
ABL1, SRC a další nereceptorové tyrozinkinázy jako nové cíle specifické protinádorové léčby
ABL1, SRC and Other Non ‑ Receptor Protein Tyrosine Kinases as New Targets for Specific Anticancer Therapy
Non ‑ receptor protein tyrosine kinases are responsible for signal transduction during many physiologic cellular processes, including cell growth and proliferation, apoptosis, differentiation, regulation of actin cytoskeleton, cell shape, adhesion, motility and migration. Aberrant activity of protein tyrosine kinases (acquired as a result of chromosomal translocation or point mutation) has been implicated in the stimulation of cancer growth and progression, the induction of drug‑resistance, tumour neovascularization, tissue invasion, extravasation and the formation of metastases. Small molecule tyrosine kinase inhibitors interfere with these pathophysiological circuits by blocking the signalling cascades triggered by the aberrantly activated protein tyrosine kinases (e. g. BCR ‑ ABL1, FIP1L1 - PDGFRA or ETV6 - PDGFRB). Tyrosine kinase inhibitors (imatinib, nilotinib, dasatinib) now belong to established anti‑cancer agents with clinical activity in patients with CML, Ph+ ALL, and myeloid neoplasms with overexpression of PDGFRA, PDGFRB and wild‑type KIT. New generation tyrosine kinase inhibitors (e. g. dasatinib) with extended activity against SRC and EPH kinases belong to promising anti‑cancer agents with documented preclinical activity in several solid tumours (e. g. prostate cancer).
Key words:
non‑receptor tyrosine kinases – SRC – BCR ‑ ABL1 – TKI – imatinib – nilotinib – dasatinib – CML – solid tumours – prostate cancer – bone metastases
Authors:
P. Klener 1,2; Klener P. Jr 1,3
Authors‘ workplace:
I. interní klinika – klinika hematologie VFN a 1. LF UK v Praze
1; Ústav hematologie a krevní transfuze, Praha
2; Ústav patologické fyziologie 1. LF UK v Praze
3
Published in:
Klin Onkol 2010; 23(4): 203-209
Category:
Reviews
Overview
Nereceptorové protein tyrozinkinázy fyziologicky regulují řadu klíčových buněčných pochodů včetně růstu, proliferace, apoptózy, diferenciace, adheze, motility či migrace. Patologicky nabytá aktivita protein tyrozinkinázy (např. následkem chromozomální translokace či bodové mutace) je pak odpovědná za růst, proliferaci, invazivitu a metastazování nádoru, za neovaskularizaci nádorové tkáně a za zvýšenou rezistenci transformovaných buněk vůči chemo/ radio/ imunoterapii. Nízkomolekulární inhibitory tyrozinkinázových proteinů vykazují protinádorový účinek do značné míry právě blokádou signálních drah spouštěných aberantně aktivovanými protein tyrozinkinázami (např. BCR ‑ ABL1, FIP1L1 - PDGFRA or ETV6 - PDGFRB). Imatinib se osvědčil v léčbě chronické myeloidní leukémie, Ph+ akutní lymfoblastové leukemie a některých neoplazií se zvýšenou expresí tyrozinkinázových proteinů, PDGFRA a PDGFRB. Širší inhibiční spektrum tyrozinkinázových proteinů nové generace (např. dasatinib), které zahrnuje kromě (BCR ‑ )ABL1, KIT a PDGFRA/ B též rodinu kináz SRC a EPH, umožňuje klinické testování protinádorového účinku kromě osvědčených hematologických diagnóz též u řady solidních tumorů (např. karcinom prostaty).
Klíčová slova:
nereceptorové tyrozinkinázy – SRC – BCR - ABL1 – TKI – imatinib – nilotinib – dasatinib – CML – solidní nádory – karcinom prostaty – kostní metastázy
Inhibitory tyrozinkinázových proteinů a počátky cílené terapie
Významným pokrokem v léčbě nádorových onemocnění bylo zavedení tzv. cílené léčby (targeted therapy). Cílená léčba specificky blokuje nitrobuněčné pochody podílející se na procesu nádorové transformace a/nebo udržení maligního fenotypu nádorových buněk [1]. Významnou kategorii cílených terapeutik představují tzv. inhibitory signálního přenosu (signal transduction inhibitors – STI), jejichž základní skupinu tvoří inhibitory tyrozinkinázových proteinů (tyrosine kinase inhibitors – TKI). Proteiny s tyrozinkinázovou aktivitou mají schopnost fosforylovat klientské proteiny na tyrozinových zbytcích, čímž významně ovlivňují jejich funkci (obvykle ve smyslu aktivace). Tyrozinkinázové proteiny hrají nezastupitelnou roli v nejrůznějších buněčných transdukčních kaskádách, a tím se významně účastní regulace buněčného růstu, proliferace, migrace, diferenciace či apoptózy [2]. Obecně lze proteiny s tyrozinkinázovou aktivitou dělit na receptorové tyrozinkinázy (RTK) a nereceptorové proteinové tyrozinkinázy (PTK). U řady nádorových onemocnění dochází k aberantní aktivaci či zvýšené expresi některých RTK (např. KIT, FLT3, EGFR/ERBB/HER, VEGFR) či PTK (např. BCR ABL1, SRC, FAK), což vede ke konstitutivní aktivaci důležitých prorůstových, proliferačních a antiapoptotických signálních drah. Mezi přípravky používané k cílené blokádě RTK patří některé monoklonální protilátky, které blokádou extracelulární domény receptoru brání vazbě ligandu, a tím spuštění transdukční kaskády (např. bevacizumab či cetuximab u nádorů s overexpresí VEGF či EGFR). Ke specifické blokádě tyrozinkinázových domén PTK se však používají výhradně nízkomolekulární látky označované jako tyrozinkinázové inhibitory (TKI). TKI blokují transdukční kaskádu buď na úrovni intracytoplazmatické části RTK, nebo na úrovni nereceptorových PTK. V tomto článku se soustředíme na nově zaváděné TKI, které specificky inhibují (mimo jiné) signální kaskádu regulovanou tyrozinkinázovým proteinem SRC (čti sark).
