
Pokroky v klinické léčbě zhoubného melanomu: inhibice kinázy B-RAF
Advances in Clinical Treatment of Malignant Melanoma: B-RAF Kinase Inhibition
Malignant melanoma is an aggressive cancer of pigment-producing cells, derivates of the neural crest. Surgical resection is the most effective form of treatment during initial phases of the disease. Advanced stages are usually treated by adjuvant immunotherapy (interferon α) or dacarbazine + multiferon. Response and survival rates are extremely poor. The emerging approach of personalized medicine brings about significant advances in the treatment of melanoma. Apart from administration of imatinib for a small subgroup of melanomas harbouring KIT mutations, the most promising approach is the use of B-RAF kinase inhibitors. The previously tested RAF inhibitors (e. g. sorafenib) did not perform better compared to conventional chemotherapy or immunotherapy. However, the results are much more promising with the recently developed inhibitor PLX4032 (Plexxikon; RG7204, Roche Pharmaceuticals; vemurafenib). This inhibitor targets tumours harbouring B-RAFV600E of B-RAFV600K activating mutations, which are present in 40–70% of malignant melanomas. An absence of the above mentioned activating mutations or parallel presence of activating RAS mutations (e. g. RASG12D) should be used as contraindications. The use of PLX4032 provides better outcome than any of the currently used therapies, including partial or complete response recorded in 81% of patients, and prolonged median survival. Currently, this drug is being tested in phase II and III trials. The incidence of PLX4032-related adverse effects is relatively high; acquired resistance repeatedly occurring within several months of treatment may also represent a significant problem. Combined therapy is probably needed to further increase the complete response rate and to prolong survival. This should either include some of the currently used chemotherapeutics, or alternatively it may employ inhibitors of some of the kinases capable of stimulating the MEK and ERK kinases independently of B-RAF (e. g. COT). Nevertheless, even PLX4032 monotherapy should be viewed as a significant improvement of the current state-of-the-art treatment of malignant melanoma.
Key words:
RAF1 – thyroid cancer – protein kinase inhibitors – antitumor agents – genetic predisposition to disease – neoplastic process – genetic markers – pharmacological biomarkers
Authors:
P. Heneberg
Authors‘ workplace:
3. lékařská fakulta Univerzity Karlovy, Praha
Published in:
Klin Onkol 2011; 24(4): 256-264
Category:
Reviews
Overview
Maligní melanom je vysoce zhoubný novotvar odvozený z pigmentových buněk, derivátů neurální lišty. V počátečních fázích onemocnění dosahuje výborných výsledků chirurgická léčba, pokročilejší stadia jsou v současné době léčena převážně adjuvantní imunoterapií interferonem α, u pacientů klinického stadia IIB-IIIC alternativně ještě kombinací dakarbazin + multiferon. Míra odpovědi a přežití pacientů s metastazujícím melanomem je extrémně nízká a nemoc je považována za prakticky neléčitelnou stávajícími, v České republice užívanými chemoterapeutickými metodami. Tento příspěvek diskutuje nové možnosti terapie melanomu dané rozvojem personalizované medicíny. Vedle aplikace imatinibu pro léčbu melanomů s mutacemi v kináze KIT (kterých je ovšem menšina) jde především o nově vyvíjené inhibitory kinázy B-RAF. S dříve vyvinutými inhibitory RAF kináz, jakými byl např. sorafenib, nebylo dosahováno průkazného zlepšení oproti stávajícím druhům terapie. Naproti tomu nový inhibitor PLX4032 (Plexxikon; RG7204, Roche Pharmaceuticals; vemurafenib) cílený na nejrozšířenější aktivující mutaci B-RAFV600E, která je přítomna ve 40–70 % zhoubných melanomů, vykazuje výrazně lepší účinnost než jakákoliv ze stávajících klinicky využívaných terapií. Léčba inhibitorem PLX4032 působí výlučně na nádory nesoucí mutaci B-RAFV600E, popř. B-RAFV600K. Kontraindikací jeho podávání je absence zmíněné aktivující mutace v kináze B-RAF anebo paralelní přítomnost aktivační mutace v G proteinu RAS, např. RASG12D. V klinické studii fáze I byla zaznamenána částečná či úplná odpověď u 81 % pacientů, prodloužil se i medián přežití. V současné době probíhají klinické studie fáze II a III. Příspěvek diskutuje i poměrně silné vedlejší efekty užívání inhibitoru PLX4032 a prokázaný vznik rezistence na toto léčivo v řádech několika měsíců. Východiskem se do budoucna zdá být kombinovaná terapie s některým ze stávajících chemoterapeutik, popř. s inhibitory kináz schopných stimulace MEK a ERK kináz nezávisle na molekule B-RAF (např. COT), nicméně i samotná monoterapie tímto inhibitorem představuje významné zlepšení oproti současnému stavu.
Klíčová slova:
RAF1 – karcinom štítné žlázy – inhibitory proteinkinázy – antitumorózní látky – genetická predispozice k nemoci – nádorové procesy – genetické markery – farmakologické biomarkery
Východiska
Maligní melanom je vysoce zhoubný novotvar odvozenýz pigmentových buněk, derivátů neurální lišty. Incidence této patologie v posledních desetiletích roste jak u nás, tak ve většině industrializovaných zemí (obr. 1). Část zdánlivého nárůstu má však patrně na svědomí zavedení více či méně plošných screeningů a osvětová činnost. Počet úmrtí následkem zhoubného melanomu zůstává v posledním desetiletí téměř nezměněn. Incidence této patologie v České republice je nejvyšší ze všech států střední a východní Evropy, mezi nejvyšší v tomto regionu se řadí i mortalita následkem zhoubného melanomu. Pro ilustraci, roku 2008 bylo v České republice zjištěno celkem 2 034 případů metastatického melanomu (3,4 % všech zjištěných zhoubných bujení); u 349 pacientů byl melanom uveden jako příčina úmrtí (1,3 % všech úmrtí na zhoubná bujení) [1]. I když incidence a mortalita melanomu jsou mnohem nižší než u některých jiných typů nádorového bujení, společenská významnost léčby melanomu je podtržena skutečností, že tato neoplazie bývá (podobně jako nádory prsu) častá již ve středním věku, což přispívá k násobnému zkrácení doby dožití oproti častěji se vyskytujícím typům nádorů, např. karcinomu plic či prostaty. Prognostika je prozatím založena zejména na hodnocení velikosti tumoru, hloubce invaze, ulceraci a mitotické aktivitě. Dané parametry však neumožňují přesnější predikci tohoto onemocnění [2].
