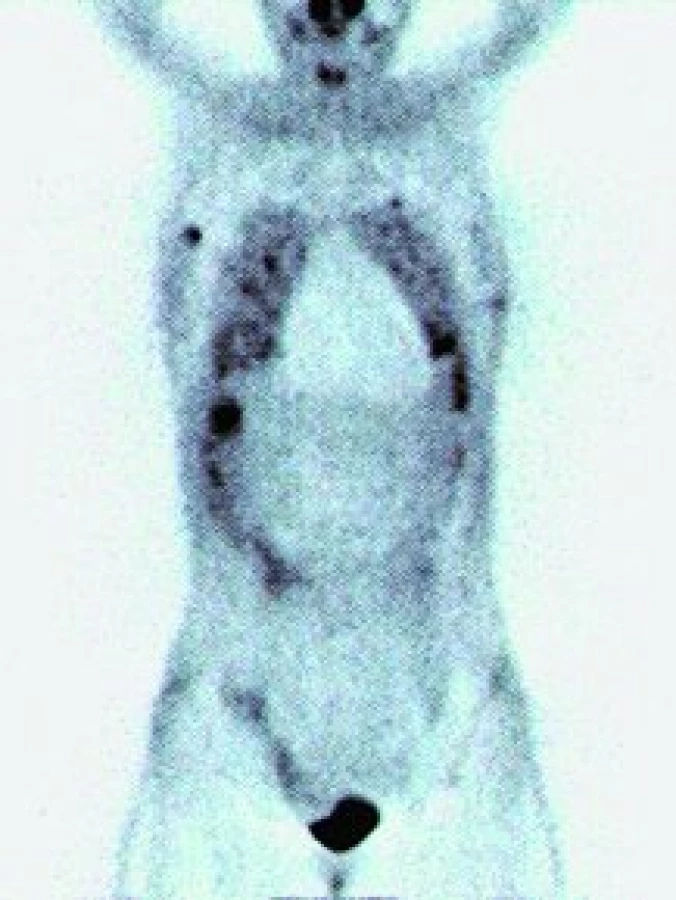
Angiosarkom
Primární plicní angiosarkom je velmi neobvyklý, většinou jsou plíce postiženy metastaticky.
Převažují dospělí jedinci, mužů je postiženo více než žen. Typické je mnohočetné, oboustranné postižení, které připomíná metastatické postižení. Vzácně se nachází solitární uzly. Tumor může invadovat do mediastina nebo hrudní stěny nebo bývá doprovázen pleurálním nebo perikardiálním výpotkem. Časné je hematogenní metastazování. Typickými klinickými příznaky jsou dušnost a hemoptýza, která až imituje obraz difuzního alveolárního krvácení. Dále může být přítomen kašel, bolesti na hrudi, celková slabost. Radiologicky jsou typické bilaterální mnohočetné nodulace, méně často neostře ohraničené infiltráty nebo solitární uzly [35].
Tumor obsahuje cévní síť, která je tvořena maligními endoteliálními buňkami, místy se nalézají shluky vřetenovitých nebo epitelioidních buněk. Imunohistochemicky jsou pozitivní faktor VIII a CD31. Většina tumorů je inoperabilních, účinek radioterapie a chemoterapie je malý. Byl popsán případ úspěšné léčby pomocí kombinace radioterapie a systémového podávání interleukinu 2 [36]. Obecně je prognóza pacientů špatná, přežití se pohybuje v řádech měsíců od stanovení diagnózy.
Sarkom plicní arterie a vény
Taktéž se jedná o velmi vzácné nádory. Častější je sarkom arterie. Mírně více postihuje ženy, průměrný věk nemocných je 50 let. Většinou roste intraluminálně s okluzí cévy, méně často prorůstá do okolních struktur. Histologicky se nádor může skládat s různých typů sarkomových buněk včetně nediferencovaného sarkomu [37]. Proto se předpokládá, že nádor vychází z pluripotentní mezenchymální buňky. Někdy mají nádorové buňky epitelioidní charakter. Klinicky se projevuje dušností, bolestmi hrudníku, kašlem, hemoptýzou, váhovým úbytkem, synkopou či teplotami. Bohužel bývá často zaměněn za plicní embolizaci [38]. Na skiagramu hrudníku je nejčastějším nálezem zvětšení hilu [39]. Dále bývají přítomny mnohočetné plicní nodulace, rozšíření srdečního stínu, snížení plicní cévní kresby, na CT plic defekt v náplni plicních arterií. Léčbou je kompletní resekce nádoru, která jako jediná modalita zvyšuje šanci na delší přežití. Role chemoterapie a radioterapie není zatím přesně definována, i když v literatuře jsou ojedinělé zmínky o pozitivním efektu kombinované chemoterapie Ifosfamid + Epirubicin [40]. Obecně je prognóza tohoto sarkomu velmi špatná a pacienti umírají do několika měsíců od stanovení diagnózy.
Sarkom plicní vény je onemocnění dospělých, více jsou postiženy ženy. Z potíží bývá přítomna dušnost, bolesti na hrudi, hemoptýza, kašel. V tumoru většinou převažuje složka leiomyosarkomu, vzácný je epitelioidní původ. Léčba je chirurgická.
