
Schnitzler-syndrom: diagnostika a léčba
Schnitzler Syndrome: Diagnostics and Treatment
Backgrounds:
The most important diagnostic criteria for Schnitzler syndrome include chronic urticaria, the presence of monoclonal IgM immunoglobulin, marked inflammation (leukocytosis, elevated CRP and erythrocyte sedimentation rate), subfebrile temperatures or fevers and bone and joint pains. It is a rare idiopathic disease that may lead to potentially life-threatening complications such as development of secondary amyloidosis or transformation into malignant lymphoproliferation. Schnitzler syndrome should be included in differential diagnostics of chronic urticaria and fevers of unknown origin. The diagnostic algorithm is based on clinical presentation and serum and urine electrophoreses to detect monoclonal components. Blockade of interleukin-1 (IL-1), key cytokine in the pathogenesis of the disease, dominates current therapeutic protocols. Anakinra (Kineret™), recombinant human IL-1 receptor antagonist, is the most widely used treatment option. According to literature, disease remission was obtained in all treated patients. Therefore, anakinra represents a significant diagnostic possibility to differentiate Schnitzler syndrome from e. g. monoclonal gammopathy of unknown significance (MGUS) associated with urticaria of different aetiology. Biological therapy with rilonacept (Arcalyst™) and canakinumab (Ilaris™) represents a new treatment alternative for patients, allowing prolonged dosing intervals of 1 and 8 weeks, respectively (compared to 24 hours with anakinra). The review article also presents findings of various imaging methods (conventional radiography, computed tomography, traditional bone scintigraphy) and photographs of patients with Schnitzler syndrome before and after anakinra therapy.
Design:
The aim of the review is to draw attention to the existence of this rare autoinflammatory and potentially pre-malignant condition, present a simple diagnostic algorithm and provide an overview of therapeutic options for the patients.
Conclusions:
Malign potential of Schnitzler syndrome, possible development into systemic amyloidosis and the fact that patients are frequently referred to oncology clinics for differential diagnostics of monoclonal gammopathy, are the main reasons why clinical oncologists should be aware of Schnitzler syndrome.
Key words:
paraproteinemias – multiple myeloma – amyloidosis – interleukins – interleukin 1 receptor antagonist protein
Authors:
P. Szturz 1; Z. Adam 1; A. Šedivá 2; Z. Fojtík 1; D. Čorbová 3; J. Neubauer 4; J. Prášek 5; R. Hájek 1; J. Mayer 1
Authors‘ workplace:
Interní hematoonkologická klinika, LF MU a FN Brno
1; Ústav imunologie, 2. LF UK a FN Motol
2; Dermatovenerologická klinika, LF MU a FN Brno
3; Radiologická klinika, LF MU a FN Brno
4; Klinika nukleární medicíny, LF MU a FN Brno
5
Published in:
Klin Onkol 2011; 24(4): 271-277
Category:
Reviews
Overview
Východiska:
Chronická kopřivka, přítomnost monoklonálního imunoglobulinu třídy IgM, vysoké zánětlivé parametry (leukocytóza, elevace CRP a zvýšená sedimentace erytrocytů), subfebrilie či febrilie, bolesti kostí a kloubů a některé další znaky jsou diagnostickými kritérii Schnitzler-syndromu. Jedná se o vzácné idiopatické onemocnění, které může vést k potenciálně život ohrožujícím komplikacím rozvojem sekundární amyloidózy či transformací do maligní lymfoproliferace. Je významnou součástí diferenciální diagnostiky chronických kopřivkových projevů a teplot nejasného původu. Diagnostický algoritmus se opírá o typické klinické příznaky a elektroforézu séra a moči k zachycení monoklonální komponenty. V terapii má dominantní úlohu biologická léčba s využitím blokády interleukinu-1 (IL-1), který je klíčovým cytokinem v patogenezi onemocnění. Nejčastěji využívaným léčivem je anakinra (Kineret™), rekombinantní antagonista humánního receptoru pro IL-1. Dle dostupných literárních údajů došlo k remisi onemocnění u všech pacientů léčených anakinrou. Anakinra proto představuje významný diagnostický nástroj umožňující odlišit Schnitzler-syndrom např. od monoklonální gamapatie nejasného významu (MGUS) asociované s urtikou jiné etiologie. Novou léčebnou alternativu pro pacienty představuje biologická terapie pomocí rilonaceptu (Arcalyst™) a kanakinumabu (Ilaris™) umožňující prodloužení dávkovacího intervalu, který je u anakinry 24 hod, na 1, resp. 8 týdnů. Tento přehledný článek je rovněž doplněn nálezy z různých zobrazovacích modalit (konvenční radiografie, výpočetní tomografie, klasické scintigrafie skeletu) a dále i fotografiemi pacientů se Schnitzler-syndromem před léčbou anakinrou a po ní.
Cíl:
Cílem práce je upozornit na existenci tohoto raritního zánětlivého a potenciálně premaligního stavu, představit jednoduchý diagnostický algoritmus a uvést přehled léčebných možností pro tyto pacienty.
Závěr:
Maligní potenciál Schnitzler-syndromu, možnost rozvoje systémové amyloidózy a skutečnost, že pacienti jsou často odesíláni na onkologické kliniky k diferenciální diagnostice monoklonální gamapatie, jsou hlavní důvody, proč by měli být kliničtí onkologové seznámeni se Schnitzler-syndromem.
Klíčová slova:
paraproteinemie – mnohočetný myelom – amyloidóza – interleukiny – receptor interleukinu 1-antagonista
Úvod
Schnitzler-syndrom je vzácné multisystémové onemocnění s nejasnou patogenezí. Od roku 1974, kdy byl francouzskou dermatoložkou Liliane Schnitzler poprvé popsán charakteristický soubor příznaků, se ve světové literatuře objevily, dominantně formou kazuistik, zprávy asi o 100 případech tohoto onemocnění [1,2]. Lze však předpokládat, že skutečný výskyt této choroby, vyznačující se typickou kombinací chronické kopřivky a monoklonální IgM gamapatie, je mnohem vyšší. Další často se vyskytující příznaky zahrnují mj. recidivující epizody subfebrilií či febrilií a bolestí kostí, kloubů a svalů. Radiograficky či histopatologicky lze v některých případech prokázat abnormální strukturu kostí a z laboratorního rozboru krve bývá patrná leukocytóza, elevace CRP a zvýšená sedimentace erytrocytů. Diagnostická kritéria jsou shrnuta v tab. 1 (upraveno podle Lipsker et al [3], de Koning et al [4] a Gilson et al [5]).