První látkou ze skupiny TKI zavedenou do klinické praxe byl v roce 2001 imatinib (STI 571). Imatinib poměrně specificky blokuje tyrozinkinázovou doménu onkoproteinu BCR ABL1 (kromě ABL1 inhibuje též PDGFR a KIT) a je v současné době lékem první volby u pacientů v chronické fázi chronické myeloidní leukemie (CP CML). Přes nesporný úspěch imatinibu dochází však u části pacientů jak k jeho intoleranci, tak k rozvoji rezistence vůči této látce. Nejčastější příčinou vzniku rezistence k imatinibu jsou bodové mutace v kinázové doméně BCR ABL1 proteinu. TKI druhé a třetí generace (např. nilotinib, dasatinib) mají ve srovnání s imatinibem výrazně vyšší afinitu vůči nemutovanému BCR ABL1 proteinu (nilotinib 20krát, dasatinib 325krát) a vykazují účinek vůči většině imatinib rezistentních klonů. Ze všech dosud popsaných mutací BCR ABL1 jeví největší rezistenci vůči imatinibu mutace T315I (tyrozin → isoleucin), která stericky brání vstupu imatinibu do hydrofobní kapsy ATP vazebného místa. Leukemické buňky s mutací T315I jsou rezistentní vůči všem třem v současnosti klinicky používaným TKI. Kromě rizika rozvoje rezistence k imatinibu se dalším pádným důvodem vývoje TKI nové generace staly hlubší poznatky o biologii maligních onemocnění. Jasně se ukázalo, že naprostá většina nádorových onemocnění nevykazuje v době diagnózy závislost pouze na jednom konkrétním onkogenním signálu (tzv. oncogene addiction), jako je tomu v případě BCR ABL1 signalizace u CML v chronické fázi. Zrušení onkogenní signalizace BCR ABL1 pomocí imatinibu vede v populaci buněk CP CML k přerušení antiapoptotické a prorůstové signalizace, následkem čehož je postupné odumření naprosté většiny leukemických buněk (pravděpodobně vyjma části leukemických kmenových buněk). Naopak u většiny solidních tumorů existuje v době stanovení diagnózy řada vzájemně redundantních onkogenních signálů, které podporují růst a přežití nádorových buněk různými způsoby, což vyžaduje odlišnou protinádorovou strategii zaměřenou na blokádu více aberantně aktivovaných signálních drah. Podobně v akcelerované fázi CML či v blastickém zvratu již obvykle nejsou leukemické buňky absolutně závislé na BCR ABL1 onkogenní signalizaci, což je molekulární příčinou výrazně nižší klinické účinnosti poměrně „úzce“ zaměřeného imatinibu. Výzkum posledních let jasně prokázal klíčovou roli tyrozinkinázového proteinu SRC v progresi různých typů nádorových onemocnění, včetně CML. Předpokládalo se proto, že duální inhibice ABL1 + SRC (např. dasatinibem) povede ve srovnání s imatinibem k účinnější eliminaci neoplastických buněk a rozšíří spektrum klinického použití TKI nové generace.
SRC kinázy a jejich postavení v transdukční kaskádě
Význam SRC kináz byl zhodnocen teprve v posledním desetiletí, ačkoliv má dlouhou historii. Téměř před 100 lety (1911) Francis Payton Rous izoloval z pokusných kuřat stižených sarkomem bezbuněčné agens, které po aplikaci zdravým kuřatům vyvolávalo rychlý růst stejného typu sarkomu [3]. Jednalo se o retrovirus nazývaný dnes Rous sarcoma virus (RSV) – historicky první retrovirus schopný maligní transformace. Analýzou genomu RSV byl následně odhalen první známý onkogen v src a popsán SRC protein jako první protein s tyrozinkinázovou aktivitou. Později byl identifikován buněčný analog onkogenu v src, protoonkogen c SRC, který představuje důležitý fyziologický regulátor řady významných signálních drah. Somatická mutace genu c SRC působící konstitutivní aberantní aktivaci proteinu SRC vede k přeměně protoonkogenu v onkogen (aktivace onkogenu SRC).
Rodina SRC nereceptorových tyrozinkináz (SRC family kinases – SFK) zahrnuje devět členů a dělí se do tří podrodin. V podrodině A jsou to kinázy SRC, YES, FYN a FGR, v podrodině B kinázy LCK, HCK, BLK a LYN. Kinázy FRK tvoří vlastní podrodinu. SRC kinázy se významně podílejí na přenosu signálů z vnějšího prostředí do nitra buňky. Tvoří tak klíčové struktury transdukčních kaskád z RTK na nitrobuněčné signální dráhy regulující proliferaci, růst a apoptózu, včetně MAPK, PI3K AKT mTOR či JAK STAT (obr. 1).SRC kinázy jsou spolu s kinázami FAK (focal adhesion kinases) také hlavním mediátorem přenosu signálů z většiny integrinových receptorů, čímž zásadním způsobem regulují buněčnou adhezi, uspořádání cytoskeletu, motilitu, migraci či neovaskularizaci [4–6]. SRC kinázy hrají také důležitou roli v regulaci osteoklastogeneze [7].