![(A) Časová křivka incidence (●) a mortality (○) zhoubného melanomu kůže (C43) v České republice [54]. (B) Incidence (■) a mortalita (□) zhoubného melanomu v odlišných regionech světa a v jednotlivých zemích střední a východní Evropy [1]. Všechny hodnoty jsou věkově standardizovány, vyjádřeny v počtu případů na 100 000 obyvatel.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/6b2b2503fd9856719a37ef0d37f358e7.jpeg)
Podobně jako u jiných typů zhoubného bujení i transformace normálního melanocytu je podmíněna proběhnutím série mutací či jiných genetických změn navozujících nekontrolovatelnou buněčnou proliferaci a autonomní růst buněk. Mezi geny, u nichž bývá v melanomech navozena změna jejich aktivity či exprese, patří mimo jiné TP53, PTEN, CDKN2A, PTPRD, NRAS a BRAF [3–8]. Poměrně dobře prozkoumanou je přímá úměra mezi incidencí melanomu a faktory vnějšího prostředí, především expozicí UV záření. Ještě donedávna se soudilo, že neexistují genetické alterace, které by podmiňovaly malignitu daného melanomu anebo jeho citlivost na chemoterapii [2]. Dnes jsou však již známy nejen první poměrně spolehlivé prediktory progrese, ale především první genetické mutace podmiňující možnost cílené personalizované léčby terapeutiky, která již úspěšně procházejí prvními fázemi klinických zkoušek [9,10].
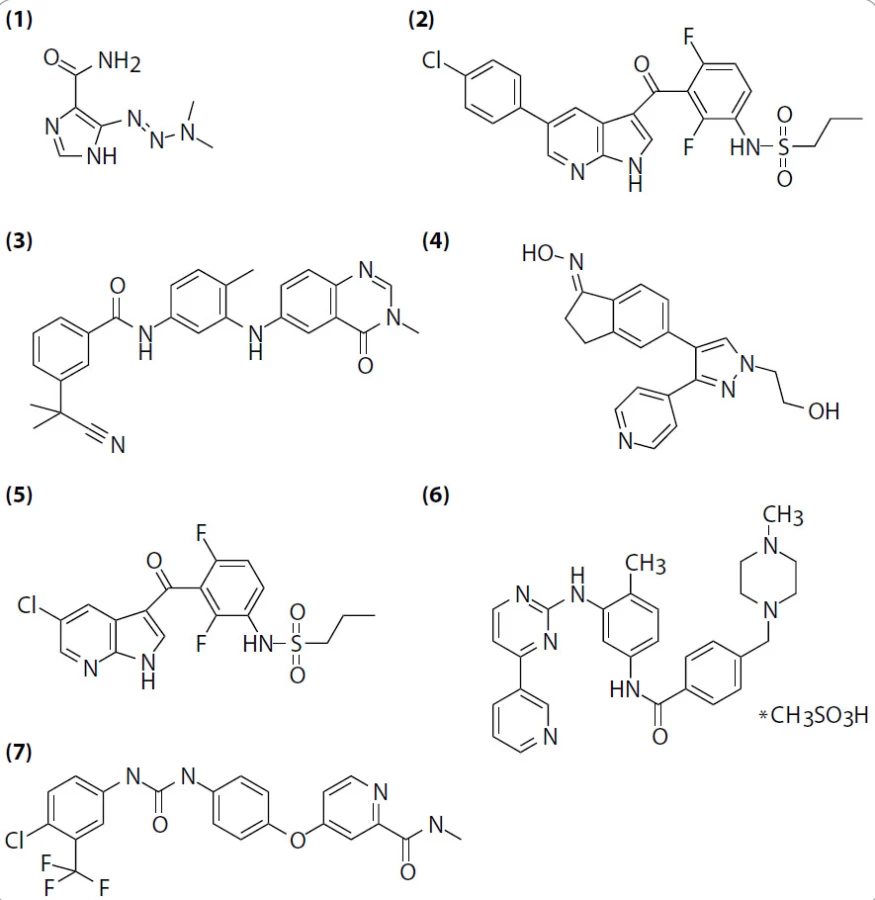
Česká onkologická společnost ČLS JEP na svých webových stránkách publikuje Zásady cytostatické léčby maligních onkologických onemocnění, vytvářené a pravidelně aktualizované pro práci lékařů léčících onkologické diagnózy. V jejich poslední aktualizaci platné k době psaní tohoto příspěvku [11] byla pro léčbu zhoubného melanomu kůže doporučena adjuvantní imunoterapie interferonem α, u pacientů klinického stadia IIB-IIIC pak ještě kombinace dakarbazin ++ multiferon (terapie multiferonem nasazována 3 týdny po zahájení podávání dakarbazinu). Neoadjuvantní chemo- či imunoterapie není u melanomu indikována. Míra odpovědi na léčbu kombinací CVD (cisplatina, vinblastin a dakarbazin) s interferonem α a interleukinem (IL)-2 se pohybuje mezi 18 a 64 %, medián přežití mezi devíti a dvanácti měsíci, dosahováno je 5–7 % kompletních remisí (u dalších chemoterapeutických metod bylo dosahováno vesměs jen 2 % kompletních remisí). Alternativní kombinace cytostatik BOLD (bleomycin, vinkristin, lomustin a dakarbazin) má míru odpovědi ještě nižší (20 %) a medián přežití jen tři měsíce. Kombinace DBD/T (dakarbazin, karmustin, cisplatina, tamoxifen), tzv. Dartmouth regimen, má míru odpovědi 46 %, medián trvání odpovědi je pod sedm měsíců. Míra odpovědi na monoterapii samotným dakarbazinem (obr. 2, (1)) je 14–20 %, fotemustinem 34 %, temozolomidem 17 % ve vybraných případech [12–15].