Maligní glomus tumor
Primární maligní glomus tumor plic je extrémně vzácný nádor, dosud bylo v literatuře popsáno je několik případů. Další nitrohrudní lokalizací může být trachea a mediastinum. Tento nádor vychází z glomových buněk, které se normálně nacházejí v parasympatických gangliích – glomus caroticum a glomus supracardiale. Jedná se o cévnatý tumor, který centrálně nekrotizuje a má tendenci invadovat do cév [41]. Je složen z epitelioidních glomových buněk, což jsou modifikované buňky hladkého svalstva a svazků vřetenovitých buněk. Nádorové glomové buňky jsou kubického tvaru s velkým jádrem a eozinofilní cytoplazmou. Je pro něj typická zvýšená mitotická aktivita. Imunohistochemicky je pozitivní vimentin, calponin, h-caldesmin a α-aktin hladkých svalů [42]. Často je bezpříznakový, někdy se projeví bolestmi na hrudi, hemoptýzou nebo pneumotoraxem. Na skiagramu hrudníku nebo CT vyšetření bývá nalézán nepravidelně ohraničený ložiskový stín s ostrými okraji. Terapeutickou volbou je chirurgické odstranění, event. doplněné chemoterapií.
Maligní tumor pochvy periferních nervů
Maligní tumor pochvy periferních nervů (Malignant Peripheral Sheath-Nerve Tumour), zvaný též maligní schwannom či neurofibrosarkom, je vzácný a agresivní sarkom. Může vycházet ze Schwannových buněk, perineurinálních buněk či fibroblastů nervových pochev. Obvykle se nachází na končetinách, hlavě a krku. Nitrohrudní lokalizace tohoto nádoru je neobvyklá a primární plicní postižení je zcela výjimečné. Je popisován jeho vztah k neurofibromatóze, u které se vyskytuje 20krát častěji než u ostatní populace [43]. Bývají postiženy osoby mladšího a středního věku. Způsobuje kašel, bolesti na hrudi a dušnost. Na skiagramu hrudníku je popisováno laločnaté, nehomogenní a dobře ohraničené ložisko různé velikosti. Skládá se z vřetenovitých do svazků uspořádaných buněk s pleomorfním jádrem. Někdy bývají přítomny i obrovské mnohojaderné nádorové buňky. Imunohistochemicky je pozitivní vimentin a S-100 protein [44]. Léčbou je resekce nádoru. Radioterapie může oddálit rekurenci onemocnění, ale nezlepšuje dobu přežití. Efekt chemoterapie je malý. Zcela raritním nálezem je primární plicní neuroblastom či ganglioneuroblastom.
Pleuropulmonální blastom (PPB)
Jde o typický dysontogenetický tumor dětí. Je to vzácný a velmi agresivní nádor, který se dělí do tří skupin: PPB1 čistě cystický tumor, PPB2 smíšený typ, PPB3 solidní tumor [45]. Je složen z primitivních blastémových buněk a maligního mezenchymálního stromatu obsahujícího různě diferencované sarkomatozní komponenty. Cystická složka obsahuje benigní metaplastické epiteliální buňky. Bývá popisována souvislost s kongenitálními malformacemi plic. Může metastazovat do mozku, kostí, lymfatických uzlin, jater, slinivky, ledvin a nadledvin. Léčba je chirurgická, doplněná radioterapií a/nebo chemoterapií. Prognóza nemoci je špatná, většina nemocných umírá do 1–2 let od zjištění diagnózy [46].
Plicní blastom
Jde o vzácný tumor dospělých, který postihuje hlavně kuřáky. Je složen z maligních žlázových epiteliálních buněk a mezenchymálních vřetenovitých, event. embryonálních buněk. Přes 80% nemocných má potíže, jako jsou kašel, bolesti na hrudi, dušnost, hemoptýza. Na skiagramu hrudníku tvoří obvykle velká, solitární periferní ložiska, hlavně v horních plicních polích. Může se šířit do lymfatických uzlin, na pleuru, bránici, do hrudní stěny, srdce, mozku, jater a měkkých tkání. Léčbou je resekce tumoru s disekcí lymfatických uzlin, efekt radioterapie a chemoterapie je nevelký. Prognóza je špatná, dvě třetiny nemocných zemřou během dvou let od zjištění nemoci.
Synoviální sarkom
Synoviální sarkom je morfologicky dobře definovaný nádor, který tvoří asi 10% všech sarkomů měkkých tkání. Přes 90% všech případů je lokalizováno periartikulárně, primární plicní postižení je vzácné. Předpokládá se, že nádor vzniká maligním zvratem pluripotentní mezenchymální buňky. Histologicky se synoviální sarkom dělí na čtyři podtypy: bifázický, monofázický fibrózní, monofázický epiteliální a málo diferencovaný [47]. V případě primárního plicního postižení se jedná většinou o monofázický fibrózní synoviální sarkom, méně často jde o bifázický podtyp. Postihuje především dospělé jedince. Většinou má charakter solidní masy v plicním parenchymu, výjimečně se nachází endobronchiálně ve formě polypoidní formace. Klinické potíže jsou nespecifické – kašel, dušnost, bolesti na hrudi, hemoptýza. Na skiagramu hrudníku tvoří laločnatá, nehomogenní, dobře ohraničená ložiska. Histologicky obraz závisí na podtypu synoviálního sarkomu. Bývají nalezeny vřetenovité buňky uspořádané do svazků a epiteliální buňky ve shlucích. Mohou být nalezeny ložiska hyalinní fibrózy, nekrózy, myxoidní tkáně a prokrvácení. Imunohistochemicky jsou sarkomové buňky pozitivní na vimentin, zatímco epiteliální na EMA a cytokeratin [48]. Léčebnou volbou je resekce tumoru s disekcí regionálních lymfatických uzlin. Radioterapii a chemoterapii lze užít jako léčbu adjuvantní nebo u inoperabilních případů, výsledky ale nejsou uspokojivé.