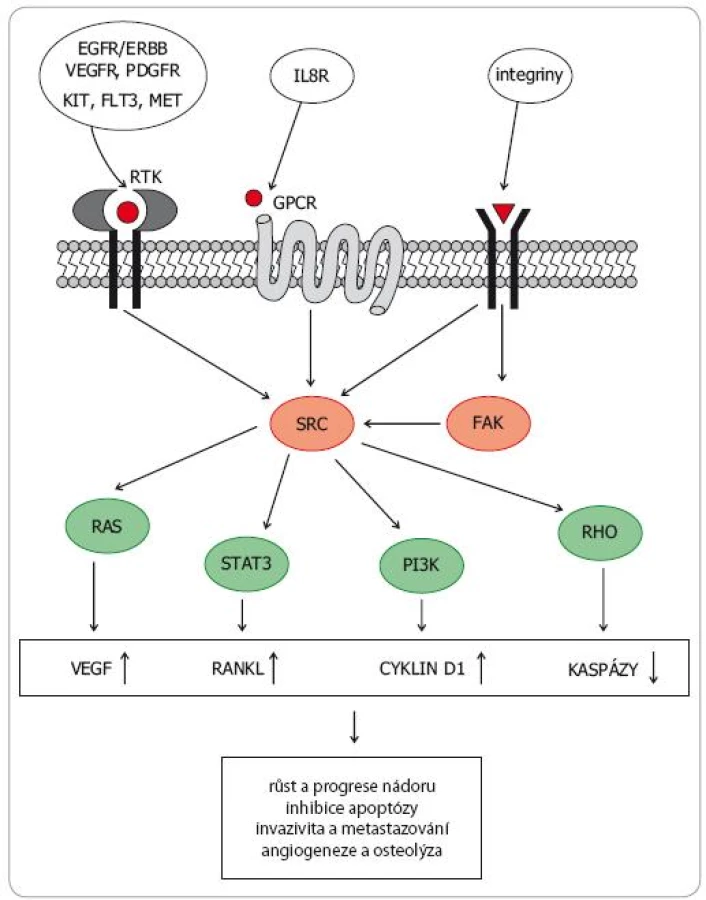
Deregulaci SRC proteinů lze detekovat u širokého spektra nádorových onemocnění. Aberantní SRC signalizace podporuje růst a proliferaci nádorových buněk a zvyšuje práh vůči apoptotickým stimulům, což vede ke ztrátě citlivosti ke konvenční chemoterapii (zejména k paklitaxelu, oxaliplatině a gemcitabinu) a k některým hormonálním přípravkům (např. k tamoxifenu). Změny v uspořádání cytoskeletu vedou k aberacím morfologie nádorové tkáně, ke změnám adhezivních vlastností neoplastických buněk a ke zvýšení jejich lokomočních a invazivních schopností. SRC indukovaná přeměna epiteliálních nádorových buněk v mezenchymální fenotyp (epithelial to mesenchymal transition – EMT) přispívá k vyšší motilitě, invazivitě, schopnosti intravazovat a následně vytvářet vzdálené metastázy [8].
Konstitutivně aktivované SRC kinázy zvyšují totiž formaci invadopodií, snižují expresi E cadherinu, stabilizují beta catenin, působí aktivaci matrixmetaloproteáz (MMP) a aktivátoru plasminogenu (uPA) a vedou k aberantní expresi a/nebo aktivaci některých integrinových receptorů. Tyto pochody významně alterují vzájemné vztahy nádorových buněk a nádorového mikroprostředí (nenádorových buněk, extracelulární matrix). Zvýšená exprese SRC tedy posiluje invazivitu, usnadňuje migraci a znamená horší prognózu onemocnění, jak bylo prokázáno např. u kolorektálního karcinomu [9]. Dysregulace SRC nacházená u většiny nádorů se také významně podílí na zvýšené schopnosti nádorové tkáně novotvořit cévy (vliv na expresi VEGF, FGF a IL8) [6,7].
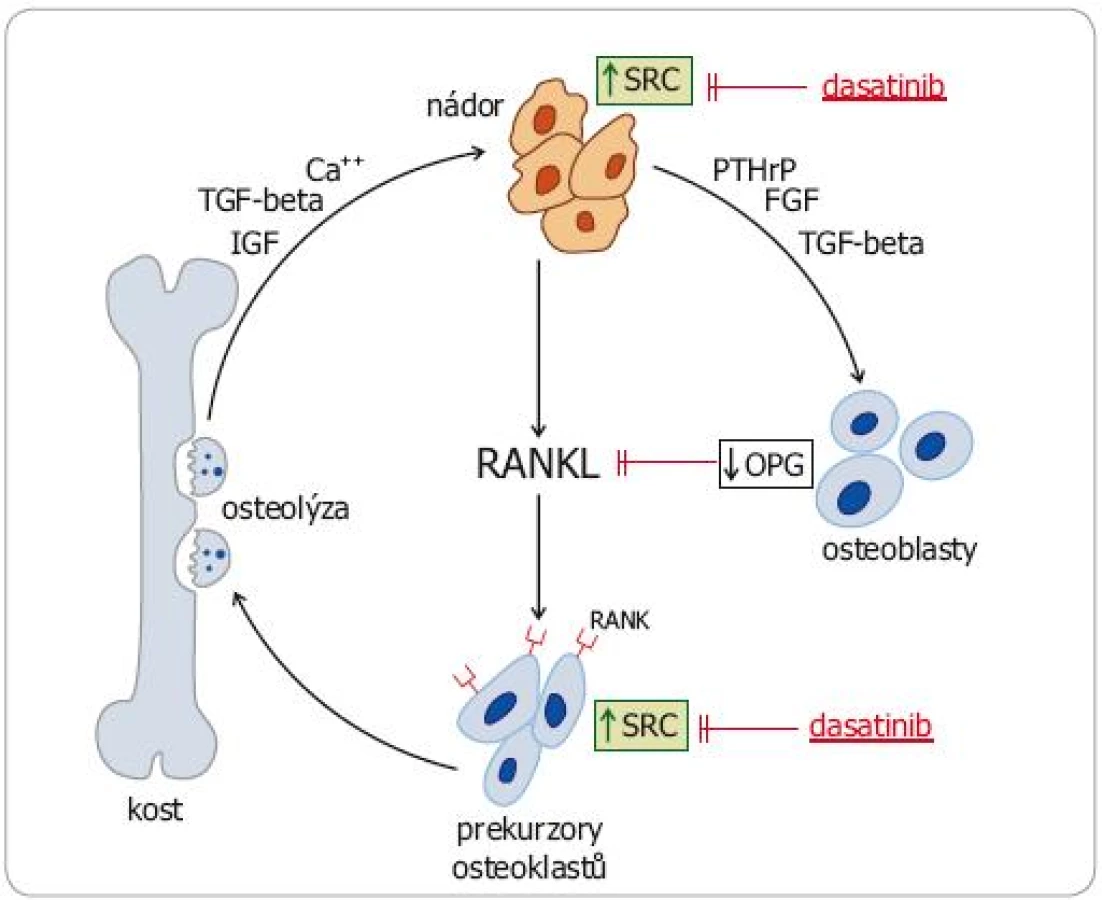
Konstitutivní aktivace SRC kináz však ovlivňuje také klíčové patofyziologické mechanizmy nádorem stimulované osteolýzy (obr. 2). Zvýšená exprese SRC v nádorových buňkách a v osteoblastech působí zvýšený výdej RANKL (receptor activator of nuclear factor kappa B ligand) a snížený výdej osteoprotegerinu (OPG), který se na RANKL váže (a inhibuje ho). Zvýšeně exprimovaný RANKL vazbou na RANK receptor aktivuje signální dráhu NFkappaB, což vede k aktivaci osteoklastů stimulací jejich proliferace a inhibici apoptózy. Také v aktivovaných osteoklastech byla zjištěna zvýšená exprese SRC. Deregulace SRC kináz u nádorů se tudíž podílí jak na vytváření kostních metastáz jako takových, tak na patologickém procesu osteoresorpce a šíření osteolytických ložisek. SRC se tudíž staly potenciálně významným cílem specifické protinádorové léčby [10,11].