Současné terapeutické přístupy pro léčbu pacientů s vysoce rizikovými maligními melanomy zůstávají u nás i ve světě stále neefektivní [12,16]. V počátečních fázích onemocnění dosahuje výborných výsledků chirurgická léčba. Ve fázi diseminace se však melanom řadí mezi nejobtížněji léčitelné diagnózy a i za užití výše zmíněných metod je téměř vždy fatální. Nemoc je považována za chemorezistentní i radiorezistentní. Medián přežití nemocných se pohybuje mezi šesti a devíti měsíci, přežití 5 a více let bývá zaznamenáno jen v 1–2 % případů [13].
B-RAF v normálních a transformovaných melanocytech
Melanocyty jsou specializované buňky naší pokožky specializované na produkci pigmentu, který nás následně chrání před negativními účinky UV záření. Jsou rezidentními buňkami pokožky, která ovlivňuje jejich biologické funkce prostřednictvím hned několika parakrinních drah. Parakrinně produkované faktory ovlivňují v melanocytech signální dráhy regulující přestavbu cytoskeletu, metabolizmus a genovou expresi. Mezi klíčové dráhy, jejichž projevy jsou u melanocytů ovlivněny parakrinní signalizací, patří RAS/RAF/MEK//ERK (MAPK) dráha a dráha cyklického AMP (cAMP).
V tomto příspěvku se zaměříme především na MAPK dráhu. První z jejích členů, RAS, je malým, na membránu vázaným G proteinem. Funguje jako GTPáza, je tedy aktivován výměnou GDP za GTP, která je v klasickém modelu zprostředkována proteinem Sos, nejčastěji po aktivaci některého z membránových receptorů mimobuněčným podnětem (typicky růstovým hormonem, interleukinem atp.). Aktivovaný RAS spouští kaskádu proteinových serin/threoninových kináz (RAF, MEK), která vyúsťuje v aktivaci MAP kináz, např. ERK1/2 či Elk. Tyto se po fosforylaci translokují do jádra buňky a stimulují genovou transkripci, typicky prostřednictvím transkripčních faktorů Fos a Jun.
Pojmy RAS a RAF ve skutečnosti označují celou rodinu proteinů. Dále se budeme zabývat rodinou RAF, která u savců (a tedy i u lidí) sestává z proteinů A-RAF, B-RAF a C-RAF. Všechny tři jsou schopny již zmíněné hlavní funkce – aktivace kinázy MEK –, ale navzájem se liší jednak v tkáňové distribuci, jednak v některých detailech svého působení, např. ve schopnosti translokace k cytoplazmatické membráně, schopnosti konformačních změn a vazby některých interakčních partnerů. Tyto tři proteiny jsou schopny tvořit diméry, ať už homodiméry (dva proteiny stejného typu), anebo heterodiméry (po jednom proteinu dvou různých typů, typicky např. jeden B-RAF a jeden C-RAF).
Ve velkém množství lidských zhoubných melanomů (přibližně ve 40–70 %) je přítomen mutantní B-RAF. Ten nadto bývá přítomen v 7–8 % všech neoplazií. Mezi další typy nádorů s vysokou frekvencí mutací kinázy B-RAF patří papilární i anaplastický karcinom štítné žlázy, kde bývají detekovány ve 40–70 % případů. Naopak zbývající dvě molekuly rodiny RAF mutovány nejsou, patrně kvůli jejich odlišným enzymatickým vlastnostem [9,17]. Jednoznačně nejčastější mutací molekuly B-RAF je substituce V600E (1799 T > A), která má za následek ~138násobné zvýšení aktivity kinázy B-RAF oproti nemutované molekule a která způsobuje konstitutivní aktivaci MAPK signální dráhy, detekovatelné například na úrovni molekul MEK a ERK [18–20]. Zde je na místě poznamenat, že mutace na obdobné pozici v molekule C-RAF má diametrálně odlišný účinek. Enzymatická aktivita kinázy C-RAF se po zavedení této mutace téměř nemění, což mimo jiné vysvětluje odlišný způsob regulace těchto dvou blízce příbuzných molekul [17]. Naopak pokud zavedeme do melanocytů molekulu B-RAFV600E, dochází k jejich okamžité transformaci asociované se zlepšeným přežíváním transformovaných buněk a zvýšenou rychlostí proliferace [21–25]. Další mutace v molekule B-RAF s podobnými účinky jsou známy (L597V, G464V, G469A), ale vyskytují se jen s velmi nízkou frekvencí [18].
PLX4032 – první úspěšný inhibitor zhoubného metastazujícího melanomu
V srpnu 2010 byly publikovány velmi zajímavé výsledky multicentrické klinické studie fáze I [9], ve které byly pacientům s melanomem podávány různé dávky inhibitoru PLX4032 (Plexxikon; RG7204, Roche Pharmaceuticals; vemurafenib; obr. 2, (2)). Tento inhibitor je zaměřen na molekuly B-RAFV600E, s nižší specifitou inhibuje i ostatní členy rodiny RAF a několik dalších kináz (tab. 1), ne však samotný nezmutovaný B-RAF, což je důležité pro selektivní léčbu nádorů se somatickými mutacemi této kinázy. Oproti jiným RAF inhibitorům, např. AZ-628 (obr. 2, (3)) či GDC-0879 (obr. 2, (4)), způsobují PLX4032 i jeho analog PLX4720 (obr. 2, (5)) tvorbu vodíkové vazby mezi NH skupinou kyseliny asparagové B-RAFD594 a dusíkem ze -SO2NH2 skupiny inhibitoru a vysunutí αC helixu molekuly B-RAFV600E [10]. Preklinické studie ukázaly, že PLX4032E a PLX4720 jsou schopny inhibice mutantní kinázy v nízkých nanomolárních koncentracích a že v souladu s předpoklady vedou k potlačení signalizace v MAP kinázové dráze. V řádově vyšších koncentracích je tento inhibitor schopen blokovat proliferaci buněk s mutantní B-RAF kinázou [26,27]. Orálně podávaný PLX4720 inhibuje růst a ve vyšších dávkách způsobuje regresi xenotransplantátů lidských melanomů do imunodeficientních myší (tab. 2). Žádné z výše popsaných efektů nebyly pozorovány u buněk exprimujících pouze nemutovanou kinázu B-RAF.


