
Identifikácia molekulárnych markerov u detí s akútnou myeloblastovou leukémiou (AML)
Identification of Molecular Markers in Children with Acute Myeloid Leukemia (AML)
Backgrounds:
AML is an aggressive, phenotypically and genetically heterogenous clonal disease of hematopoietic progenitor cells with a great molecular variability. New WHO classification 2008 divides de novo AML according to cytogenetic and molecular prognostic and predictive markers. Recently, it is increasingly possible to identify a subgroup of poorer prognosis patients among those with normal karyotype AML. The aim of our study was to identify prognostically important molecular markers in children with AML, to stratify patients with normal karyotype and to monitor the disease according the genetic findings.
Material and Methods:
In 2008–2010, we analyzed bone marrow and peripheral blood samples of 20 children with de novo AML by conventional cytogenetic analysis, fluorescence in situ hybridisation and molecular diagnostics. The molecular analysis was performed on the cDNA level, with the restriction analysis of PCR products (FLT3-TKD), conventional PCR (MLL-PTD, NPM1mut, FLT3-ITD) and quantification RT-PCR method (expression of fusion transcripts, BAALC, WT1).
Results:
Samples from 20 children with AML were analyzed using the conventional cytogenetics, FISH and molecular methods. Abnormal karyotype was identified in 13 patients (65%). Further analysis revealed FLT3-ITD in 5/20 (25%), FLT3-TKD in 3/20 (15%), NPM1mut in 2/20 (10%) and MLL-PTD in 1/20 (5%), overexpression of WT1 gene in 15/20 (75%) and overexpression of BAALC in 13/20 (65%) patients.
Conclusion:
Wide cytogenetic and molecular screening helped to find at least one genetic marker in all 20 patients for later follow-up and risk stratification. 4/20 (20%) patients died of the disease progression.
Key words:
acute myeloid leukemia – genetic methods – mutation screening of FLT3 – NPM1 and MLL – WT1 and BAALC expression
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
10. 5. 2011
Accepted:
22. 11. 2011
Authors:
D. Ilenčíková 1; J. Sýkora 2; Z. Mikulášová 2; Vanda Repiská 3
![]()
Authors‘ workplace:
2. Detská klinika, LF UK a DFNsP, Bratislava, Slovenská republika
1; Oddelenie onkologickej genetiky, Národný onkologický ústav, Bratislava, Slovenská republika
2; Ústav lekárskej biológie, genetiky a klinickej genetiky, LF UK a UNB, Bratislava, Slovenská republika
3
Published in:
Klin Onkol 2012; 25(1): 26-35
Category:
Original Articles
Overview
Východiská:
AML je agresívne, fenotypovo a geneticky heterogénne klonálne ochorenie krvotvorných progenitorových buniek s veľkou molekulárnou variabilitou. Nová klasifikácia WHO 2008 delí de novo AML podľa cytogenetických a molekulárnych prognostických a prediktívnych markerov. V poslednej dobe je stále väčšia možnosť určiť podskupinu rizikových pacientov so zlou prognózou medzi tými s normálnym karyotypom. Cieľom našej štúdie bolo zistiť prognosticky významné molekulárne markery u detí s AML na stratifikáciu pacientov s normálnym karyotypom a na sledovanie ochorenia podľa genetického nálezu.
Súbor pacientov a metódy:
V rokoch 2008–2010 sme analyzovali vzorky kostnej drene a periférnej krvi u 20 detí s de novo AML konvenčnou cytogenetickou analýzou, fluorescenčnou in situ hybridizáciou a molekulovou analýzou. Molekulárna analýza bola vykonaná na úrovni cDNA restrikčnou analýzou PCR produktov (FLT3-TKD), konvenčnou PCR (MLL-PTD, NPM1mut, FLT3-ITD) a kvantifikačnou RT-PCR (expresia fúznych génov, génov BAALC a WT1).
Výsledky:
Vzorky 20 detí s AML boli analyzované využitím konvenčnej cytogenetiky, FISH a molekulovo-genetickými metódami. Abnormálny karyotyp bol zistený u 13 pacientov (65 %). Ďalšia analýza ukázala FLT3-ITD v 5/20 (25 %), FLT3-TKD v 3/20 (15 %), NPM1mut 2/20 (10 %) a MLL-PTD v 1/20 (5 %), nadmernej expresie WT1 génu v 15/20 (75 %) a nadmernej expresie BAALC v 13/20 (65 %) pacientov.
Záver:
Široký cytogenetický a molekulárny skríning pomohol nájsť aspoň jeden genetický marker u všetkých 20 pacientov pre ďalšie sledovanie a stratifikáciu rizika detí s AML. Na progresiu ochorenia umreli 4/20 (20 %) detí.