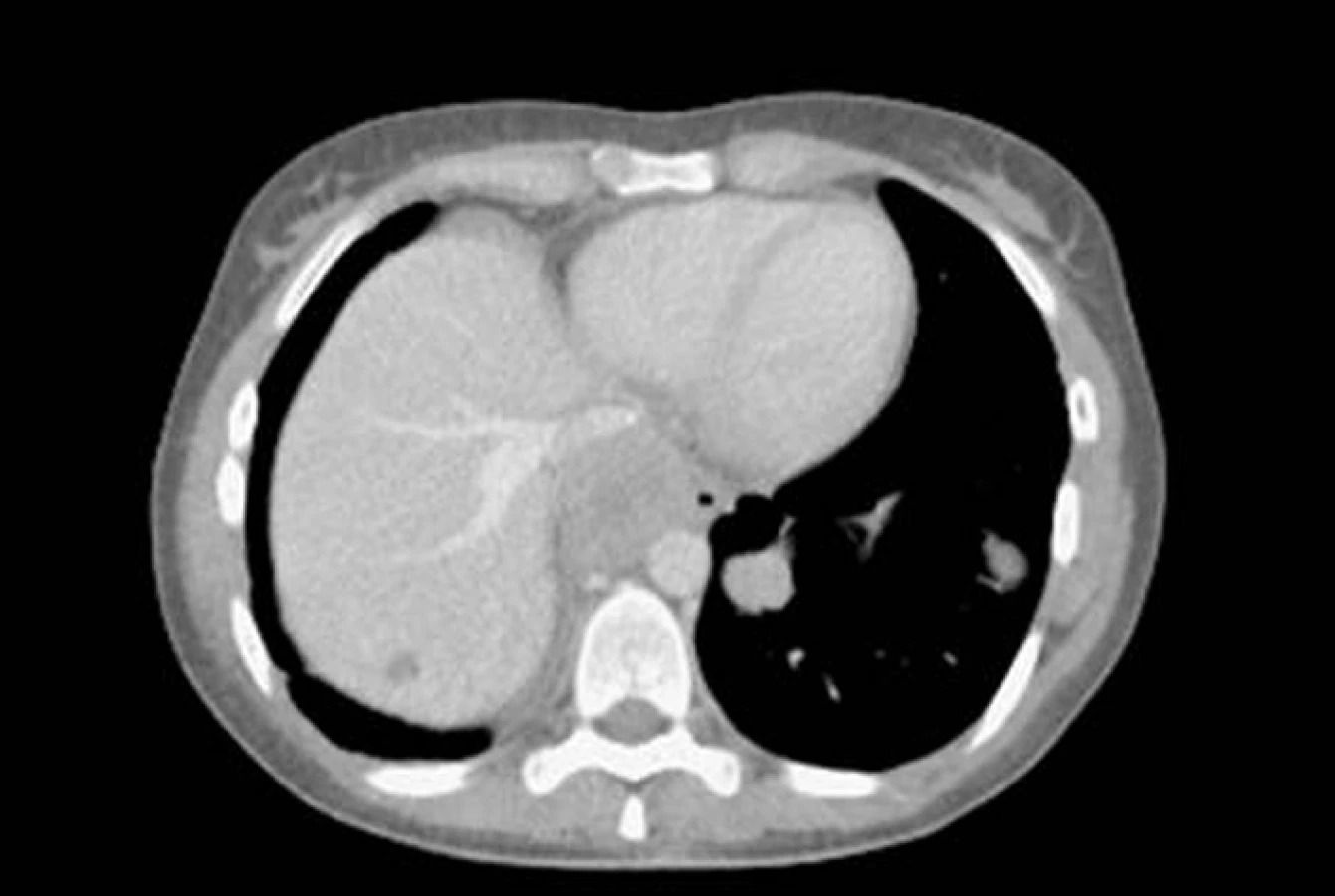
Primitivní neuroektodermální tumor
Primitivní neuroektodermální tumor (Primitive Neuroectodermal Tumour) plic je velmi vzácný nádor, patřící do rodiny Ewingova sarkomu. Nejčastěji se nachází v měkkých tkáních nebo kostech, ale může se vyskytovat i v ovariích, varlatech, děloze, ledvinách, slinivce a myokardu. Postihuje hlavně děti a mladé dospělé. V histologickém obraze jsou přítomny těsně nahloučené primitivní malé kulaté nádorové buňky. Imunohistochemicky je přítomna pozitivita glykogenu, neuron-specifické endolázy, S-100 proteinu a MIC-2 markeru, což je p30/32 povrchový buněčný antigen [49]. Cytogenetickou abnormalitou je translokace dlouhých ramének chromozomů 11 a 22. Nejčastějším radiologickým nálezem je heterogenní hyperdenzní masa v plicním parenchymu, někdy i s centrálním hypodenzním projasněním. Léčba je agresivní a spočívá v chirurgické resekci kombinované s chemoterapií a radioterapií.