Proteiny s tyrozinkinázovou aktivitou jako specifické cíle protinádorové léčby
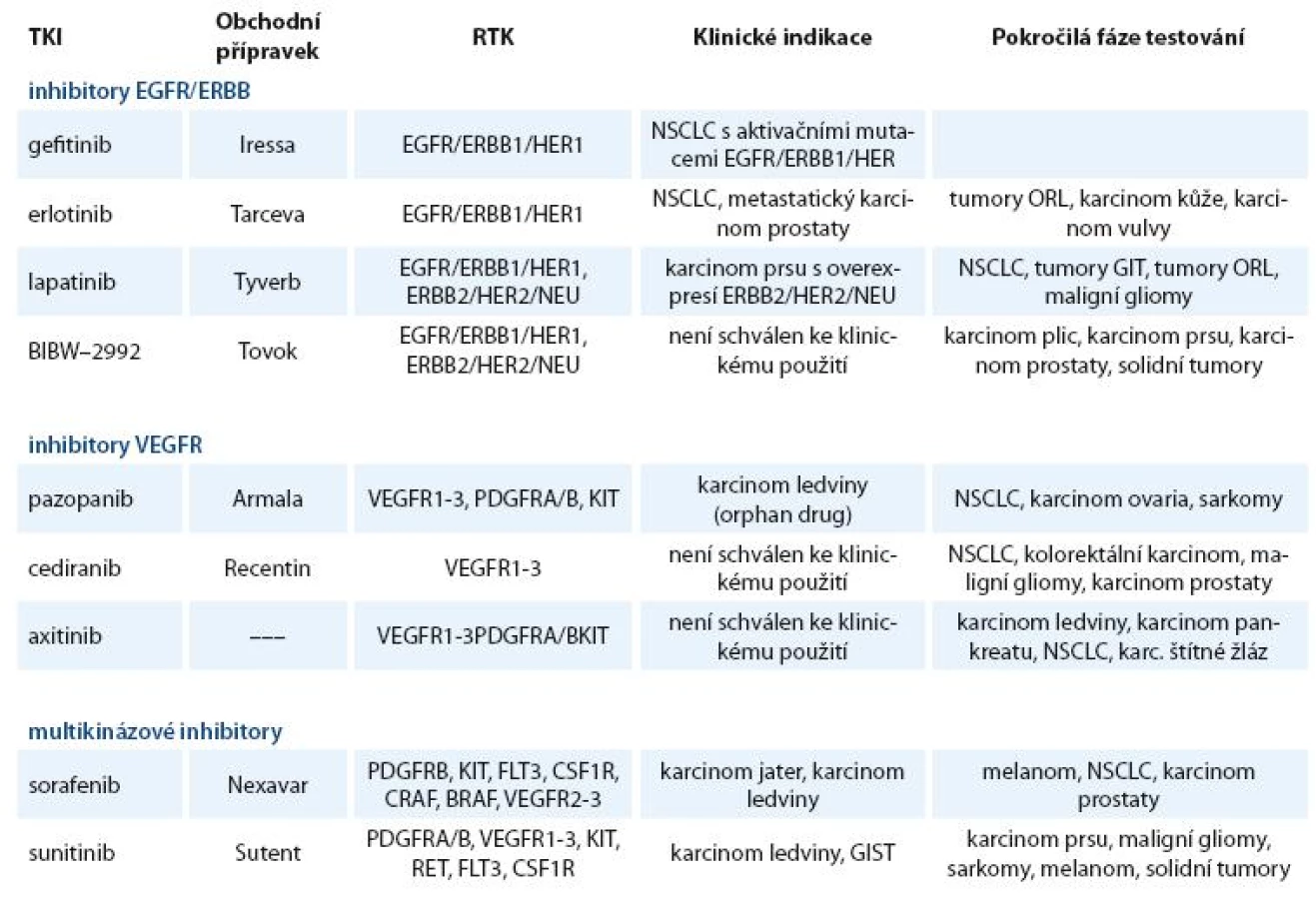
Inhibitory RTK našly své uplatnění v léčběrůzných nádorových onemocnění. V klinické praxi nebo v klinickém zkoušení je větší počet těchto inhibitorů, z nichž některé se již staly pravidelnou součástí léčebných schémat. Používané a nejvíce studované inhibitory receptorových TKI jsou uvedeny v tab. 1.