Etiopatogeneze Schnitzler-syndromu je značně komplexní a doposud není objasněna. Podle některých hypotéz a podle klinických i laboratorních příznaků je klíčovým bodem patogeneze zánět způsobený interleukinem-1 (IL-1), který je považován za hlavní prozánětlivý cytokin a zodpovídá zřejmě za typické příznaky Schnitzler-syndromu [2,6]. S ohledem na rekurentní charakter zánětlivé odpovědi organizmu u těchto pacientů a na výše zmíněnou roli cytokinů je Schnitzler-syndrom některými autory řazen mezi získaná autoinflamatorní onemocnění [4,7,8].
Chronická kopřivka někdy provázená intenzivním pruritem bývá u nemocných obvykle hlavním příznakem, který je přivede na dermatologickou ambulanci nebo k praktickému lékaři. Jelikož je diferenciální diagnostika urtikariálních morf značně rozsáhlá, bývá při sporadických zprávách o Schnitzler-syndromu v odborném tisku toto onemocnění diagnostikováno až s různě dlouhým časovým odstupem. Bez znalosti správné diagnózy bývá léčba frustrující a její výsledky jsou přinejlepším nekonstantní. Dosud nejúčinnější terapie spočívá v blokádě IL-1 většinou pravidelnou denní aplikací anakinry, receptorového antagonisty IL-1 (firemní název Kineret™).
Tento článek si klade za cíl seznámit čtenáře s touto raritní jednotkou, představuje jednoduchý diagnostický algoritmus vedoucí ke stanovení správné diagnózy a uvádí přehled terapeutických možností, včetně představení některých nových molekul s léčebným potenciálem. Publikována je rovněž série fotografií dokumentujících naše léčebné výsledky.
Klinické, paraklinické a laboratorní nálezy
Chronická kopřivka
Kožní změny (obr. 1 a 2) charakteru chronicko-recidivující až chronicko-kontinuální kopřivky jsou popisovány jako erytematózní makulopapulózní ložiska až splývající geografické plochy velikosti od 0,5 až po více než 10 cm v průměru. Všechna ložiska mají stejnou růžovou až červenou barvu i charakter. Denně se mohou objevovat nové výsevy, trvající 12–36 hod a potom pomalu mizící. U některých pacientů vznikají každý den nové morfy, zatímco u jiných se vyskytují i několik týdnů dlouhé klidové pauzy; u většiny popsaných případů však byly patologické kožní erupce přítomny kontinuálně. K výsevu