Desmoplastický malokulatobuněčný tumor
Desmoplastický malokulatobuněčný tumor (Desmoplastic Small Round Cell Tumour) plic je extrémně vzácný. Častěji je tento typ sarkomu lokalizován v břiše, pánvi a na omentu. Jedná se o primitivní polyfenotypický sarkom. Je vysoce maligní a jeho prognóza je velmi špatná. Nejčastěji postihuje adolescenty a mladé muže ve věku 15–35 let. Klinicky se obvykle projevuje bolestmi na hrudi a pleurálním výpotkem. Histologicky jsou nalézána zašpičatělá hnízda malých kulatých nádorových buněk uložená ve fibroblastickém stromatu. Imunohistochemicky je typickým nálezem pozitivní barvení na desmin ve formě perinukleárního tečkování. Specifické jsou i cytogenetické abnormality – t(11, 22)p(13, q12) [50]. Diferenciálně diagnosticky je nutno vyloučit jiné tumory z malých kulatých buněk, ke kterým patří malobuněčný mezoteliom, primitivní neuroektodermální tumor, non hodgkinský lymfom a malobuněčný karcinom plic. Chirurgické odstranění tumoru doplněné chemoterapií a radioterapií je léčbou u lokalizovaného nádoru [51]. U pokročilých forem nemoci je možná již jen paliativní léčba.
Maligní tumor ze zrnitých buněk
Maligní tumor ze zrnitých buněk (Malignant Granular Cell Tumour – MGCT) je poměrně vzácné onemocnění vycházející z mezenchymálních buněk. Bylo popsáno postižení kůže, dutiny ústní, prsu, močového měchýře a hrudníku. Plicní MGCT se vyskytuje ve všech věkových skupinách a více jsou postiženy ženy. Více než 90% všech tumorů je umístěno endobronchiálně s tendencí k peribronchiálnímu šíření. U několika procent případů je popsáno mnohočetné endobronchiální postižení. Z klinických potíží je nejčastější kašel, dále bolesti na hrudi, dušnost, hemoptýza, váhový úbytek a teploty. Častá je obstrukční pneumonie, naopak vzácnými komplikacemi jsou bronchiektázie a plicní absces. Na skiagramu hrudníku bývá obvykle nacházena atelektáza nebo obraz obstrukční pneumonie, ale u asymptomatických případů se nalézá okrouhlá léze. Nádor obsahuje jemné fibrózní stroma. V něm jsou rozptýleny nádorové buňky, které jsou většinou okrouhlého nebo oválného tvaru. Obsahují velké množství zrnité eozinofilní cytoplazmy, zatímco jádra jsou malá. Nádorové buňky jsou pozitivní na S-100 protein, catepsin B, neuron specifickou enolázu. Léčba je většinou chirurgická, ale u malých endobronchiálních tumorů velikosti do 1cm lze zkusit endoskopickou léčbu [52].
Intimální sarkom
Intimální sarkom je vzácný tumor postihující velké cévy, jako jsou aorta, dutá žíla a plicní arterie. Ačkoliv nádor postihuje cévy, není řazen mezi vaskulární sarkomy ale mezi sarkomy nejasného původu. Důvodem je, že tumor je složen z nediferencovaných nádorových buněk nejasného původu. Tento sarkom se šíří podél intimálního povrchu a často vytváří intraluminální polyp, který obturuje průsvit cévy. Uvolněné nádorové buňky se pak šíří krevním řečištěm do dalších částí těla. Tumor taktéž prorůstá do okolních mediastinálních struktur a plicního parenchymu. Klinicky dominuje dušnost, kašel a hemoptýza. Při angio-CT vyšetření bývá nalézán defekt nebo úplná okluze plicních arterií a nádor může být zaměněn za trombembolickou nemoc. Histologicky je tumor složen z vřetenovitých buněk s příměsí polygonálních a obrovských mnohojaderných buněk. Imunohistochemicky je pozitivní vimentin a CD31. Ideální léčbou je kompletní resekce tumoru, u které je popisováno tříleté přežití 69%. U inoperabilních případů se ale střední doba přežití pohybuje mezi 1,5–5,5 měsíců od stanovení diagnózy [53]. V literatuře jsou ojedinělé zmínky o úspěšné léčbě tohoto sarkomu chemoterapií ve složení ifosfamid + epirubicin.
Maligní mezenchymom
Tento sarkom je charakterizován přítomností dvou nebo více různých maligních mezenchymálních buněčných linií. Může se jednat o buňky lipo-, leiomyo-, rabdomyo-, chondro , osteo-, fibrosarkomu. Předpokládá se, že tento nádor vzniká maligní diferenciací pluripotentní mezenchymální buňky. Maligní mezenchymom se nejčastěji vyskytuje v retroperitoneu a stehnu. V hrudníku může postihovat srdce, hrudní stěnu, pleuru a plíce [54]. Plicní maligní mezenchymom je často spojen s kongenitálními plicními cystami. Klinicky se projevuje nespecifickými příznaky, jako je kašel, dušnost, bolest na hrudi. Nádor má poměrně velkou tendenci recidivovat a metastazovat. Kompletní resekce tumoru je nejefektivnější terapií, ale i paliativní resekce přináší symptomatické zlepšení. Chemoterapie a radioterapie má jen malý efekt. Prognosticky nepříznivé faktory jsou věk nad 40 let a přítomnost rabdomyomové složky v tumoru [55].