Ve zmíněné studii [9] byl inhibitor podáván původně v krystalické formě, 200 mg p. o. denně. Oproti předpokladům se ukázala neúčinná, detailnější analýza výsledků ale poukázala na skutečnost, že v krevním séru byly hladiny inhibitoru jen velmi nízké a důvodem nedostatečné účinnosti nebyla jeho malá aktivita, ale nízká prostupnost inhibitoru do krevního řečiště. Nábor pacientů byl proto zastaven a byla vyvinuta nová forma inhibitoru, mikroprecipitovaný prášek podávaný opět perorálně ve formě kapsulí či tablet.
Do studie byli zahrnuti pacienti nad 18 let s pevnými nádory histologicky neodpovídající na konvenční formy terapie. ECOG skóre (Eastern Cooperative Oncology Group score) všech pacientů bylo 0 nebo 1, tzn. pacienti byli schopni nejméně lehké práce, očekávaná doba přežití u nich byla tři nebo více měsíců, byli bez zvětšujících se či nestabilních metastáz v mozku, s dobrou funkcí jater a ledvin a s normálním hematologickým obrazem. Naprostá většina kohorty pacientů, u nichž byl zkoumán vliv zvyšující se dávky léku (89 %, 49 z 55), měla diagnostikován metastazující melanom, tři ze zbývajících šesti pacientů měli papilární karcinom štítné žlázy s mutací B-RAFV600E. Screening na tuto mutaci nebyl pro zahrnutí do studie požadován, ale mutace byla v průběhu studie identifikována u většiny zkoumaných subjektů. Pacientům byl podáván inhibitor v koncentracích 160–1 120 mg 2× denně. Dávka 160 mg 2× denně neměla žádný vliv na růst nádoru. Odpověď na podávání léku se zvyšující se dávkou rostla: při 240 mg 2× denně byl lék účinný u jednoho z 16 pacientů s B-RAFV600E mutací, při 320–360 mg 2× denně u dvou ze čtyř pacientů, při 720 mg 2× denně u čtyř ze šesti pacientů. Při dávce 1 120 mg inhibitoru 2× denně byla pozitivní odpověď zaznamenána u čtyř z pěti testovaných pacientů. Průměrná doba odpovědi byla dva až > 18 měsíců. Částečná či kompletní odpověď byla zaznamenána i u tří pacientů s papilárním karcinomem štítné žlázy, kteří byli do studie také zahrnuti. Naopak žádná odpověď nebyla zaznamenána u pěti pacientů s metastazujícím melanomem, jejichž nádory nenesly mutaci B-RAFV600E a kteří obdrželi ≥ 240 mg 2× denně. Při dávkách do 720 mg 2× denně nebyly u pacientů pozorovány žádné nežádoucí vedlejší efekty. Dávka 960 mg 2× denně byla pozorována jako tolerovaná u skupiny prvních šesti pacientů v části studie zkoumající vliv zvyšujících se dávek léčiva. Vyšší dávky, 1 120 mg 2× denně, způsobovaly u většiny pacientů vyrážku (stupně 3), u některých únavu (stupně 3) a artralgii (rovněž až stupně 3) [9].
Na základě výše popsaných výsledků byla dávka 960 mg 2× denně (AUC0-24 1741 µM × h při poločasu 50 hod) použita pro rozšíření studie. Do tohoto rozšíření byli pro lepší vypovídací schopnost zahrnuti jen pacienti s mutací B-RAFV600E, na které je lék cílen. Tato dávka léčiva byla během 15 dnů schopna velmi výrazně snížit hladinu cyklinu D1, proteinu Ki-67 a hladinu fosforylace ERK kinázy. Zajímavé je, že ERK v buněčném jádře byl působením inhibitoru defosforylován rychleji než cytoplazmatický ERK. Nicméně změny ve fosforylaci cytoplazmatického ERK kinázy velmi dobře korelovaly s odpovědí na léčbu, zatímco změny ve fosforylaci ERK kinázy v buněčném jádře s odpovědí na léčbu nekorelovaly vůbec [10]. U 81 % pacientů (26 z 32) byla zaznamenána odpověď na léčbu – u dvou z nich úplná, u 24 částečná. Medián přežití ještě nebylo možno v době psaní práce stanovit, neboť 16 ze 32 pacientů přežívalo po více než 18 měsíců (bude se pohybovat nad sedm měsíců). Během prodlouženého podávání léčiva se ukázaly poměrně silné vedlejší efekty i u použité dávky 960 mg 2× denně, tedy nejen u vyššího dávkování, jak naznačoval předchozí experiment. 41 % pacientů (13 z 32) si vyžádalo snížení dávky na 720, řidčeji až na 600 nebo 480 mg 2× denně. Nejčastějšími vedlejšími efekty druhého či třetího stupně byly opět artralgie, vyrážka, nevolnost, fotosenzitivita, únava, spinocelulární karcinomy, svědění a dysestezie. Spinocelulární karcinomy byly zaznamenány u 31 % pacientů, kterým byla podávána dávka 960 mg inhibitoru 2× denně. Medián doby, která uplynula od začátku podávání léčiva do objevení se spinocelulárních karcinomů, byl osm týdnů [9]. Tyto změny nebyly pozorovány v rámci preklinických studií na potkanech a psech, u kterých koncentrace inhibitoru dosahovaly podobných či vyšších hodnot než u lidí – AUC0-24 2 600 µM × h (potkani) a AUC0-24 820 µM × h (psi), přičemž podáváno jim bylo až 1 000 mg/ kg/den [28].