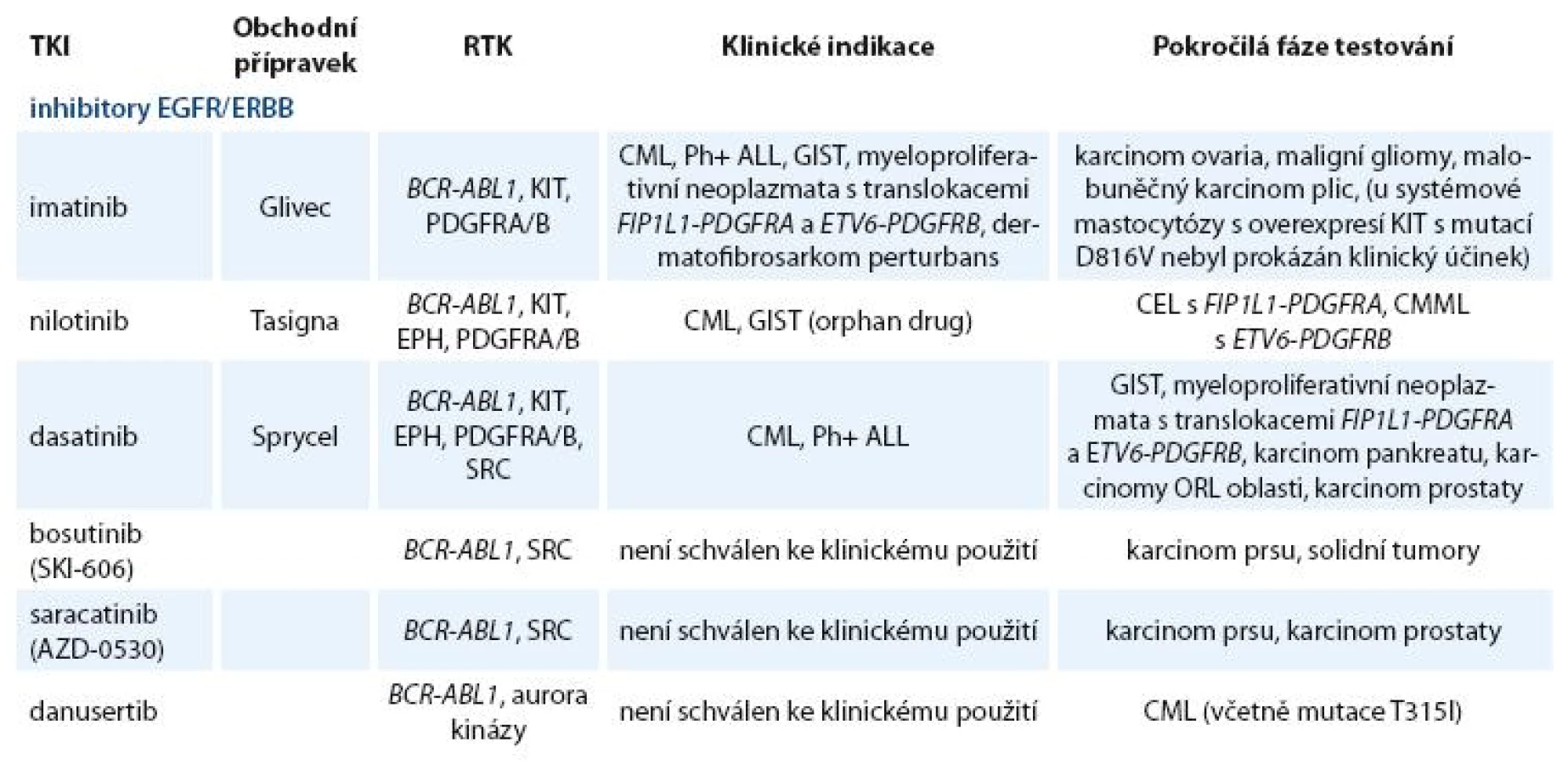
Zavedení prvního inhibitoru PTK BCR ABL1 (imatinibu) do klinické praxe před deseti lety znamenalo počátek éry cílené terapie. Od té doby byla vyvinuta a testována řada TKI nové generace [12]. Kromě „klasických“ tyrozinkinázových inhibitorů jsou vyvíjeny látky blokující serin treoninové kinázy (např. inhibitory aurora kináz) a řada dalších látek, jejichž výčet není předmětem tohoto článku [13–15]. Přehled klinicky používaných a nejvíce studovaných TKI zacílených na BCR ABL1, SRC a jiné tyrozinkinázové proteiny je uveden v tab. 2.
Inhibitory (BCR )ABL1
Imatinib (Glivec), derivát fenylaminopyrimidinu, je nízkomolekulární perorální TKI s inhibičním účinkem vůči tyrozinkinázové doméně fúzního onkogenu BCR ABL1. Kromě toho inhibuje i další tyrozinkinázové proteiny KIT a PDGFRA/B (včetně fúzních genů FIP1L1/PDGFRA a ETV6/PDGFRB). Imatinib (400mg/den) je indikován k primoterapii všech pacientů s CP CML [16]. Kromě pacientů s CML je fúzní gen BCR ABL1 detekován také u části nemocných s akutní lymfoblastickou leukemií (Ph+ ALL). U části pacientů s hypereosinofilním syndromem (HES) byl popsán rekurentní výskyt fúzního genu FIP1L1 PDGFRA, který vzniká kryptickou delecí malé části chromozomu 4q12. Tento typ onemocnění je dle WHO klasifikace 2008 nyní řazen mimo HES do skupiny „myeloidní a lymfoidní neoplazie s eozinofilií a abnormalitami PDGFRA, PDGFRB a FGFR1“. Gen FIP1L1 PDGFRA je stokrát citlivější vůči imatinibu než BCR ABL1. Proto u myeloidních neoplazií s FIP1L1-PDGFRA plně dostačuje léčba imatinibem v dávce 100mg/den (léčba je účinná až u 96% nemocných). U části pacientů s CMML lze detekovat translokaci t(5;12), která dává vznik fúznímu proteinu ETV6 PDGFRB. I tato aberantně aktivovaná tyrozinkináza vykazuje citlivost vůči inhibičním účinkům imatinibu. Zvýšená exprese nemutované receptorové tyrozinkinázy KIT je zase nalézána u části nemocných s gastrointestinálními stromálními tumory (GIST), u nichž je léčba imatinibem také indikována. Naopak nádorové buňky pacientů se systémovou mastocytózou, u nichž lze téměř vždy detekovat aktivační mutaci receptorové tyrozinkinázy KIT (D816V), jsou vůči imatinibu rezistentní [16]. Imatinib se v současnosti testuje u dalších nádorů, jako jsou gliomy, SCLC, karcinom ovaria aj. Při léčbě imatinibem se může postupně vyvinout rezistence, která má různé příčiny. Kromě mutací kinázové domény BCR ABL1 proteinu (viz výše) vzniká rezistence následkem zvýšené exprese glykoproteinu P nebo amplifikace BCR ABL1 genu (tyto příčiny lze do určité míry překonat eskalací dávky imatinibu 600mg/den).