Závěr
Primární plicní sarkomy jsou vzácná maligní onemocnění. Nejčastější jsou leiomyosarkom, maligní fibrózní histiocytom, fibrosarkom, hemangiopericytom a rabdomyosarkom [3]. Podezření na možný sarkom může vzbudit už patologický plicní nález v rámci vyšetřování různými zobrazovacími metodami včetně pozitronové emisní tomografie. Neexistuje ale žádný jednoznačný diagnostický ani laboratorní marker sarkomů. Definitivní potvrzení diagnózy přináší jedině histologické a imunohistochemické vyšetření vzorku tumoru. Z bioptických metod lze dle lokalizace tumoru použít endoskopické metody – forceps biopsii, transbronchiální plicní biopsii, dále transparietální punkci plic. Přesnější jsou chirurgické metody – torakoskopie nebo torakotomie. Nezřídka je ale definitivní diagnóza zjištěna až při pitvě. Kardinální a často obtížně zodpověditelnou otázkou je, zda se jedná o primární plicní tumor či metastatické postižení.
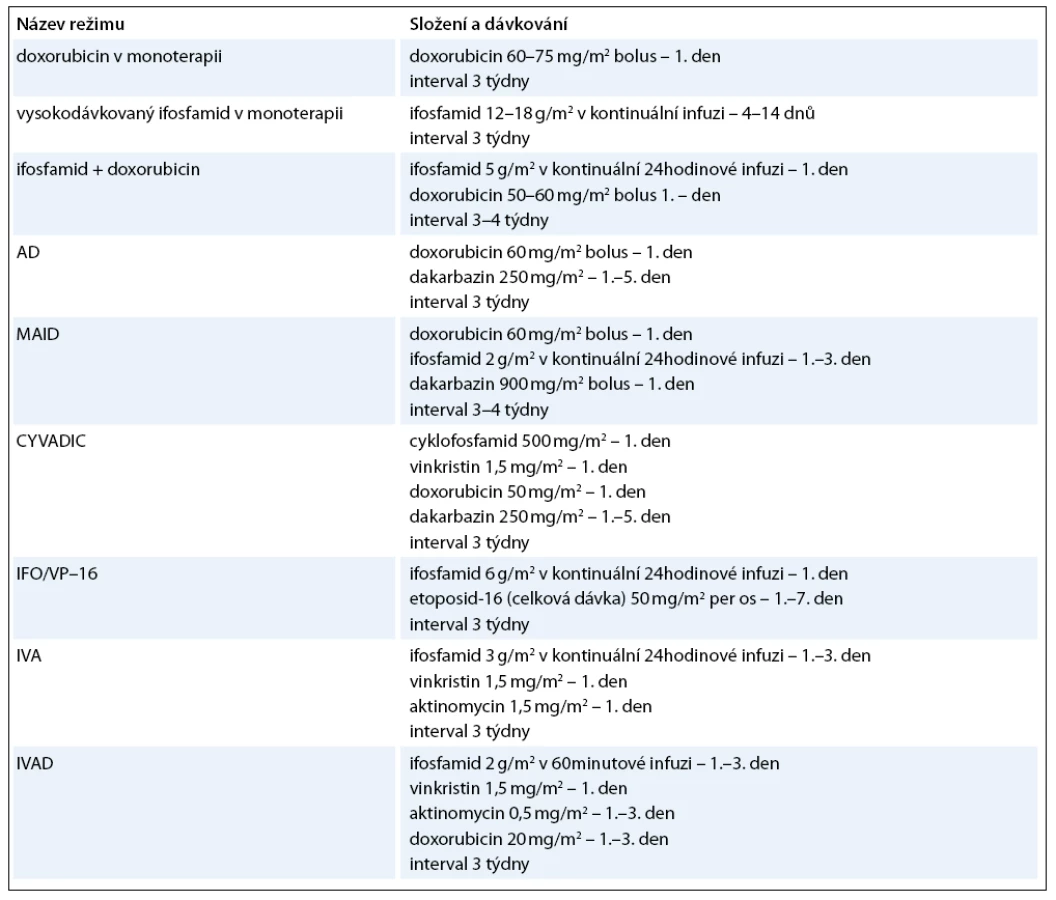
Optimální terapeutickou modalitou u primárních plicních sarkomů je samozřejmě kompletní chirurgické odstranění (nejlépe i s disekcí regionálních lymfatických uzlin), ale mnohdy je možná pouze paliativní resekce nebo je nádor primárně inoperabilní. V těchto případech nastupují další léčebné modality – radioterapie a chemoterapie. Radioterapie je užívána hlavně jako adjuvantní léčba k lokální kontrole se snahou snížit výskyt lokálních recidiv. Celková dávka záření je 50–60 grayů [56]. Neoadjuvantní radioterapie má za cíl zmenšit rozsah tumoru, a umožnit tak lepší chirurgické odstranění nádoru. V indikovaných případech je možno použít brachyterapii. Role chemoterapie je méně přesně definována. Podle posledních standardů České onkologické společnosti není chemoterapie indikována u klinických stadií I–IIA. U stadií IIB a III je indikována adjuvantní chemoterapie u nádorů větších 5cm a špatně diferencovaných nebo nediferencovaných sarkomů. Neoadjuvantní chemoterapie může být v indikovaných případech přidána k radioterapii. U primárně inoperabilních případů je přípustná paliativní chemoterapie [57]. Problémem chemoterapie je její vysoká toxicita a poměrně nízká léčebná odpověď, která se pohybuje kolem 20% [56]. Existují různé monoterapeutické nebo kombinované režimy, nejčastější jsou uvedeny v tab. 5. Mezi používaná cytostatika patří doxorubicin, ifosfamid, dakarbazin, vinkristin, etoposid, epirubicin, daunorubicin, cyklofosfamid, metotrexát, aktinomycin D, mitoxantron, bleomycin, paclitaxel, vinorelbin. V poslední době se studuje efekt kombinované léčby gemcitabinem a docetaxelem [58]. Probíhají již první pokusy s biologickou léčbou a byla popsána úspěšná terapie angiosarkomu pomocí interleukinu 2. Velmi nadějně se jeví dle zveřejněných studií léčba sarkomů měkkých tkání trabectedinem (látka izolovaná z mořského pláštěnce Ecteinascidia turbinata), jež prodlužuje období bez nemoci a medián přežití [59]. Z dalších biologických preparátů jsou testovány anti VEGF monoklonální protilátka bevacizumab, inhibitory tyrozinkinázy imatinib, sunitinib, pazopanib a sorafenib [60,61]. Nezřídka je ale rozsah onemocnění takový, že je možná již jen paliativní symptomatická léčba.