nových morf může dojít v souvislosti s konzumací alkoholu, kořeněných jídel, po tělesném vypětí nebo jiném stresu, ale i bez zjevné příčiny. Postiženy bývají obvykle končetiny a trup, zatímco hlava a krk, stejně tak jako chodidla a ruce, zůstávají ušetřeny. Svědění je nekonstantním příznakem, který může zpočátku chybět a objevit se až s odstupem několika měsíců či roků [4,9,10].
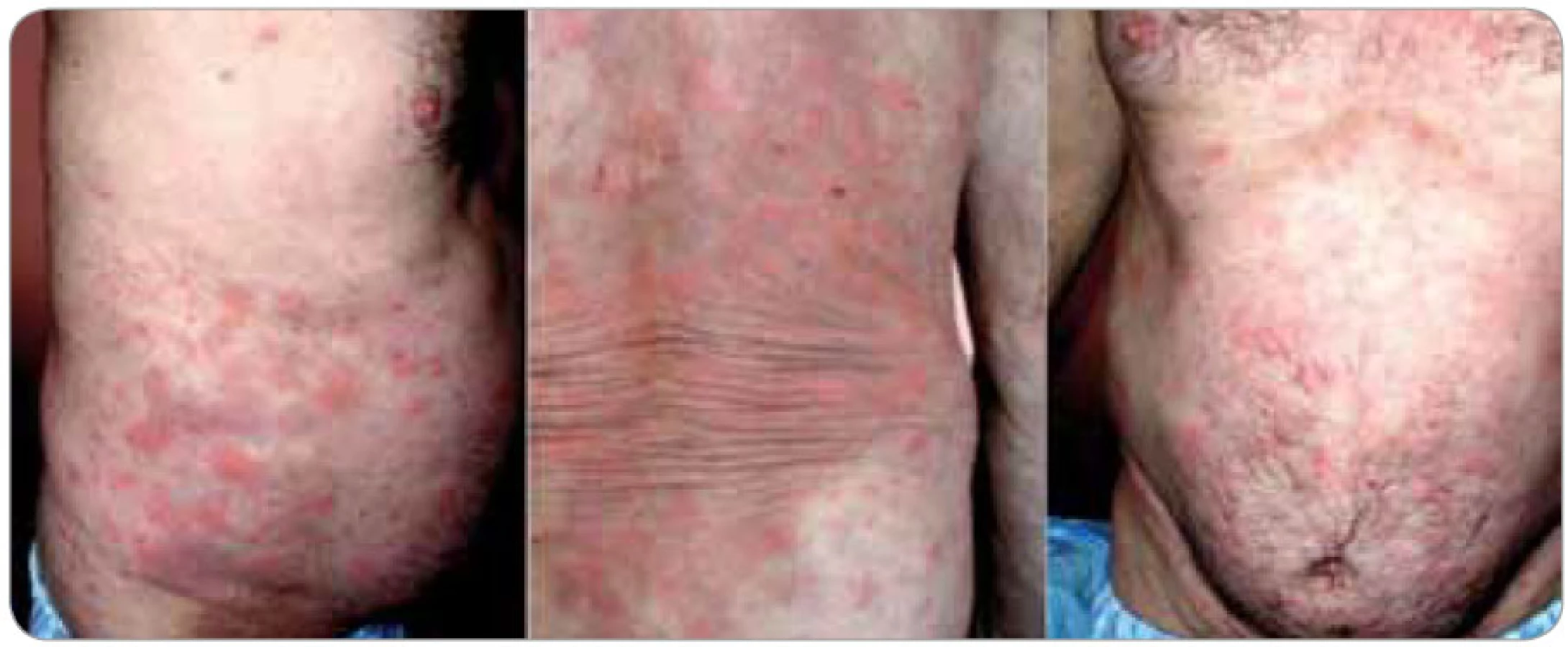
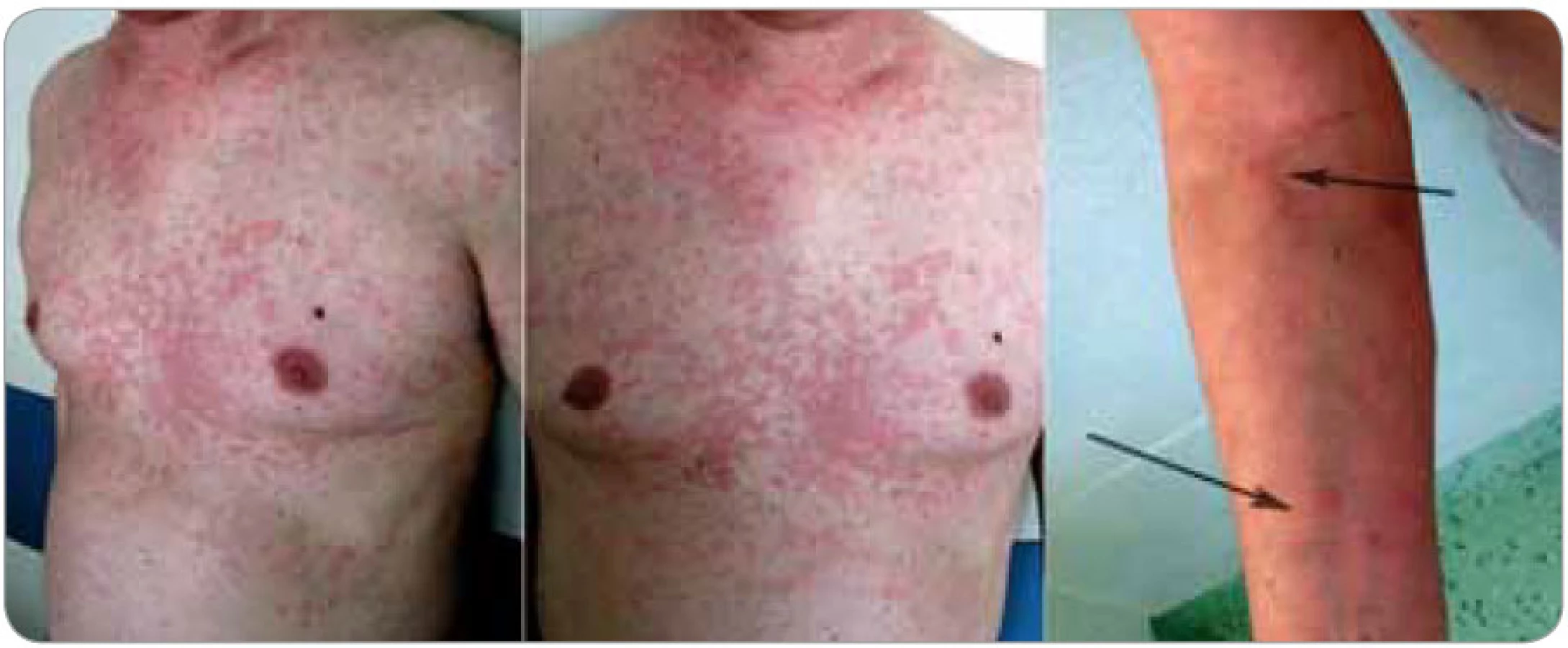
Kieffer et al [11] vyčlenili z nepřesně vymezené skupiny tzv. neutrofilních urtikarií klinickopatologickou jednotku, kterou označili jako neutrofilní urtikariální dermatózu (neutrophilic urticarial dermatosis). U takto postižených pacientů se vyskytují kožní eflorescence charakteru kopřivky (plošně papulózně vyvýšené, ostře ohraničené tuhé morfy) s histopatologickými znaky neutrofilní dermatózy, tedy s perivaskulárními a intersticiálními neutrofilními infiltráty s intenzivní leukocytoklasií, ale bez přítomnosti vaskulitidy a bez edému dermis. Odlišit je nutné urtikariální vaskulitidu s histopatologickými známkami vaskulitidy (prosáknutí endotelu, extravazace erytrocytů, fibrinoidní nekróza cévní stěny) a nález u Sweetova syndromu, kde se typicky uvádí přítomnost dermálního edému. Neutrofilní urtikariální dermatóza se vyskytuje jako součást Stillovy nemoci vzniklé v dospělosti, systémového lupusu erythematodu, Schnitzler--syndromu a zřejmě i některých hereditárních autoinflamatorních nemocí (skupina kryopyrin asociovaných syndromů). S ohledem na silnou asociaci se systémovými chorobami má znalost této patologie význam nejen pro dermatology a revmatology, ale i pro internisty, infekcionisty a onkology. Pro tuto jednotku je rovněž typická absence léčebné odpovědi na antihistaminika.
Jako nejpravděpodobnější mechanizmus vzniku kožních změn u pacientů se Schnitzler-syndromem se jeví hypotéza, podle níž depozita monoklonálního imunoglobulinu typu IgM v dermálně/epidermálním spojení spouštějí lokální zánětlivou reakci, která vede k erupci kopřivkové morfy na kůži. Tato depozita lze prokázat imunofluorescenčním vyšetřením [12–14] a jedná se o stejné izotypy protilátek IgM, které cirkulují v krvi [15].