Nilotinib (Tasigna) je syntetický kompetitivní inhibitor BCR ABL1 strukturálně odvozený od imatinibu. Má podobné spektrum účinnosti (kromě BCR ABL1 inhibuje též KIT a PDGFRA/B). Blokáda BCR ABL1 je 20–50krát účinnější než blokáda imatinibem a působí i na některé mutace BCR ABL1 rezistentní k imatinibu (nikoli ovšem na mutaci T315I) [17]. V současné době je nilotinib indikován k léčbě CP CML a AP CML (400mg 2krát denně) u pacientů s rezistencí či intolerancí k imatinibu či dasatinibu.
Duální inhibitory (ABL1 a SRC)
Dasatinib (Sprycel) patří mezi tzv. duální inhibitory, neboť kromě inhibice BCR ABL1 (325krát vyšší inhibiční aktivita v porovnání s imatinibem), KIT a PDGFRA/B inhibuje též nereceptorové tyrozinkinázy rodiny SRC a kinázové domény receptorů pro ephrin (EPH) [18]. Z toho pramení i jeho širší spektrum účinnosti. Dasatinib je účinný u většiny mutovaných forem kinázy BCR ABL1 (s výjimkou mutace T315I). Proniká hematoencefalickou bariérou, což by teoreticky připouštělo jeho účinnost v léčbě leukemické infiltrace CNS u Ph pozitivních lymfoblastických leukemií [19]. V současné době je dasatinib indikován k léčbě CP CML (100mg/den), AP CML a Ph+ ALL (70mg 2krát denně) u pacientů s rezistencí či intolerancí k imatinibu či nilotinibu. Pravidelný záchyt zvýšené exprese SFK u různých typů solidních nádorů vytvořil teoretický předpoklad pro terapeutické použití dasatinibu v této indikaci [20,21]. Preklinický výzkum i pilotní klinické studie tento předpoklad zdá se potvrzují.
U karcinomu prostaty je zatím s použitím SRC inhibitorů nejvíce zkušeností a v současné době probíhá i největší počet klinických studií jak s dasatinibem v monoterapii, tak v kombinaci s docetaxelem [21]. U tohoto nádoru je z rodiny SRC kináz nejvíce exprimována kináza LYN. Zvýšená exprese SFKs (SRC family kinases) koreluje s omezením citlivosti na androgen deprivační léčbu [22]. Inhibice src kináz dasatinibem se totiž ukázala jako efektivní léčebný přístup i po ztrátě hormonální závislosti („castration refractory forms“). Studie fáze I/II kombinující docetaxel a dasatinib u kastračně rezistentního karcinomu prostaty byly prezentovány na kongresu ASCO 2009. Odpověď v PSA antigenu byla zaznamenána u 13/32 (41%) pacientů, klinický benefit (PR + SD) u pacientů hodnotitelných dle RECIST kritérií byl zaznamenán u všech sledovaných pacientů [23]. Dasatinib také signifikantně omezuje incidenci metastáz karcinomu do mízních uzlin, které se považují za prognosticky nepříznivý faktor [24]. Kromě toho inhibice SRC kináz utlumí aktivitu osteoblastů a omezí vznik osetoblastických metastáz [25]. U karcinomu prsu byla prokázána zvýšená exprese a aktivita SFKs [26]. Na experimentálních buněčných liniích karcinomu prsu byl demonstrován inhibiční účinek dasatinibu na růst nádoru. Za povšimnutí stojí experimentální data u ER/PR/HER2 negativních buněčných linií (tzv. „triple negative“) [27], která vedla ke studiím fáze IIdasatinibu u pacientek s „triple negativním“ karcinomem prsu (www.clinicaltrials.gov). U gliomů má dysregulace signalizační kaskády SRC prokazatelnou souvislost s proliferační aktivitou. Bylo prokázáno, že dasatinib účinně blokuje proliferaci a migraci gliomových buněk a indukuje jejich apoptózu [28]. Klinické ověřování účinnosti dasatinibu je teprve v počátcích. U kolorektálního karcinomu se účinnost dasatinibu prověřuje ve větším počtu klinických studií [29]. Studuje se posílení účinnosti cetuximabu nebo kombinace FOLFOX. Dasatinib se dále zkouší u většího počtu solidních nádorů. Na myším modelu bylo prokázáno, že dasatinib potlačil progresi pankreatického duktálního adenokarcinomu a omezil jeho metastazování. Následně bylo potvrzeno, že inhibice SRC potlačuje invazivitu tohoto nádoru [30]. Dasatinib může též obnovit citlivost nádoru na gemcitabin. Očekávají se první výsledky účinnosti dasatinibu u sarkomů [31], u mezoteliomu pleury, NSCL, nádorů ORL oblasti nebo u ovariálního karcinomu.
Ovlivnění metastáz. Kostní metastázy jsou častou komplikací u různých solidních nádorů. Bez ohledu na to, zda se jedná o osteoblastické či osteolytické metastázy, rozhodující pro destrukci kosti je aktivita osteoklastů. Při úloze SFKs v patofyziologickém procesu metastazování je zcela logické, že útlum aktivity SFKs dasatinibem může do tohoto procesu účinně zasáhnout. Významně inhibuje proliferaci osteoklastů na zvířecích modelech [32]. Tento antimetastatický účinek může být potencován současným užíváním bisfosfonátů nebo použitím monoklonální protilátky denosumabu.