K prognostickým faktorům patří velikost tumoru, histologický typ, grading, klinické stadium sarkomu a rozsah operačního výkonu [2–4]. Dle různých literárních údajů je medián přežití 48 měsíců a pětileté přežití se pohybuje mezi 38 a 48% [2,3,56,62]. Problémem jsou ale malé počty případů publikované v jednotlivých pracích. Obecně lze říci, že primární plicní sarkomy mají ve velké části případů agresivní průběh a jejich léčba je svízelná a ne příliš úspěšná. Nádory často recidivují a prognóza bývá nejistá [63].
Na našem pracovišti jsme během posledních let diagnostikovali a dále sledujeme a léčíme několik pacientů s různými typy primárních plicních sarkomů. V obrazové dokumentaci uvádíme skiagramy hrudníku, skeny CT vyšetření hrudníku a skeny celotělového PET vyšetření pacientů s epitelioidním hemangioendoteliomem (obr. 1,2,3), synoviálním sarkomem (obr. 4,5,6) a špatně diferencovaným sarkomem (obr. 7,8,9).
Práce je podpořena grantem Ministerstva zdravotnictví ČR: NS9959-3.
Autoři
deklarují, že v souvislosti s předmětem studie nemají žádné
komerční zájmy.
The
authors declare they have no potential conflicts of interest
concerning drugs, pruducts, or services
used in the study.
Redakční
rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro
publikace zasílané do biomedicínských
časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform
requirements” for biomedical papers.
MUDr. Tereza Jakubcová
Klinika
plicních nemocí a tuberkulózy
FN
a LF UP Olomouc
I. P. Pavlova 6
775
20 Olomouc
e-mail:
tereza.jakubcova@fnol.cz
Sources
1. Zatloukal P, Petruželka L. Karcinoid plic. 1. vyd. Praha: Grada Publishing 2001.
2. Régnard JF, Icard P, Guibert L et al. Prognostic Factors and Results After Surgical Treatment of Primary Sarcomas of the Lung. Ann Thorac Surg 1999; 68 : 227–231.
3. Porte HL, Metois DG, Leroy X et al. Surgical treatment of primary sarcoma of the lung. Eur J Cardiothorac Surg 2000; 18 : 136–142.
4. Shidham VB. Benign and Malignant Soft Tissue Tumors. Available from http://www.emedicine.com/orthoped/TOPIC377.HTM. [updated 2006].
5. Gladish GW, Sabloff BM, Munden RF et al. Primary Thoracic Sarcomas. Radiographics 2002; 22 : 621–637.
6. Krygier G, Amado A, Salisbury S et al. Primary lung liposarcoma. Lung Cancer 1997; 17 : 271–275.
7. Said M, Migaw H, Hafsa C et al. Imaging features of primary pulmonary liposarcoma. Australasian Radiology 2003; 47 : 313–317.
8. Sin HL, Shim JJ, Shin JS et al. Primary Endobronchial Leiomyosarcoma. Respiration 2001; 68 : 99–102.
9. Dong Shang L, Ko-Huang L, Jang-Ming S et al. Primary Bronchopulmonary Leiomyosarcoma of the Left Main Bronchus in a Child Presenting With Wheezing and Atelectasis of the Left Lung. Pediatric Pulmonology 2002; 33 : 318–321.
10. Wyatt JM, Matsubara O. Update in pulmonary vascular diseases. Pathology International 2004; 54 : 469–489.
11. Yekeler E, Dursun M, Yildirim A et al. Diffuse pulmonary lymphangiomatosis: imaging findings. Diagn Intervent Radiology 2005; 11 : 31–34.
12. Ozcan C, Celik A, Ural Z et al. Primary Pulmonary Rhabdomyosarcoma Arising Within Cystic Adenomatoid Malformation: A Case Report and Review of the Literature. J Pediatric Surg 2001; 36 : 1062–1065.
13. Conquest HF, Thornton JL, Massie JR et al. Primary Pulmonary Rhabdomyosarcoma. Ann Surg 1965; 161 : 688–692.
14. Shan ND, Diwanji SR. Primary chondrosarcoma of the lung with cutaneous and skeletal metastase. Singapure Med 2007; 48 : 196–199.
15. Watanabe A, Ito M, Nomura F. Primary Chondrosarcoma of the Lung – A Case Report with Immunohistochemical Study. Jpn J Med 1990; 29 : 616–619.
16. Yamazaki K, Okabayashi K, Hamatake D et al. Primary Osteosarcoma of the Lung: A Case Report. Ann Thorac Cardiovasc Surg 2006; 12 : 126–128.