Kľúčové slová:
akútna myeloidná leukémia – genetické metódy – mutačný skríning FLT3 – NPM1 a MLL – expresia WT1 a BAALC
Východiská
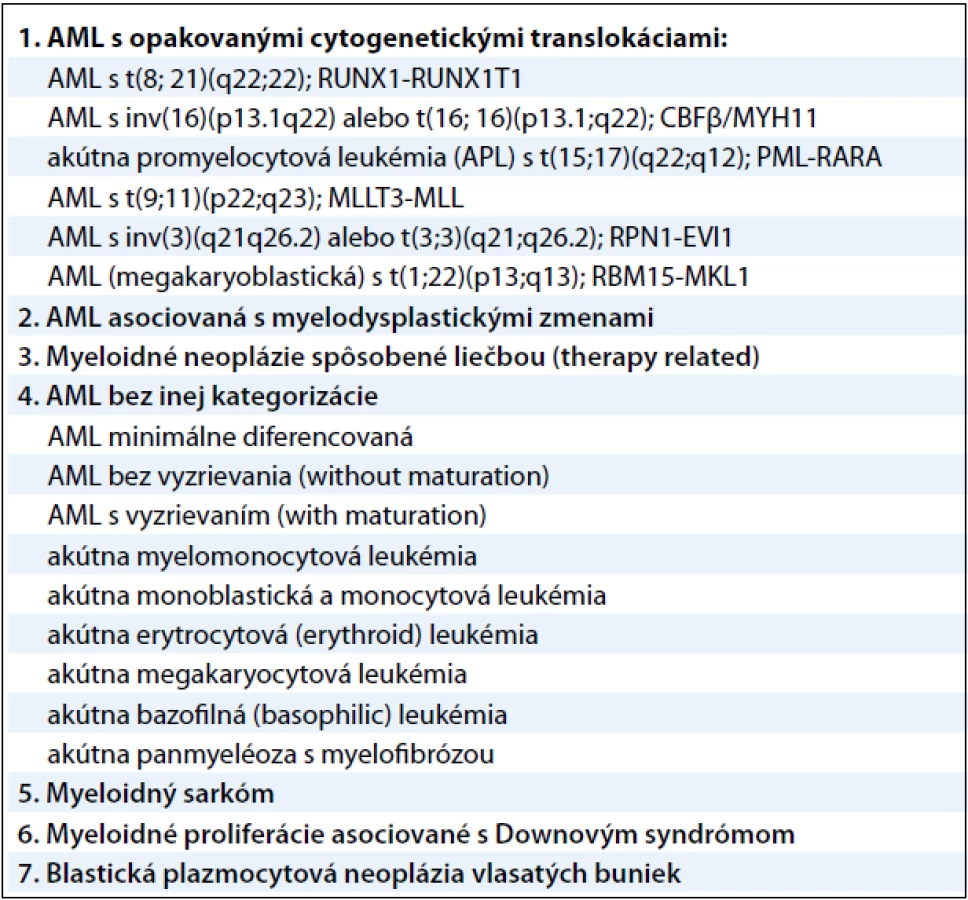
Akútna myeloblastová leukémia je veľmi agresívne, fenotypicky a geneticky heterogénne klonálne ochorenie s veľkou variabilitou patogenézy na cytogenetickej, ako aj na molekulárnej úrovni. AML je charakteristická malígnou transformáciou myeloidnej línie bielych krviniek a predstavuje 2–4 % všetkých malignít. Jej incidencia je 2–3/100 000 obyvateľov a zvyšuje sa vekom (v skupine nad 65 rokov je incidencia 15–27/100 000 obyvateľov) a je mierne vyššia u mužov ako u žien [1]. Príčina vzniku nie je doteraz dostatočne ozrejmená. Predpokladá sa, že ionizujúce žiarenie, rôzne chemické látky (benzén, organické rozpúšťadlá, organické farbivá, herbicídy a pesticídy), fajčenie a z fyzikálnych vplyvov napríklad elektromagnetické polia sa uplatňujú ako podporné faktory pri leukemogenéze. Z liekov majú významnú úlohu predovšetkým cytostatiká, z nich hlavne alkylačné látky a inhibítory topoizomerázy II. Predpoklad, že genetická predispozícia zohráva dôležitú úlohu v patogenéze AML, potvrdzuje vyšší výskyt AML v spojení s rôznymi vrodenými genetickými poruchami – Bloomov syndróm, Downov syndróm, Klinefelterov syndróm, ataxia teleangiectasia, xeroderma pigmentosum a ďalšie. Dôležitú úlohu zohrávajú aj imunologické defekty, prípadne vírusy. Malígna transformácia vzniká vo včasnom štádiu myelocytových prekurzorov [2]. V roku 2008 revidovala Svetová zdravotnícka organizácia klasifikáciu myeloidných neoplázií a akútnych leukémií. Klasifikácia využíva všetky dostupné poznatky – morfologické, cytochemické, imunofenotypyzáciu, genetické a klinické vlastnosti ochorení. Pre AML platia tieto skupiny uvedené v tab. 1 [3].
Pre potreby presnej klasifikácie biologicky rozdielnych podtypov AML využívame morfologické, cytochemické metódy, imunofenotypizáciu a genetické metódy [2,4].