Monoklonální komponenta, zánět a další nálezy
Přítomnost monoklonálního imunoglobulinu třídy IgM je pro stanovení diagnózy Schnitzler-syndromu nezbytná. U 89 % nemocných se jedná o IgM typu kappa. Koncentrace monoklonálního imunoglobulinu je obvykle při stanovení diagnózy nízká (pod 10 g/ l) a zůstává stabilní nebo se pozvolna v průběhu času zvyšuje. Vyšší hodnoty vyvolávají podezření na transformaci ve Waldenströmovu makroglobulinemii. Zánět je vyjádřen zvýšenou sedimentací erytrocytů, CRP a leukocytózou s neutrofilií. Zánětlivé markery někdy dosahují velmi vysokých hodnot. Trombocytóza a anémie chronických chorob jsou popisovány u 10 % nemocných [3,12].
Intermitentně zvýšená tělesná teplota, obvykle bez pocitu zimnice a třesavky, je jedním z hlavních příznaků Schnitzler--syndromu. U některých pacientů může dosáhnout až 40 °C s variabilní tolerancí i reakcí na nesteroidní antiflogistika.
Dalším důležitým znakem je muskuloskeletální postižení, vyskytuje se asi u 80 % nemocných [3,4]. Schnitzler-syndrom způsobuje změny kostního metabolizmu. Zvýšení kostní denzity je nejčastějším radiologickým nálezem. V oblastech zvýšené kostní denzity jsou často pociťovány bolesti [16–18]. Bolesti kostí, nejčastěji v oblasti pánve a holenních kostí, jsou popisovány u 59 % nemocných a často k nim přistupují ještě bolesti kloubů (kyčle, kolena, zápěstí, lokte), někdy i myalgie [4]. V průkazu osteolyticko-osteosklerotických nebo hyperostotických ložisek se může uplatnit konvenční radiografie, výpočetní tomografie, magnetická rezonance, ale i scintigrafie skeletu pomocí technecium pyrofosfátu, jak dokládá obr. 3.
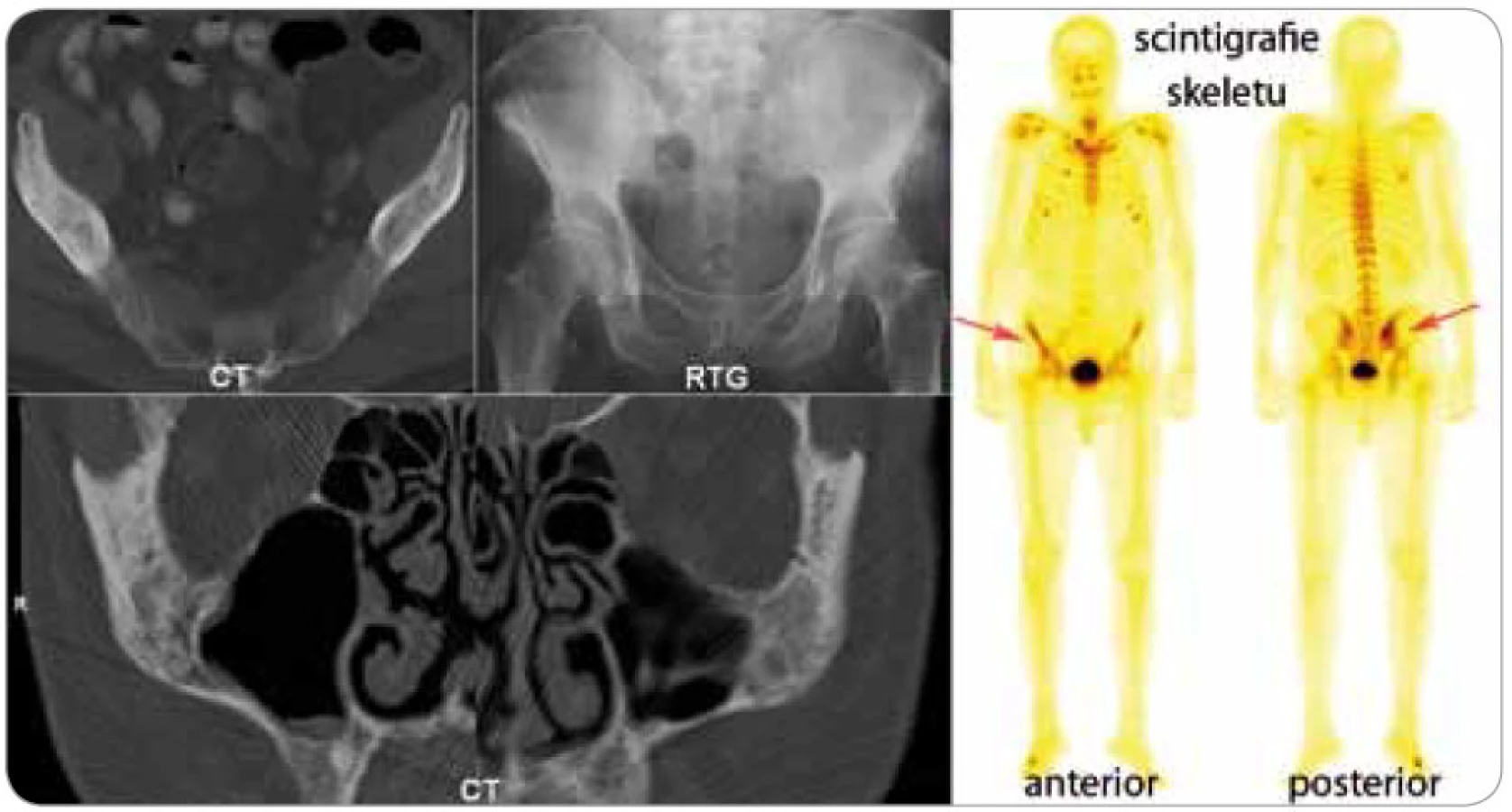
Palpačně zvětšené uzliny byly popsány u 50 % nemocných a hepatosplenomegalie u 33 % nemocných [3].