17. Kadowaki T, Hamada H, Yokoyama A. Two Cases of Primary Pulmonary Osteosarcoma. Internal Medicine 2005; 4 : 632–637.
18. Sagawa M, Ueda Y, Matsubara F et al. Intrapulmonary Solitary Fibrous Tumor Diagnosed by Immunihistochemical and Genetic Approaches: Report of a Case. Surg Today 2007; 37 : 423–425.
19. Kanamori Y, Hashizume K, Sugiyama M et al. Intrapulmonary Solitary Fibrous Tumor in an Eight-Year-Old Male. Pediatr Pulmonol 2005; 40 : 261–264.
20. Sakurai H, Hasegawa T, Watanabe S et al. Inflammatory myofibroblastic tumor of the lung. Eur J Card Thorac Surg 2004; 25 : 155–159.
21. Melloni G, Carretta A, Ciriaco P et al. Inflammatory Pseudotumor of the Lung in Adults. Ann Thorac Surg 2005; 79 : 426–432.
22. Findik S, Erkan ML, Kandemir B. A Case report: Primary Malignant Fibrous Histiocytoma of the Lung. Turkish Respiratory Journal 2001; 2 : 44–46.
23. Maeda J, Ohta M, Inoue M et al. Surgical Intervention for Malignant Fibrous Histiocytoma of the Lung: Report of a Case. Surg Today 2007; 37 : 316–319.
24. Myung SS, Kang-Jey H. Primary Hemangiopericytoma of Lung: Radiography and Pathology. AJR 1979; 133 : 1077–1083.
25. Marec-Bérard P. Malignant Hemangiopericytoma. Available from http://www.orpha.net/data/patho/GB/uk-HCP.pdf.
26. Kim L, Yoon YH, Choi SJ et al. Hyalinizing spindle cell tumor with giant rosettes arising in the lung: Report of a case with FUS-CREB3L2 fusion transcripts. Pathology International 2007; 5 : 153–157.
27. Bejarano PA, Padhya TA, Smith R et al. Hyalinizing Spindle Cell Tumor With Giant Rosettes – A Soft Tissue Tumor With Mesenchymal and Neuroendocrine Features. Arch Pathol Labor Med 2000; 124 : 1179–1184.
28. Logrono R, Filipowicz EA, Eyzaguirre EJ et al. Diagnosis of primary fibrosarcoma of the lung by fine-needle aspiration and core biopsy: A case report and review of the literature. Arch Pathol Lab Med 1999; 123 : 731–735.
29. Savas C, Candir O, Ozguner F. Acute Respiratory Distress Due to Fibrosarcoma of the Carina in a Child. Pediatr Pulmonol 2004; 38 : 355–357.
30. McGinnis M, Jacobs G, el-Naggar A et al. Congenital peribronchial myofibroblastic tumor (so-called „congenital leiomyosarcoma“). A distinct neonatal lung lesion associated with nonimmune hydrops fetalis. Mod Pathol 1993; 6 : 487–492.
31. Horikoshi T, Kikuchi A, Matsumoto Y et al. Fetal hydrops associated with congenital pulmonary myofibroblastic tumor. J Obstet Gynaecol Res 2005; 31 : 552–555.
32. Aboulafia DM. The Epidemiologic, Pathologic and Clinical Features of AIDS-Associated Pulmonary Kaposi’s Sarkoma. Chest 2000; 117 : 1128–1145.
33. Snopková S, Chalupa P. Postižení plic u infekce HIV/AIDS. In: Ševčík P, Skřičková J, Šrámek V et al. Záněty plic v intenzivní medicíně. 1. vyd. Praha: Galén 2004.
34. Jakubec P, Jakubcová T, Hutyrová B et al. Epitelioidní hemangioendoteliom. Kazuistiky v alergologii, pneumologii a ORL 2007; 4 : 7–14.
35. Atasoy C, Fitoz S, Yigit H et al. Radiographic, CT, and MRI findings in primary pulmonary angiosarcoma. J Clin Imaging 2001; 25 : 337–340.
36. Kojima K, Okamoto I, Ushijima S et al. Successful Treatment of Primary Pulmonary Angiosarcoma. Chest 2003; 124 : 2397–2400.
37. Colby TV, Koss MN, Travis WD. Tumors of the Lower Respiratory Tract (Atlas of Tumor Patology, series 3, fasc. 13). Washington: Armed Forces Institute of Patology 1995.
38. Matoo A, Fedullo PF, Kapelanski D et al. Pulmonary Artery Sarcoma. A Case Report of Surgical Cure and 5-Year Follow up. Chest 2002; 122 : 745–747.
39. Bhatia K, Ellis S. Unusual lung tumours: an illustrated review of CT features suggestive of this diagnosis. Cancer Imaging 2006; 6 : 72–82.
40. Uchida A, Tabata M, Kiura K et al. Successful Treatment of Pulmonary Artery Sarcoma by a Two-drug Combination Chemotherapy Consisting of Ifosfamide and Epirubicin. Jpn J Clin Oncol 2005; 35 : 417–419.
41. Majer I, Haruštiak S, Janík P et al. Glomus trachey jako neobvyklý prípad dýchavice. Kazuistiky v alergologii, pneumologii a ORL 2007; 4 : 4–6.
42. Hishida T, Hasegawa T, Asamura H et al. Malignant glomus tumor of the lung. Pathol Int 2003; 53 : 632–636.
43. Chao BH. Intrathoracic Malignant Peripheral Nerve Sheath Tumor in Neurofibromatosis 1. J Clin Oncol 2008; 26 : 2216–2218.