Inhibitor PLX4032 byl předmětem již sedmi studií zahrnutých v databázi Clinicaltrials.gov. Některé z nich ještě probíhají, část již byla ukončena. Proběhly či probíhají hned čtyři studie fáze I. V první z nich (NCT00405587) byly u 75 pacientů s kolorektálním karcinomem či melanomem zkoumány vedlejší efekty a farmakodynamická aktivita v biopsiích předmětných nádorů kvantifikací markeru buněčné proliferace Ki67 a kvantifikací hladiny fosforylace kinázy ERK. Druhá ze studií fáze I (NCT01107418) se zabývá podáváním inhibitoru PLX4032 pacientům s melanomem, kteří již prodělali předchozí léčbu klasickými metodami. Tato studie byla zahájena teprve v květnu 2010, bylo již nabráno cílových 55 pacientů. Vyhodnocována bude farmakokinetika (koncentrace inhibitoru v plazmě), indicence nežádoucích vedlejších efektů, budou provedena běžná laboratorní vyšetření na bezpečnost a tolerabilitu, sledována bude míra odpovědi a přežití. V třetí a čtvrté ze studií fáze I je zkoumána farmakokinetika a metabolizmus tohoto léčiva u pacientů s metastazujícím melanomem v monoterapii (NCT01164891) a v kombinaci s jinými léčivy (NCT01001299). V první ze zmíněných studií bude léčivo označeno radioaktivním uhlíkem 14C a budou zjišťovány mechanizmy eliminace léčiva, samozřejmě vedlejší účinky, laboratorní parametry a ECG, míra odpovědi a velikost nádorů pomocí CT, MRI, scintigrafie kostí a délka přežití. Ve druhé ze zmíněných studií je zjišťován vliv inhibitoru PLX4032 na farmakokinetiku substrátů CYP450 v kombinaci kofein, warfarin + vitamin K, omeprazol, dextrometorfan a midazolam. Mezi výstupy budou patřit hodnoty koncentrace PLX4032 a dalších výše uvedených pěti sloučenin v krvi včetně jejich metabolitů, míra odpovědi, délka trvání odpovědi, čas do začátku odpovědi, přežití, přežití bez progrese a běžné laboratorní hodnoty. Od roku 2009 probíhá v USA a Austrálii studie fáze II (NCT00949702) podávající inhibitor PLX4032 pacientům s metastazujícím melanomem. Jde o open-label studii za účelem potvrzení efektivity léčby. Hlavními výstupy je míra odpovědi (kritéria RECIST), trvání odpovědi, přežití bez progrese, míra zlepšení fyzického stavu, přežití, vedlejší efekty, laboratorní hodnoty, farmakokinetika, kvalita života vyjádřená pomocí FACT-M a ověření metodiky zvané CoDx (Hoffmann-La Roche) detekující mutaci B-RAFV600E. Běží již i studie fáze III (NCT01006980) zaměřující se na monoterapii inhibitorem PLX4032 u pacientů s metastatickým melanomem bez předchozí léčby (porovnání s monoterapií dakarbazinem). Léčba prvního pacienta v této studii byla zahájena v lednu 2010, studie samozřejmě ještě stále pokračuje. Jde bohužel o open-label studii; výstupními daty bude celkové přežití vyhodnocované dva roky po zahájení léčby, vedlejšími výstupy pak přežití bez progrese, míra odpovědi, trvání odpovědi, zaznamenány samozřejmě budou vedlejší efekty. Bude zjišťována maximální dosažená koncentrace PLX4032 v plazmě a farmakokinetika tohoto léčiva. Podmínkou zařazení do studie je kromě jiného pozitivita na mutaci B-RAFV600E – jde tedy o klasický příklad personalizované medicíny vyžadující předvýběr pacientů, u nichž je očekávána dobrá odpověď na podávané léčivo, podobně jako by v ČR bylo žádoucí specifikovat uživatelské skupiny pro podávání již schválených inhibitorů kinázy EGFR [29]. Na základě předběžných výsledků zmíněné studie fáze III FDA schválila, že PLX4032 může již být podáván v režimu „expanded access“ (viz NCT01248936), tedy že je již dostupný i pacientům, kteří se sice nekvalifikují pro studii, ale mohli by z léčby benefitovat. PLX4032 je prvním inhibitorem nádorového bujení, který se Plexxiconu podařilo protlačit až do třetí fáze klinických zkoušek (ve spolupráci s Roche Pharmaceuticals). Velmi impresivní skupina dalších je ale již ve stadiu preklického testování a prvních dvou fázích klinického testování.
Kontraindikace PLX4032
I když to autoři ve své klíčové studii [9] nezmiňují, podávání inhibitoru PLX4032 je velmi problematické u pacientů nesoucích nejen mutaci B-RAFV600E, ale i aktivační mutaci některého z genů rodiny RAS [28,30–32]. Ač PLX4032 již prošel díky slibným výsledkům až do fáze III klinických zkoušek v léčbě maligního melanomu, vznikl opakovaně problém: přibližně u čtvrtiny pacientů se vyvinuly hyperproliferativní léze v kůži, keratoacanthomy, u některých i spinocelulární karcinomy [9]. Tvorba kožních lézí je známa i v případě podávání jiných RAF inhibitorů (sorafenib, XL281, GSK118436) [31–35]. Ve všech případech ale zmíněné patologie vymizely, když bylo podávání inhibitoru ukončeno. Inhibitor PLX4032 totiž podobně jako jiné inhibitory aktivity kinázy B-RAF efektivně potlačuje její enzymatickou činnost. V nádorech nesoucích zmíněnou onkogenní mutaci skutečně dochází k naprostému utlumení signalizace přes její signální dráhu. Pokud ale cílová buňka nese i mutaci v G proteinu RAS (k čemuž dochází velmi často), inhibitor PLX4032 na potlačení růstu nádoru nestačí, a naopak jej stimuluje. Sice potlačuje kinázovou aktivitu svého substrátu, ale vedlejším efektem je stimulace membránové lokalizace tohoto substrátu, jeho homo- či heterodimerizace s dalšími molekulami kinázy B-RAF nebo blízce příbuzné C-RAF a následná transaktivace dimérů. Důvodem jsou změny konformace substrátu po navázání inhibitoru, které jsou nezávislé na potlačení samotné kinázové aktivity. Ke vzniku nádoru tedy vedou potenciálně hned tři cesty (obr. 3). Samotný PLX4032 je schopen se vázat i na C-RAF, za podmínek in vitro byla IC50 této vazby jen o dvě třetiny vyšší oproti vazbě na B-RAFV600E, což naznačuje poměrně značný potenciál k paralelní inhibici molekuly C-RAF. Ve většině buněk k tomu skutečně dochází, ale v buňkách s hyperaktivní kinázou RAS bylo experimentálně prokázáno, že při používaném dávkování není PLX4032 signalizaci přes C-RAF v těchto buňkách schopen zabránit. Možným mechanizmem, kterým by se této nežádoucí aktivitě v buňkách s hyperaktivním proteinem RAS dalo zamezit, je paralelní inhibice kinázy C-RAF pomocí PKA a/nebo cAMP. Pokud by bylo možno roli PKA nebo samotného cAMP v cílových buňkách posílit, kombinovanou terapií by bylo možné dosáhnout vymizení nežádoucích efektů pozorovaných při monoterapii inhibitorem PLX4032 u pacientů s nádory nesoucími mutace v genech pro B-RAF i RAS protein [36].