Diferenciální diagnostika
Erytémem a urtikou se mohou projevovat různá hematoonkologická onemocnění (monoklonální gamapatie nejasného významu, POEMS syndrom, Waldenströmova makroglobulinemie, mnohočetný myelom, lymfomy), která mají rozdílná diagnostická kritéria a některá lze odlišit histologickým vyšetřením kostní dřeně nebo lymfatických uzlin [19–23]. Monoklonální gamapatie nejasného významu může v některých případech koincidovat s urtikou jiné etiologie, případně i s dalšími znaky Schnitzler-syndromu, chybí však léčebná odpověď na biologickou léčbu anakinrou, která zatím dle literárních údajů u pacientů se Schnitzler-syndromem prokazuje 100% účinnost [2].
Diferenciálně diagnosticky je třeba od Schnitzler-syndromu rovněž odlišit autoimunitní nemoci [24]. Podobné kožní a systémové projevy (zejména febrilie) jako Schnitzler-syndrom může způsobovat Stillova nemoc vznikající v dospělosti. Kožní manifestací je v tomto případě makulopapulózní prchavá vyrážka lososově růžové barvy typicky v oblasti trupu a na končetinách. Odlišení těchto dvou chorobných stavů je možné pomocí laboratorní metodiky, kdy u Schnitzler-syndromu dominuje monoklonální imunoglobulin IgM, zatímco u Stillovy choroby vysoké hodnoty feritinu. Urtikariální vyrážka, teploty, artralgie a anémie jsou průsečíkem Schnitzler-syndromu a systémového lupus erythematodes. V rámci lupusu se však kožní projevy vyskytují v typických lokalizacích, jako je obličej, a jsou trvalejšího charakteru. Přítomny jsou rovněž orgánově nespecifické protilátky (ANA, anti-dsDNA, anti-SM, antinukleozomální protilátky). U získaného C1q-inhibitoru jsou charakteristickým nálezem angioedémy a laboratorně zjišťujeme nízké hladiny složek komplementového systému (zejména C1q-inhibitoru).
Hereditární autoinflamatorní onemocnění (kryopyrinopatie, familiární středozemní horečka, periodické horečky spojené s defektem receptoru pro tumor necrosis factor, hyperIgD syndrom, PAPA syndrom) jsou charakterizovány epizodami opakujících se horečnatých stavů, které trvají dny až týdny, kdy dochází ke vzplanutí zánětlivého stavu bez jasné vnější příčiny. Jedná se o autozomálně dominantně nebo recesivně dědičné syndromy s poznanými genetickými mutacemi a dalšími znaky, které umožňují odlišení od Schnitzler-syndromu. Podobně jako u Schnitzler-syndromu se v jejich léčbě uplatňuje mj. anakinra [25].
Existují i další onemocnění, která je nutné zahrnout do diferenciální diagnostiky Schnitzler-syndromu: idiopatická chronická urtika (přítomna léčebná odpověď na antihistaminika, chybí paraprotein), hypokomplementemická urtikariální vaskulitida (hypokomplementemie, glomerulonefritida, uveitida), pozdní tlaková urtikarie (chybí paraprotein, zánět i anémie), kryoglobulinemie (přítomnost kryoglobulinu, závislost projevů na teplotě), morbus Behçet (slizniční ulcerace, uveitida, neurologické příznaky), mastocytóza (histologický průkaz mastocytů v morfě), infekční nemoci jako hepatitida B a C a chronická meningokokcemie (mikrobiologický průkaz infekčního agens) [24].
Diagnostický algoritmus
Chronická kopřivka představuje mnohdy nelehký diferenciálně diagnostický úkol a v některých případech se může jednat o Schnitzler-syndrom. Diagnostický algoritmus u pacienta přicházejícího k lékaři s chronickou kopřivkou přehledně uvádí tab. 2. Klíčová je přitom role elektroforézy a imunofixace, které odhalí přítomnost monoklonální komponenty v séru či v moči, a bývají tak často tím prvním, co lékaře nasměruje ke správné diagnóze. Pacienti bývají následně odesíláni na pracoviště s hematoonkologickou specializací pro nově zjištěnou monoklonální gamapatii, někdy při současně přítomných bolestech kostí s podezřením na mnohočetný myelom.

Prokážeme-li u pacienta přítomnost monoklonálního imunoglobulinu, následuje série dalších vyšetření (zejména vyšetření kostní dřeně a skeletu, případně biopsie zvětšených lymfatických uzlin) jako součást diferenciálně diagnostického procesu každé nově zjištěné monoklonální gamapatie. V případě, že se jedná o pacienta s chronickou kopřivkou a s přítomností monoklonálního imunoglobulinu třídy IgM (event. IgG), měli bychom na existenci Schnitzler-syndromu vzpomenout a zahrnout jej do naší diagnostické rozvahy.
Prognóza onemocnění a terapeutické možnosti
Někteří autoři pohlížejí na Schnitzler-syndrom jako na premaligní stav, a to s ohledem na jeho možný přechod do lymfoproliferativního onemocnění (lymfom marginální zóny, Waldenströmova makroglobulinemie, mnohočetný myelom). Riziko transformace se odhaduje asi na 15 % a dochází k ní nejdříve po 10–20 letech od prvních příznaků nemoci. Pro předpověď přechodu do maligní lymfoproliferace nebyly popsány žádné prognostické znaky ani dosud nebyl zdokumentován žádný případ spontánní remise onemocnění. Z hlediska mortality je však průběh příznivý, 91 % nemocných žije déle než 15 let. Další potenciálně závažné komplikace zahrnují těžký průběh anémie chronických chorob a rozvoj sekundární amyloidózy, jejíž výskyt je však ojedinělý [2,3,26].
Do roku 2005, kdy se objevila první zpráva o úspěšné biologické terapii preparátem anakinra [27], byla léčba této choroby svízelná a frustrující a lékem první volby se na dlouhou dobu staly nesteroidní antiflogistika a kortikosteroidy. Blokátory cyklooxygenázy sice účinkovaly při symptomatické léčbě teploty a bolestí kostí a kloubů, neměly však většinou žádný vliv na kožní projevy [8,9,18]. Glukokortikoidy snižují intenzitu kožních projevů, ale pro dosažení léčebného efektu je zapotřebí dostatečně vysokých dávek, které prohlubují jejich nežádoucí účinky.