44. Tashiro T, Kawakita C, Takai C. Primary pulmonary malignant peripheral nerve sheath tumor: a case report. Acta Cytol 2007; 51 : 820–824.
45. Hill DA, Sadeghi S, Schultz MZ et al. Pleuropulmonary Blastoma in an Adult. Cancer. 1999; 85 : 2368–2374.
46. Abdelmohsen MH, Amr RS, Shereif LB. Pleuropulmonary blastoma. Available from http://www.ispub.com/journal/the_internet_journal_of_thoracic_and_cardiovascular_surgery.html. Version 6 [ 2004]. ISSN 1524–0724.
47. Niva H, Masuda S, Kobayashi C et al. Pulmonary synovial sarcoma with polypoid endobronchial growth: A case report, immunohistochemical and cytogenetic study. Patology International 2004; 54 : 611–615.
48. Hosono T, Hironaka M, Kobayashi A et al. Primary Pulmonary Synovial Sarcoma Confirmed by Molecular Detection of SYT-SSX1 Fusion Gene Transcripts: a Case Report and Review of the Literature. Jpn J Clin Oncol 2005; 35 : 274–279.
49. Yoon YL, Do HK, Ji HL et al. Primary Pulmonary Ewing’s Sarcoma/Primitive Neuroectodermal Tumor in a 67-year-old Man. J Korean Med Sci 2007; 22 : 59–63.
50. Wang Z, Xiao W, Zheng S. Desmoplastic small round cell tumor of the lung: case report. Chin Med J 2007; 120 : 2327–2328.
51. Stuart-Buttle CE, Smart CJ, Pritchard S. et al. Desmoplastic small round cell tumour: a review of literature and treatment options. Surg Oncol 2008; 17 : 107–112.
52. Jiang M, Anderson T, Nwogu C et al. Pulmonary malignant granular cell tumor. World J Surg Oncol 2003; 1 : 22–26.
53. Nakajima J, Morota T, Matsumoto J et al. Pulmonary Intimal Sarcoma Treated by a Left Pneumonectomy with Pulmonary Arterioplasty Under Cardiopulmonary Bypass: Report of a Case Surg Today 2007; 37 : 496–499.
54. Deslée G, Guillou PJ, Baehrel B et al. Malignant mesenchymoma of the pleura. Interac Cardiovasc Thorac Surg 2003; 2 : 376–378.
55. Adachi T, Oda Y, Sakamoto A. Prognostic factors in the so-called malignant mesenchymoma: a clinicopathological and immunohistochemical analysis. Oncol Rep 2003; 10 : 803–811.
56. Mastroianni BE, Falchero L, Chalabreysse L et al. Primary sarcomas of the lung. A clinicopathologic study of 12 cases. Lung Cancer 2002; 38 : 283–289.
57. Česká onkologická společnost ČLS JEP. Kapitola 22: Zhoubné nádory měkkých tkání. In: Zásady cytostatické léčby maligních onkologických onemocnění. 6. vyd. Brno: 2008. Dostupné z http://www.linkos.cz/info_praxe/standardy_htlm.php?t=26.
58. Maki RG, Wathen JK, Patel SR et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma aliance for research through collaboration study 002 /corrected/. J Clin Oncol 2007; 25 : 2755–2763.
59. Le Cesne A, Domont J, Cioffi A. The new era of trabectedin in soft tissue sarcomas. EJHP Practice 2008; 14 : 72–75.
60. Schoffski P, Dumez H, Wolter P et al. Clinical impact of trabectedin (ecteinascidin 743) in advanced/metastatic soft tissue sarcoma. Expert Opin Pharmacother 2008; 9 : 1609–1618.
61. Ray-Coquard I, Blay JY. Adult soft tissue sarcomas. EJHP Practice 2008; 14 : 68–70.
62. Petrov DB, Vlasov VI, Kalaydjiev GT et al. Primary pulmonary sarcomas and carcinosarcomas – postoperative results and comparative survival analysis. Eur J Cardiothorac Surg 2003; 23 : 461–466.
63. Zatloukal P. Nádory plic. In: Zatloukal P, Fiala P, Votruba J et al. Vnitřní lékařství, díl IIIa Pneumologie. 1. vyd. Praha: Galén 2001.
64. Travis WD, Colby TV, Corrin B et al. In Collaboration with Sobin LH and Pathologists from 14 Countries. World Health Organization International Histological Classification of Tumours. Histological Typing of Lung and Pleural Tumours. 3rd ed. Berlin: Springer-Verlag 1999.
65. Fletcher CDM, Unni KK, Mertens F (eds). World Health Organization: Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press 2002.
66. Kotilingam D, Lev DC, Lazar AJ et al. Staging Soft Tissue Sarcoma: Evolution and Change. CA Cancer J Clin 2006; 56(5): 282–291.
67. Fleming ID, Cooper JS, Henson DE et al (eds). AJCC Cancer Staging Manual. 5th ed. Philadelphia: Lippincott-Raven 1997.
68. Greene FL, Page DL, Fleming ID et al (eds). AJCC Cancer Staging Manual. 6th ed. New York: Springer 2002.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2009 Issue 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Primární plicní sarkomy
- Paliativní a hospicová péče v České republice a v Evropě
- Radikální operační výkon a intenzivní chemoterapie jsou podmínkou úspěšné léčby osteosarkomu
- Liečba sunitinibom a hypotyreóza – kazuistika a prehľad literatúry