Bosutinib je tyrozinkinázový inhibitor 2. generace, s převážným účinkem na ABL1 a SRC. Prověřované indikace jsou CML a metastatický karcinom prsu. Bosutinib stabilizuje E cadherin beta catenin komplex, což přispívá k jeho antiinvazivním schopnostem [33].
Saracatinib (AZD 0530) jako inhibitor SRC se zkouší v podobných indikacích jako dasatinib [34,35]. Brání fosforylaci paxilinu a FAK, a působí tak proti vzniku metastáz [36]. V preklinické studii působil inhibici čtyř z pěti linií nediferencovaného karcinomu štítné žlázy [37], ale klinická účinnost v této indikaci čeká na ověření.
Bafetinib (INO 406) je další z prověřovaných inhibitorů, účinný u CML i v případě různých mutací, s výjimkou mutace T3151 [38]. Zkouší se v léčbě metastatického karcinomu prsu a u maligních gliomů.
Danusertib (PHA 739358) je nízkomolekulární inhibitor BCR ABL1 a aurora kináz, který jeví účinnost u CML včetně forem rezistentních na imatinib, nilotinib a dasatanib. Je totiž účinný i v případech mutace T3151. Zkouší se u rezistentní CML a u pokročilých metastatických forem solidních nádorů [39].
Inhibitory FAK jsou vedle inhibitorů SRC dalším prověřovaným cílem pro protinádorovou léčbu. Za tímto účelem bylo vyvinuto několik nízkomolekulárních inhibitorů (TAC 544, TAE 226 či PF 228). Farmakologická inhibice FAK sice snižovala v preklinických modelech motilitu a invazivitu nádorových linií, nicméně se ukázalo, že tyto inhibitory působí kompenzatorní zvýšení exprese PTK zvané PTK2/PYK2. Proto byl nedávno navržen duální inhibitor FAK/PYK2 s kódovým označením PF 562271, který v preklinických modelech vykazuje výrazný protinádorový účinek [40].
Kombinace TKI s jinými modalitami protinádorové léčby
Experimentální studie naznačují, že aktivace onkogenu SRC není sama o sobě schopna vyvolat maligní transformaci. Protinádorový účinek dasatinibu a dalších inhibitorů SRC je proto testován též v kombinaci s jinými léčivy.
Kombinace s konvenční chemoterapií se ukázala výhodná zejména s některými cytostatiky. Je to docetaxel (při léčbě karcinomu prostaty), dále kapecitabin (u karcinomu prsu) nebo gemcitabin (v léčbě karcinomu pankreatu jiných solidních nádorů). Bylo prověřováno i současné podání dasatinibu s chemoterapeutickou kombinací FOLFOX.
S hormonální léčbou lze dasatinib kombinovat u karcinomu prsu, kde potencuje účinek tamoxifenu. Za efektivní se považuje kombinace s inhibitory receptorových TK. U NSCLC a maligních gliomů se zkouší kombinace s erlotinibem. Dasatinib lze kombinovat i s monoklonálními protilátkami, např. s cetuximabem. Bylo prokázáno, že dasatinib může resenzitizovat nádorové buňky k cetuximabu [41]. Kombinace s denosumabem se nabízí v léčbě kostních metastáz.
Nežádoucí účinky inhibitorů nereceptorových kináz
Léčba je většinou velmi dobře tolerována, nežádoucí účinky jsou zpravidla jen 1. až 2. stupně. Inhibitory BCR ABL1 působí retenci tekutin, projevující se periferními otoky, méně často pleurálním výpotkem. Dále se popisuje mírná neutropenie a trombocytopenie, nevolnost, bolesti ve svalech a bolesti břicha. U inhibitorů SRC kináz jsou nežádoucí účinky podobné, limitující toxicitou je nejčastěji průjem a únava.
Závěr
Zavedení inhibitorů nereceptorových protein tyrozinkináz otevřelo novou možnost v posílení účinnosti protinádorové léčby nejen u hematologických malignit, ale i v terapii solidních nádorů. První zkušenosti s inhibitorem SRC kináz dasatinibem potvrzují jeho účinnost zejména u karcinomu prostaty. Posouzení dalších vhodných indikací, nalezení optimálního dávkování a účelných kombinací s konvenční léčbou si však nepochybně vyžádá ještě další, časově náročné úsilí.
Práce
byla podpořena společností BMS a Výzkumným záměrem MSM
0021620808.
This
work was supported in part by the Czech Ministry of Education under
project No. MSM 0021620808 and by BMS (Bristol-Myers Squibb).
Autoři
deklarují, že v souvislosti s předmětem studie nemají
žádné komerční zájmy.
The
authors declare they have no potential conflicts of interest
concerning drugs, products, or services
used in the study.
Redakční
rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro
publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform
requirements” for biomedical papers.
prof.
MUDr. Pavel Klener, DrSc.
I.
interní klinika - klinika hematologie VFN a 1. LF UK v Praze
U nemocnice 2
128
08 Praha 8
e-mail:
pavel.klener@ruk.cuni.cz
Sources
1. Pierotti MA, Negri T, Tamborini E et al. Targeted therapies: The rare cancer paradigm. Molecular Oncology 2010; 4(1): 19 – 37.
2. Klener P, Klener P Jr. Nová protinádorová léčiva a léčebné strategie v onkologii. Praha: Grada Publishing; , 2010.
3. Rous P. A sarcoma of the fowl transmissible by an agent separable from tumor cells. J Exp Med 1911; 13(4): 397 – 411.
4. Guarino M. Src signaling in cancer invasion. J Cell Physiol 2010; 223(1): 14 – 26.
5. Tsygankov AY, Shore SK. Src: regulation, role in human carcinogenesis and pharmacological inhibitors. Curr Pharm Des 2004; 10(15): 1745 – 1756.
6. Eliceiri BP, Paul R, Schwartzberg PL et al. Selective requirement for src kinase during VEGF‑induced angiogenesis and vascular permeability. Mol Cell 1999; 4(6): 915 – 924.
7. Summy JM, Gallick GE. Src family kinase in tumor progression and metastasis. Cancer Metastasis Rev 2003; 22(4): 337 – 358.