Vyzkoušeny byly také kolchicin [28] či dapson [13], hydroxychlorochin a chlorochin [12], dále plazmaferéza [13,15] a nitrožilní imunoglobuliny [29], cytostatika (metotrexát, cyklofosfamid, chlorambucil) a imunosupresiva (azathioprin, cyklosporin) [3], rituximab [30], anti-TNF [31], s významnějším léčebným efektem i interferon alfa [27], thalidomid [32] a fototerapie metodou PUVA [3]. Žádný z těchto léčebných postupů nevedl u všech případů k žádoucí léčebné odpovědi nebo bylo podávání limitováno nežádoucími účinky. Antihistaminika, jak již bylo v textu zmíněno, jsou u Schnitzler-syndromu na kožní projevy neúčinná [9,12,14,16]. Bolesti kostí však mírní či zcela odstraňují bisfosfonáty [33].
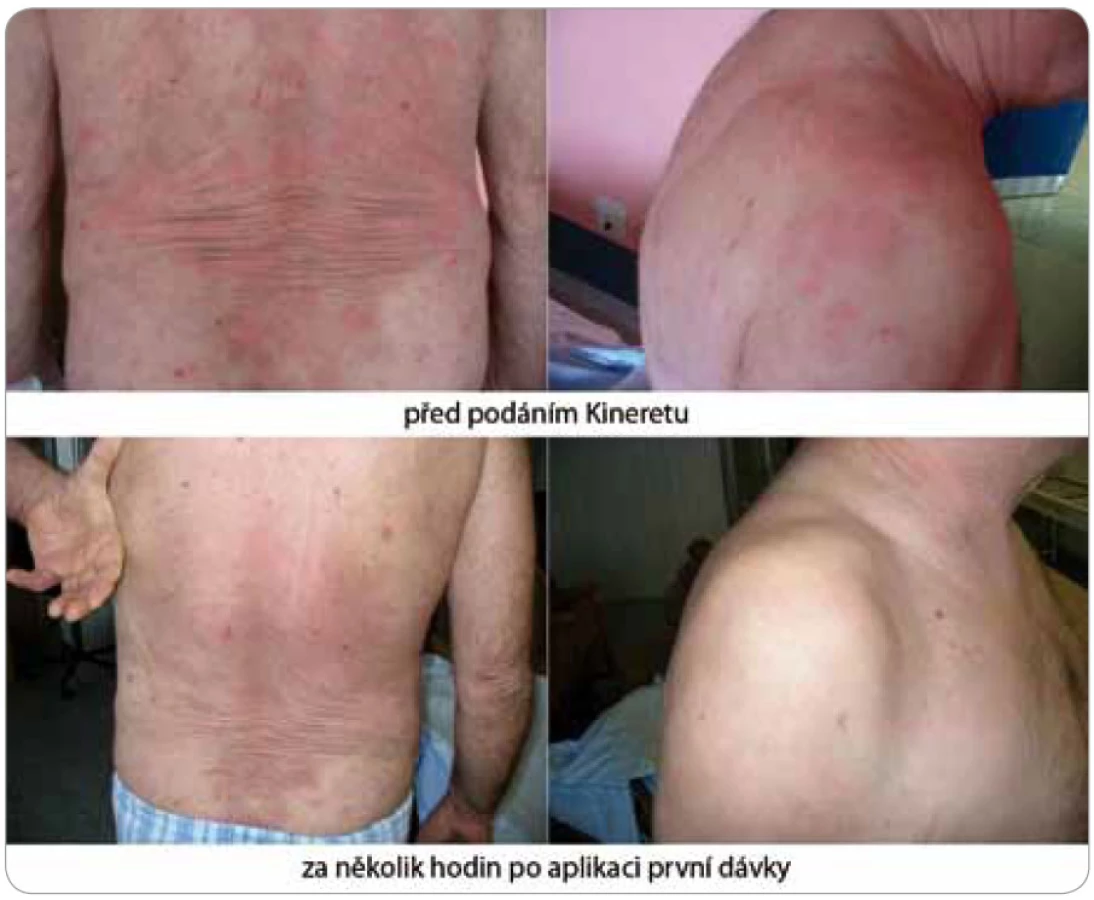
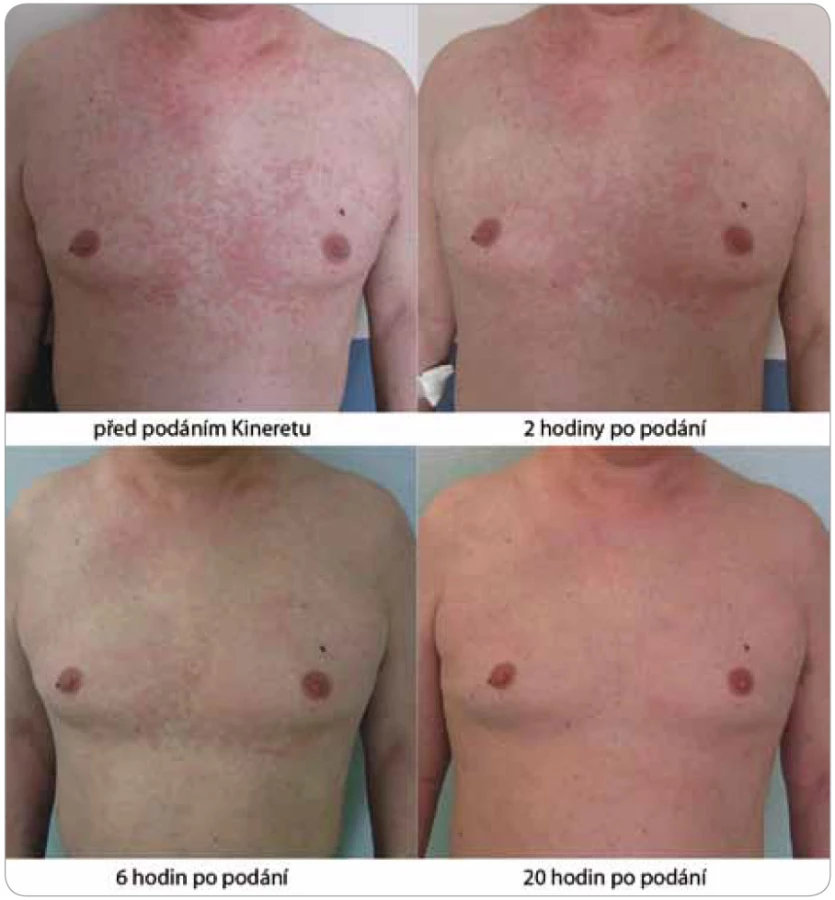
Zásadní změnu přinesl až nověji zkoušený preparát anakinra, rekombinantní antagonista lidského receptoru pro IL-1 (obr. 4 a 5). Ve všech dosud popsaných případech navodila první injekce (100 mg s. c.) během několika hodin kompletní remisi nemoci [2,28]. Anakinra zcela odstranila svědění kůže a kožní projevy. Léčebný účinek anakinry se projeví také v laboratorních nálezech, kde je patrný pokles zánětlivé odpovědi organizmu. Vzhledem ke krátkému biologickému poločasu anakinry je její aplikace každodenní a trvalá a po vysazení se příznaky dostavují do 24–48 hod [4]. Přes evidentní a zásadní příznivý vliv na zánět nemá však léčba blokádou IL-1 vliv na monoklonální komponentu.