![Schematické znázornění vedlejších účinků inhibitoru PLX4032 v buňkách s mutacemi v genech RAS a BRAF. Podle [30,39], upraveno.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/5fae188321ba94d8e8d0b31511b77d19.jpeg)
V pozorovaných patologiích indukovaných podáváním PLX4032 mutantní hyperaktivní RAS způsobuje aktivaci RAF kináz, jejich vazbu na membránu a dimerizaci, a tím stimuluje buňky k růstu. Protože PLX4032 stimuluje vznik těchto dimérů, nechtěně podporuje také vznik benigních i maligních nádorů [30,37]. Mutace RAS kinázy se nevyskytují jen v benigních keratoacanthomech, ale často také způsobují nádory plic, které jsou ve svých raných stadiích (odpovídajících délce trvání dosud provedených studií PLX4032) mnohem obtížněji detekovatelné a zároveň mnohem nebezpečnější.
Vznik rezistence na PLX4032 byl recentně popsán i prostřednictvím mechanizmu zahrnujícího receptorovou tyrozinovou kinázu PDGFRβ. Zvýšená exprese této kinázy vede ke vzniku rezistence na PLX4032 nezávisle na kináze MEK a při absenci rezistenci způsobujících mutací v molekule B-RAF. Tato nová data byla ale získána sekvencováním vzorků z pouhých 12 pacientů, z nichž pět získalo rezistenci právě prostřednictvím overexprese PDGFRβ [38]. Podobně byla na jiné kohortě pacientů vystavených PLX4032 zjištěna zvýšená exprese inzulinového receptoru IGF-1R a s ní související zvýšená fosforylace Akt kinázy při absenci mutací v molekulách B-RAF, N-RAS a PTEN [39]. Pro posouzení klinické relevance vztahu exprese a aktivity receptorových kináz PDGFRβ a IGF-1R k podávání inhibitoru PLX4032 bude zapotřebí rozsáhlejších kohort pacientů umožňujících jednoznačnější závěry s dopady na klinickou praxi.
Je možné, že RAF inhibitory využívané v klinickém a preklinickém výzkumu způsobují i jiné změny buněčné signalizace. Inhibitor PLX4032 má schopnost modulovat aktivitu poměrně široké plejády molekul s IC50 v podobném rozmezí jako pro B-RAFV600E. Patří mezi ně kinázy SRMS, ACK1, C-RAF, MAP4K5 (KHS1) a FGR (tab. 2). MAPK dráha je nadto regulována i jinými enzymy a adaptorovými proteiny, jmenovitě fosfatázami [40–42] či adaptorovými proteiny, jakými je 14-3-3 [43].
Jedním z novátorských přístupů je recentní studie autorů ze známého Institutu Broadových [44], kdy autoři exprimovali 600 kináz a kinázám příbuzných otevřených čtecích rámců a testovali jejich vliv na rezistenci buněk nesoucích mutaci B-RAFV600E k podávanému RAF inhibitoru. Zjistili, že pomocí genu MAP3K8 kódujícího proteinový produkt COT/Tpl2 jsou schopni tuto rezistenci ovlivnit. Protein COT aktivuje kinázu ERK prostřednictvím mechanizmu závislého na kináze MEK, ale nezávislého na signalizaci přes RAF. Aby toho nebylo málo, exprese proteinu COT je asociována s de novo rezistencí jak buněčných linií s B-RAFV600E, tak i melanomů z pacientů po relapsu onemocnění po léčbě inhibitory kináz MEK nebo RAF [44]. Kombinovaná terapie inhibitory proteinů COT a RAF je tak do budoucna jednou z možností, jak dále prodloužit přežívání pacientů se zhoubným melanomem.
Závěr
Personalizovaná medicína se stává pro vývoj nových protinádorových léčiv v posledních letech typickou (obr. 4). Pouze molekulárně-biologická charakterizace maligních onemocnění totiž často umožňuje rozlišit histologicky identické malignity vzniklé odlišnými patogenetickými procesy. Do personalizované terapie melanomu v současnosti neřadíme pouze inhibici kinázy B-RAF diskutovanou v tomto článku, ale i inhibitory kinázy KIT. Imatinib (obr. 2, (6)) je totiž schopen způsobit regresi 33 % melanomů nesoucích mutace v kináze KIT [45]. Je ale nutno podotknout, že oproti BRAF mutacím jsou mutace v kináze KIT v melanomech poměrně vzácné.