8. Roodman GD. Mechanisms of bone metastasis. New Engl J Med 2004; 350(16): 353 – 259.
9. Aligayer H, Boyd DD, Heiss MM et al. Activation of src in primary colorectal carcinoma: an indicator of poor prognosis. Cancer 2002; 94(2): 344 – 351.
10. Kopetz S, Shah AN, Gallick GE. Src continues aging: Current and future clinical directions. Clin Cancer Res 2007; 13(24): 7232 – 7236.
11. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol 2009; 6(10): 587 – 595.
12. Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev 2009; 28(1 – 2): 35 – 49.
13. Provenzano PP, Keely PJ. The role of focal adhesion kinase in tumor initiation and progression. Cell Adh Mgr 2009; 3(4): 347 – 350.
14. Bruton VG, Frame MC. Src and focal adhesion kinase as therapeutic targets in cancer. Curr Opin Pharmacol 2008; 8(4): 1 – 6.
15. Naomomto HH, Watanabe BX, Sakurama K et al. Focal ahesion kinase as potential target for cancer therapy (Review). Oncol Rep 2009; 22(5): 973 – 979.
16. Klener P, Klamová H. Imatinib – nová perspektiva v léčbě nádorových onemocnění. Čas Lék čes 2004; 143 : 577 – 581.
17. Deremer DL, Ustun C, Natarajan K. Nilotinib: a second ‑ generation tyrosin kinase inhibitor of chronic myelogenous leukemia. Clin Ther 2008; 30(11): 1956 – 1975.
18. Kim LC, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19(3): 415 – 425.
19. Hu Y, Swerdlow S, Duffy TM et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. PNAS 2006 : 103(45): 16870 – 16875.
20. Araujo J, Logothetis C. Dasatinib: A potent SRC inhibitor in clinical development for treatment of solid tumors. Cancer Treat Rew 2010; Epub ehaed of print.
21. Fizazi K. The role of src in prostate cancer. Ann Oncol 2007; 18(11): 1765 – 1773.
22. Nam S, Kim D, Cheng JQ et al. Action of the Src family kinase inhibitor, Dasatinib (BMS ‑ 354825), on human prostate cancer cells. Cancer Res 2005; 65(20): 9185 – 9189.
23. Araujo et al. Dasatinib and docetaxel combination treatment for patients with castration‑resistant progressive prostate cancer: A phase I/ II study (CA180086). J Clin Oncol 2009; 27 (Suppl): 15.
24. Park SI, Zhang J, Phillips KA et al. Targeting Src family kinases inhibits growth and lymph nodes metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res 2008; 68 : 3323 – 3333.
25. Koreckij T, Nguyen H, Brown LG et al. Dasatinib inhibits the growth of prostate cancer in bone and provides additional protection from osteolysis. Brit J Cancer 2009; 101(2): 263 – 268.
26. Finn RS. Targeting src in breast cancer. Ann Oncol 2008; 19(8): 1379 – 1386.
27. Dizdar O, Dede DS, Bulut N et al. Dasatinib may inhibit c ‑ kit in triple negative breast cancer cell lines. Breast Cancer Res Treat 2009; 107(2): 303 – 305.
28. Du J, Bernasconi P, Clauser KR et al. Bead‑based profiling of tyrosine kinase phosphorylation identifies src as a potential target for glioblastoma therapy. Nat Biotechnol 2009; 27(1): 77 – 83.
29. GIG Media Group. Selected clinical trials in colorectal cancer. Clin Colorectal Cancer 2008; 7 : 69.
30. Ito H, Gardber ‑ Thorpe J, Zinner MJ et al. Inhibition of tyrosinkinase src suppress pancreatic cancer invasiveness. Surgery 2003; 134(2): 221 – 226.
31. Shor AC, Keschman EA, Lee FY et al. Dasatinib inhibits migration and invasion in divers human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on Src kinase for survival. Cancer Res 2007; 67(6): 2800 – 2808.
32. Saad F. Src as therapeutic target in men with prostate cancer and bone metastases BJU Int. 2009; 103(4): 434 – 440.
33. Keller G, Schafhausen P, Brummendorf TH. Bosutinib. Recent Results Cancer Rev 2010; 184 : 119 – 127.
34. Dulsat C, Mealy N, Castaner R. Saracatinib. Drugs Fut 2009; 34(2): 106.
35. Tabernero J, Cervantes A, Hoekman K et al. Phase I study of AZD0530, an oral potent inhibitor of src kinase: First demonstration of inhibition of Src activity in human cancers. J Clin Oncol 2007; ASCO Ann Meeting Proc; 23 : 3520.
36. Green TP, Fennell M, Whittaker R. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol 2009; 3 : 248 – 261.
37. Schweppe RE, Kerege AA, French JD et al. Inhibition of Src with AZD0530 reveals the Src ‑ Focal adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endokrin Metab 2009, 94(6): 2199 – 2203.
38. Kantarjian H, Coutre PL, Cortes J et al. Phase 1 study of INNO ‑ 406, a dual Abl/ Lyn kinase inhibitor, in Philadelphia chromosome ‑ positive leukemias after imatinib resistance or intolerance. Cancer 2010 (epub ahead of print).
39. Cohen RB, Jones SF, Aggarwal C et al. A phase I dose‑escalation study of danusertib (PHA ‑ 739358) administered as a 24 - hour infusion with and without granulocyte colony ‑ stimulating factor in a 14 - day cycle in patients with advanced solid tumors. Clin Cancer Res 2009; 15 : 6694 – 6701.
40. Roberts WG, Ung E, Ehalen P et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF ‑ 562, 271. Cancer Res 2008; 68(6): 1935 – 1944.
41. Wheeler DL, Lida M, Kruser TJ et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther 2209; 8(8): 696 – 703.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2010 Issue 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Kožní karcinom z Merkelových buněk
- Diseminovaný karcinóm prsníka u 28- ročného muža
- Malignity žlučových cest
- Cílená léčba bronchioloalveolárního karcinomu inhibitory tyrozinkinázové aktivity EGFR: přehled literatury a kazuistika klinicky promptní a výrazné odpovědi na léčbu erlotinibem