K dispozici jsou však již další preparáty blokující IL-1 s delším biologickým poločasem, než má anakinra. Rilonacept (firemní název Arcalyst™) je dimerický fúzní protein, který se skládá z extracelulární části receptoru pro IL-1 (IL-1R1) a z jeho akcesorního proteinu (IL-1R-AcP), které jsou oba navázány na Fc fragment IgG protilátky. Vzniklý komplex účinně blokuje IL-1 [34]. Výhodou je možnost aplikovat 160 mg podkožně v intervalu 1 týdne. Další účinnou molekulou může být i kanakinumab (firemní název Ilaris™), humánní monoklonální protilátka proti lidskému IL-1 beta, která po navázání na tento ligand brání jeho interakci s receptory pro IL-1 a následnému spuštění zánětlivé kaskády [35]. Aplikuje se 150 mg podkožní injekcí a doporučený dávkovací interval je 8 týdnů. Oba přípravky jsou indikovány pro léčbu kryopyrinopatií. Zajímavou alternativou v léčbě Schnitzler-syndromu může být i antagonista humánního receptoru pro interleukin-6, tocilizumab (firemní název RoActemra™) [8], jehož terapeutický potenciál se opírá o opakovaně nalézané zvýšené sérové hladiny interleukinu-6 u nemocných se Schnitzler-syndromem. Účinnost uvedených tří preparátů v indikaci Schnitzler-syndromu je nutné ověřit dalšími studiemi.
Závěr
Vzácné choroby lze rozpoznat, pokud na jejich existenci vzpomeneme, zahrneme je do naší diferenciálně diagnostické rozvahy a správně indikujeme nezbytná klinická, paraklinická a laboratorní vyšetření. V některých případech se může jednat o natolik výjimečné choroby, že informace je nutné hledat v publikovaných popisech jednotlivých případů nebo malých souborů pacientů.
Schnitzler-syndrom je třeba mít na zřeteli při diferenciální diagnostice chronických kopřivkových projevů a teplot nejasného původu. Jedná se o vzácné onemocnění s potenciálně život ohrožujícími komplikacemi při přechodu do lymfoproliferativního onemocnění nebo při rozvoji sekundární amyloidózy. Základní vyšetření u této nemoci by proto mělo zahrnovat elektroforézu a imunofixaci séra a moči, vyšetření kostní dřeně a biopsii z kožních lézí. Obligátním požadavkem je vyloučení infekční nebo nádorové etiologie s případnou exstirpací zvětšených lymfatických uzlin na histologické vyšetření. Monoklonální imunoglobulin by měl být pravidelně sledován a při výrazném vzestupu doporučujeme opakované přešetření pacienta včetně odběru kostní dřeně a provedení nových zobrazovacích vyšetření s cílem odhalit případnou transformaci do maligní lymfoproliferace. Maligní potenciál Schnitzler-syndromu, možnost rozvoje systémové amyloidózy a skutečnost, že pacienti jsou často odesílání na onkologické kliniky k diferenciální diagnostice monoklonální gamapatie, jsou hlavní důvody, proč by kliničtí onkologové měli být s tímto onemocněním seznámeni.
Tato publikace byla připraven v rámci aktivity následujících grantů: grantu IGA ČR NT 12215-4 a dále pak grantů MŠMT MSM0021622434, LC06027 a grantů IGA MZd NT11154, NT12130, NT12215 a NS10408.
This publication was prepared as part of the Internal Grant Agency of the Czech Republic grants NT 12215-4, the Ministry of Education, Youth and Sports of the CR grants MSM0021622434, LC06027 and the Ministry of Health of the Czech Republic’s Internal Grant Agency grants IGA MZd NT11154, NT12130, NT12215 and NS10408.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
MUDr. Petr Szturz
Interní hematoonkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: petr.szturz@fnbrno.cz
Obdrženo/Submitted: 9. 12. 2010
Přijato/Accepted: 4. 1. 2011
Sources
1. Schnitzler L, Schubert B, Boasson M et al. Urticaire chronique lésions osseuses macroglobulinémie IgM: maladie de Waldenström? Bull Soc Fr Dermatol Syph 1974; 81 : 363–366.
2. Besada E, Nossent H. Dramatic response to IL1-RA treatment in longstanding multidrug resistant Schnitzler’s syndrome: a case report and literature review. Clin Rheumatol 2010; 29(5): 567–571.
3. Lipsker D, Veran Y, Grunenberger F et al. The Schnitzler syndrome: four new cases and review of the literature. Medicine (Baltimore) 2001; 80(1): 37–44.
4. de Koning HD, Bodar EJ, van der Meer JW et al. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum 2007; 37(3): 137–148.