![Bibliometrická analýza dokumentující rychlý vzestup zájmu o témata diskutovaná v tomto příspěvku, ať již jde o samotnou inhibici aktivity B-RAF kinázy, či obecně o personalizovanou medicínu jako takovou. Analýza byla provedena dle protokolu popsaného detailněji dříve [55]. Krátce, v databázové platformě WOS byl vyhledán počet publikací o (A) inhibitoru PLX4032, (B) mutaci B-RAFV600E, (C) RAF kinázách ve vztahu ke vzniku a progresi nádorového bujení, (D) melanomech a (E) personalizované medicíně. Použité vyhledávací fráze a celkový počet nalezených záznamů byl následující: (A) Topic=(plx4032); n = 29 záznamů; (B) Topic=(V600E); n = 586 záznamů; (C) Title=(raf) AND Title=(cancer* OR tumor* OR tumour*); n = 339 záznamů; (D) Title=(melanoma); n = 45,192 záznamů; (E) Title=(personalized medicine); n = 606 záznamů. Vyhledávání reprezentuje pouze záznamy zahrnuté v databázové platformě WOS a obsahující v názvu (C–E), popř. i v abstraktu či klíčových slovech (A–B) předmětnou frázi. Vyhledávání bylo provedeno 9. ledna 2011.](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d3a568d8a2b7a93e4eadae184ad73286.jpeg)
Výše popsané výsledky s inhibitorem PLX4032 jsou výrazně slibnější než předchozí testy s jinými RAF inhibitory. Např. sorafenib (Nexavar, Bayer; obr. 2, (7)), inhibitor kinázy B-RAF (jak nemutované, tak té se substitucí B-RAFV600E) a zároveň inhibitor příbuzné kinázy C-RAF, jednak postrádá selektivitu pro B-RAFV600E mutaci a jednak v klinických zkouškách jak v monoterapii, tak v kombinaci s chemoterapeutiky nevedl k průkaznému zlepšení oproti stávajícím druhům terapie [46–49]. Důvody nejsou příliš zřejmé, patrně jde o vedlejší účinky inhibice nemutované kinázy B-RAF a C-RAF znemožňující zvýšení koncentrace sorafenibu na hladinu dostatečnou k potlačení růstu nádoru.
Do budoucna bude nutno vyřešit problém vzniku rezistence na PLX4032, který Flaherty et al [9] zaznamenali v řádech měsíců po zahájení podávání inhibitoru u pacientů, kteří k němu byli zpočátku senzitivní. Tento problém je nutno vzít v potaz především v perspektivě budoucího prodloužení doby přežívání pacientů s melanomem. Velmi recentně, 25. března 2011, schválil americký FDA klinické použití protilátky ipilimumab (Bristol-Myers Squibb) cílené proti inhibiční molekule CTLA-4 v terapii melanomu [50]. Tato terapie ovlivňuje imunitní systém mimo jiné i u pacientů rezistentních k B-RAF inhibitorům, a dává tak do budoucna velkou naději pro další prodloužení přežití pacientů s maligním melanomem.
Inhibitor PLX4032 nepůsobí na nádory nesoucí nemutovaný B-RAF [26,51]. V současné době představuje nejefektivnější metodu léčby melanomu u pacientů s aktivační mutací B-RAFV600E (viz výše), popř. i u pacientů s obdobnou mutací B-RAFV600K[52]. Samotná mutace B-RAFV600E způsobuje pouze vznik benigních névů [53], do budoucna bude nejen proto zcela jistě žádnoucí zaměřit se na vývoj kombinované terapie zahrnující více signálních drah typických pro vznik zhoubného melanomu, případně kombinovat inhibitor PLX4032 s imunoterapií či chemoterapií.
Tato práce byla podpořena výzkumným záměrem 3. lékařské fakulty Univerzity Karlovy v Praze MSM0021620814.
This research was supported by a research goal of the Third Faculty of Medicine, Charles University in Prague No. MSM0021620814.
Autor deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
The author declares he has no potential conflicts of interest concerning draugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
RNDr. Petr Heneberg, Ph.D.
Univerzita Karlova v Praze
Ruská 87
100 00 Praha 10
e-mail: Petr.Heneberg@lf3.cuni.cz
Obdrženo/Submitted: 20. 1. 2011
Přijato/Accepted: 22. 2. 2011
Sources
1. Ferlay J, Shin HR, Bray F, Forman D et al. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 10. International Agency for Research on Cancer. France: Lyon 2010. [aktualizováno 9. ledna 2011; citováno 9. ledna 2011]. Available from: http://globocan.iarc.fr.
2. Brychtová S, Fiurášková M, Sedláková E et al. Molekulární změny ovlivňující progresi maligního melanomu. Klin Onkol 2005; 18(4): 145–148.
3. Pavey S, Johansson P, Packer L et al. Microarray expression profiling in melanoma reveals a BRAF mutation signature. Oncogene 2004; 23(23): 4060–4067.
4. Solomon DA, Kim JS, Cronin JC et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res 2008; 68(24): 10300–10306.
5. Hussussian CJ, Struewing JP, Goldstein AM et al. Germline p16 mutations in familial melanoma. Nat Genet 1994; 8(1): 15–21.
6. Curtin JA, Fridlyand J, Kageshita T et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005; 353(20): 2135–2147.
7. Guldberg P, Thor Straten P, Birck A et al. Disruption of the MMAC1/PTEN gene by deletion or mutation is a frequent event in malignant melanoma. Cancer Res 1997; 57(17): 3660–3663.
8. Stretch JR, Gatter KC, Ralfkiaer E et al. Expression of mutant p53 in melanoma. Cancer Res 1991; 51(21): 5976–5979.
9. Flaherty KT, Puzanov I, Kim KB et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363(9): 809–819.
10. Bollag G, Hirth P, Tsai J et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010; 467(7315): 596–599.
11. Zásady cytostatické léčby maligních onkologických onemocnění. Česká onkologická společnost ČLS JEP. 11. vydání [aktualizováno 1. srpna 2010; citováno 4. října 2010]. Dostupné z: http://www.linkos.cz/odbornici/info_praxe/standardy.php? t=1.
12. Michálek J, Matějková E, Smejkalová J et al. Využití tumor infiltrujících lymfocytů u pacientů s metastazujícím melanomem. Klin Onkol 2007; 20(4): 298–301.
13. Poprach A, Michalová E, Pavlík T et al. Současný stav testování chemorezistence nádorů ex vivo v Masarykově onkologickém ústavu v Brně. Klin Onkol 2008; 21(3): 116–121.
14. Keiholz U, Goey SH, Punt CJ et al. Interferon alfa-2a and interleukin-2 with or without cisplatin in metastatic melanoma: a randomized trial of the European Organization for Research and Treatment of Cancer Melanoma Cooperative Group. J Clin Oncol 1997; 15(7): 2578–2588.