5. Gilson M, Abad S, Larroche C et al. Treatment of Schnitzler’s syndrome with anakinra. Clin Exp Rheumatol 2007; 25(6): 931.
6. Loock J, Lamprecht P, Timmann C et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol 2010; 125(2): 500–502.
7. Eiling E, Schröder JO, Gross WL et al. The Schnitzler syndrome: chronic urticaria and monoclonal gammopathy – an autoinflammatory syndrome? J Dtsch Dermatol Ges 2008; 6(8): 626–631.
8. Kluger N, Bessis D, Guillot B. Tocilizumab as a potential treatment in Schnitzler syndrome. Med Hypotheses 2009; 72(4): 479–480.
9. Adam Z, Krejčí M, Pour L et al. Schnitzlerův syndrom – popis čtrnáctiletého průběhu nemoci a přehled informací o této nemoci. Vnitř Lék 2008; 54(12): 1140–1153.
10. Sanmartín O, Febrer I, Botella R et al. Urticarial lesions and monoclonal IgM gammopathy. Schnitzler’s syndrome. Arch Dermatol 1994; 130(9): 1193–1198.
11. Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore) 2009; 88(1): 23–31.
12. Berdy SS, Bloch KJ. Schnitzler’s syndrome: a broader clinical spectrum. J Allergy Clin Immunol 1991; 87(4): 849–854.
13. Borradori L, Rybojad M, Puissant A et al. Urticarial vasculitis associated with a monoclonal IgM gammopathy: Schnitzler’s syndrome. Br J Dermatol 1990; 123(1): 113–118.
14. Janier M, Bonvalet D, Blanc MF et al. Chronic urticaria and macroglobulinemia (Schnitzler’s syndrome): report of two cases. J Am Acad Dermatol 1989; 20(2 Pt 1): 206–211.
15. Olsen E, Førre O, Lea T et al. Unique antigenic determinants (idiotypes) used as markers in a patient with macroglobulinemia and urticaria. Similar idiotypes demonstrated in the skin and on peripheral blood lymphocytes. Acta Med Scand 1980; 207(5): 379–384.
16. Lecompte M, Blais G, Bisson G et al. Schnitzler’s syndrome. Skeletal Radiol 1998; 27(5): 294–296.
17. De Waele S, Lecouvet FE, Malghem J et al. Schnitzler’s syndrome: an unusual cause of bone pain with suggestive imaging features. AJR Am J Roentgenol 2000; 175(5): 1325–1327.
18. Flórez AF, Gallardo-Agromayor E, García-Barredo R et al. Radiological aid to clinical diagnosis of Schnitzler’s syndrome: multimodality imaging approach. Clin Rheumatol 2008; 27(1): 107–110.
19. Krejčí M, Adam Z, Hájek R. Mnohočetný myelom. Klin Onkol 2008; 21 (Suppl 1): 187–189.
20. Pujol RM, Barnadas MA, Brunet S et al. Urticarial dermatosis associated with Waldenström’s macroglobulinemia. J Am Acad Dermatol 1989; 20(5 Pt 1): 855–857.
21. Lipsker D, Cribier B, Maloisel F et al. Chronic urticaria and IgA myeloma. Acta Derm Venereol 1998; 78(5): 395.
22. Karakelides M, Monson KL, Volcheck GW et al. Monoclonal gammopathies and malignancies in patients with chronic urticaria. Int J Dermatol 2006; 45(9): 1032–1038.
23. Ščudla V, Budíková M, Petrová P et al. Analýza sérových hladin vybraných biologických ukazatelů u monoklonální gamapatie nejistého významu a mnohočetného myelomu. Klin Onkol 2010; 23(3): 171–181.
24. Fojtík Z, Adam Z, Krejčí M et al. Diferenciální diagnostika syndromu Schnitzlerové a dalších chorob způsobujících urtiku, kostní bolesti a subfebrilie. In: Rovenský J, Pavelka K, Plank L et al (eds). Vybrané kazuistiky v reumatológii. Bratislava: SAP 2009 : 81–101.
25. Šedivá A. Poruchy regulace zánětu a periodické horečky. Alergie 2006; 8(1): 36–41.
26. Claes K, Bammens B, Delforge M et al. Another devastating complication of the Schnitzler’s syndrome: AA amyloidosis. Br J Dermatol 2008; 158(1): 182–184.
27. Martinez-Taboada VM, Fontalba A, Blanco R et al. Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum 2005; 52(7): 2226–2227.
28. Šedivá A, Poloučková A, Podrazil M et al. Characterization of the B-cell compartment in a patient with Schnitzler syndrome. Scand J Rheumatol 2010; 40(2): 158–160.
29. Lebbe C, Rybojad M, Klein F et al. Schnitzler’s syndrome associated with sensorimotor neuropathy. J Am Acad Dermatol 1994; 30(2 Pt 2): 316–318.
30. Eiling E, Möller M, Kreiselmaier I et al. Schnitzler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol 2007; 57(2): 361–364.
31. Thonhoffer R, Uitz E, Graninger W. Schnitzler’s syndrome-exacerbation after anti-TNF treatment. Rheumatology 2007; 46(6): 1041–1042.
32. de Koning HD, Bodar EJ, Simon A et al. Beneficial response to anakinra and thalidomide in Schnitzler’s syndrome. Ann Rheum Dis 2006; 65(4): 542–544.
33. Obořilová A, Adam Z. Schnitzler’s syndrome. Vnitř Lék 1998; 44(7): 423–427.
34. Stahl N, Radin A, Mellis S. Rilonacept-CAPS and beyond. Ann N Y Acad Sci 2009; 1182 : 124–134.
35. Church LD, McDermott MF. Canakinumab: a human anti-IL-1β monoclonal antibody for the treatment of cryopyrin-associated periodic syndromes. Expert Rev Clin Immunol 2010; 6(6): 831–841.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2011 Issue 4
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Mukozitida dutiny ústní a faryngu – možnosti ovlivnění bolesti
- Schnitzler-syndrom: diagnostika a léčba
- Zinek – molekulární mechanizmy u karcinomu prostaty
- Využití prokalcitoninu v diferenciální diagnostice febrilií u pacientů s mnohočetným myelomem