15. Balch CA, Atkins MB, Sober AJ. Cutaneous melanoma. In: DeVita VT Jr, Hellman S, Rosenberg SA et al (eds). Cancer: Principles and Practice of Oncology. 7th ed. Philadelphia: Lippincott Williams & Wilkins Philadelphia 2005.
16. Uldrijan S, Nenutil R, Fait V et al. Melanomové fragmenty a jejich krátkodobá kultivace jako nástroj pro sledování odezvy na cytotoxické látky s rozdílným mechanismem účinku. Klin Onkol 2001; 14(1): 25–30.
17. Emuss V, Garnett M, Mason C et al. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res 2005; 65(21): 9719–9726.
18. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417(6892): 949–954.
19. Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004; 6(4): 313–319.
20. Wan PT, Garnett MJ, Roe SM et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004; 116(6): 855–867.
21. Ikenoue T, Hikiba Y, Kanai F et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res 2003; 63(23): 8132–8137.
22. Hingorani SR, Jacobetz MA, Roberson GP et al. Suppression of BRAFV599E in human melanoma abrogates transformation. Cancer Res 2003; 63(17): 5198–5202.
23. Wellbrock C, Ogilvie L, Hedley D et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res 2004; 64(7): 2338–2342.
24. Sumimoto H, Miyagishi M, Miyoshi H et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene 2004; 23(36): 6031–6039.
25. Karasarides M, Chiloeches A, Hayward R et al. B-RAF is a therapeutic target in melanoma. Oncogene 2004; 23(37): 6292–6298.
26. Tsai J, Lee JT, Wang W et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA 2008; 105(8): 3041–3046.
27. Søndergaard JN, Nazarian R, Wang Q et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med 2010; 8: 39.
28. Hatzivassiliou G, Song K, Yen I et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010; 464(7287): 431–435.
29. Heneberg P. Zpřesněme indikaci podávání inhibitorů kinázové aktivity EGFR. Klin Onkol 2011; 24(2): 87–93.
30. Cichowski K, Jänne PA. Drug discovery: Inhibitors that activate. Nature 2010; 464(7287): 358–359.
31. Heidorn SJ, Milagre C, Whittaker S et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010; 140(2): 209–221.
32. Poulikakos PI, Zhang C, Bollag G et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464(7287): 427–430.
33. Kong HH, Cowen EW, Azad NS et al. Keratoacanthomas associated with sorafenib therapy. J Am Acad Dermatol 2007; 56(1): 171–172.
34. Schwartz GK, Robertson S, Shen A et al. A phase I study of XL281, a selective oral RAF kinase inhibitor, in patients (Pts) with advanced solid tumors. J Clin Oncol 2009; 27: 15. Abstract 3513.
35. Kefford R, Arkenau H, Brown MP et al. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol 2010; 28. Abstract 8503.
36. Dumaz N, Hayward R, Martin J et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res 2006; 66(19): 9483–9491.
37. Nazarian R, Shi H, Wang Q et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468(7326): 973–977.
38. Villanueva J, Vultur A, Lee JT et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18(6): 683–695.
39. Heneberg P. Inhibitory, které aktivují. Vesmír 2010; 89: 356.
40. Nunes-Xavier C, Romá-Mateo C, Ríos P et al. Dual-specificity MAP kinase phosphatases as targets of cancer treatment. Anticancer Agents Med Chem 2011; 11(1): 109–132.
41. Jailkhani N, Chaudhri VK, Rao KV. Regulatory cascades of protein phosphatases: implications for cancer treatment. Anticancer Agents Med Chem 2011; 11(1): 64–77.
42. Heneberg P. Use of protein tyrosine phosphatase inhibitors as promising targeted therapeutic drugs. Curr Med Chem 2009; 16(6): 706–733.
43. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 2005; 6(11): 827–837.
44. Johannessen CM, Boehm JS, Kim SY et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468(7326): 968–972.
45. Carvajal RD, Chapman PB, Wolchok JD et al. A phase II study of imatinib mesylate (IM) for patients with advanced melanoma harboring somatic alterations of KIT. J Clin Oncol 2009; 27. Abstract 9001.
46. Wilhelm SM, Carter C, Tang L et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64(19): 7099–7109.
47. Eisen T, Ahmad T, Flaherty KT et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer 2006; 95(5): 581–586.
48. McDermott DF, Sosman JA, Gonzalez R et al. Double-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study Group. J Clin Oncol 2008; 26(13): 2178–2185.
49. Hauschild A, Agarwala SS, Trefzer U et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol 2009; 27(17): 2823–2830.
50. Ledford H. Melanoma drug wins US approval. Nature 2011; 471(7340): 561.
51. Yang H, Higgins B, Kolinsky K et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res 2010; 70(13): 5518–5527.
52. Rubinstein JC, Sznol M, Pavlick AC et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med 2010; 8: 67.
53. Michaloglou C, Vredeveld LC, Soengas MS et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436(7051): 720–724.
54. Dušek L, Mužík J, Kubásek M et al. Epidemiologie zhoubných nádorů v České republice, verze 7.0. Masarykova univerzita, Brno 2007. [aktualizováno 14. ledna 2011; citováno 14. ledna 2011]. Dostupné z: http://www.svod.cz.
55. Heneberg P. Supposed steep increase in publications on cruciate ligament and other topics. Eur J Orthop Surg Traumatol 2011; 21(6): 401–405.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2011 Issue 4
- Metamizole vs. Tramadol in Postoperative Analgesia
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
- Spasmolytic Effect of Metamizole
- Obstacle Called Vasospasm: Which Solution Is Most Effective in Microsurgery and How to Pharmacologically Assist It?
Most read in this issue
- Mukozitida dutiny ústní a faryngu – možnosti ovlivnění bolesti
- Schnitzler-syndrom: diagnostika a léčba
- Zinek – molekulární mechanizmy u karcinomu prostaty
- Využití prokalcitoninu v diferenciální diagnostice febrilií u pacientů s mnohočetným myelomem