Genetické metódy používané v diagnostike AML
V diagnostike AML sa stále viac využívajú cytogenetické metódy a metódy molekulovo-genetickej analýzy. V súčasnosti sa najviac využíva kombinácia klasickej a molekulárnej cytogenetiky (FISH) a z metód molekulárnej genetiky PCR (polymerázová reťazová reakcia) a jej modifikácie.
Klasická cytogenetika slúži na stanovenie celkového karyotypového obrazu ochorenia a monitorovanie priebehu choroby. Poskytuje celkový prehľad o kvalitatívnych a kvantitatívnych zmenách v karyotype. Fluorescenčná in situ hybridizácia (FISH) patrí k metódam molekulovej cytogenetiky a umožňuje detegovať špecifické nukleotidové sekvencie s rozsahom od jednej po niekoľko sto kilobáz. Využitím špecifických sond sa dá identifikovať počet chromozómov, jednotlivé chromozómy alebo iba niektoré oblasti chromozómov, prípadne len špecifické lokusy [5,6]. PCR patrí k metódam molekulárnej genetiky a umožňuje amplifikáciu špecifických úsekov DNA in vitro [7,8]. Dovoľuje amplifikáciu aj malého množstva DNA. V súčasnej dobe sa používa variant PCR umožňujúci priamu kvantifikáciu PCR-produktu v priebehu reakcie, a to „real-time PCR“. Kvantifikácia množstva molekúl nukleových kyselín je dôležitá pri detailnom štúdiu génovej expresie. Napríklad, real-time PCR môže byť použitá k detekcii a kvantifikácii fúzneho génu, produktu chromozómových translokácií. Kvantitatívne stanovenie fúzneho génu vo vyšetrovanej vzorke sa využíva na určenie minimálneho reziduálneho ochorenia (MRD) alebo progresie, či relapsu ochorenia [9]. Ďalším variantom PCR je reverzná transkriptázová PCR (RT-PCR). Je vhodná, pokiaľ nie je známa vnútorná organizácia exónov a intrónov v géne.
Cytogenetický a molekulárny nález u pacientov s AML
V čase diagnózy sa u 55–60 % pacientov s AML zisťujú klonálne chromozómové aberácie spojené s rôznou prognózou. Podľa cytogenetického nálezu môžeme chromozómové aberácie rozdeliť do 3 skupín:
- s priaznivou prognózou – t(8;21), inv(16), t(15;17),
- so strednou prognózou – normálny karyotyp, del(7q), del(9q), t(9;11), del(11q), del(20q), +8, +11, +13, +21,
- s nepriaznivou prognózou – komplexný karyotyp (≥ 3 aberácie), inv(3)/t(3;3),-7, -5, del(5q), t(6;9), t(6;11), t(t11;19).
Zastúpenie jednotlivých chromozómových aberácií je znázornené v grafe 1. Približne 40–45 % pacientov nemá pri vyšetrení konvečnou cytogenetikou žiadnu chromozómovú aberáciu. U takmer 80 % pacientov s normálnym karyotypom bola v publikovaných multicentrických štúdiách dokázaná prítomnosť mutácie v minimálne jednom z nasledovných génov: NPM1, CEBPA, MLL, FLT3 a NRAS [12]. Na základe týchto poznatkov sa v súčasnosti zameriava veľká pozornosť na vyšetrenie mutačného stavu hore uvedených génov a študuje sa ich prognostický význam. Významné prognostické ukazovatele boli rozdelené do 3 skupín:
- mutácie (FLT3 ITD, FLT3 D835, Ras) aktivujúce signálne dráhy, čím dochádza k zvýšeniu proliferácie a inhibícia apoptózy buniek kostnej drene,
- mutácie (MLL TD, NPM1, CEBPA) ovplyvňujúce procesy transkripcie, čo vedie k oslabeniu diferenciácie buniek,
- zvýšená expresia génov WT1, BAALC a ERG (ETS-related gene).
Mimoriadny prognostický význam nadobudli zvlášť genotypy asociované s priaznivou prognózou: NPM1 a CEBPA. S nepriaznivou prognózou sú často spájané mutované gény MLL - ITD a FLT3-ITD.

Charakteristika molekulovo--genetických markerov
FLT3 – FMS-like tyrozín kináza 3 – je transmembránový proteín. Gén pre tento proteín je lokalizovaný na 13q12. Receptor génu je exprimovaný na hemopoetických progenitorových bunkách a hrá dôležitú úlohu v prežívaní a diferenciácii kmeňových buniek. Doposiaľ boli identifikované dva dôležité typy mutácií FLT3 génu – vnútorná tandemová duplikácia (internal tandem duplication = ITD) a bodová mutácia kyseliny asparágovej D835. Pri FLT3 ITD dochádza k duplikácii génovej sekvencie. Dĺžka aj pozícia sekvencie varíruje od 18 do 100 bp. Vyskytuje sa u 15 % detských AML pacientov [12]. V dôsledku duplikácie nastáva konštitutívna aktivácia FLT3 receptora, čím dochádza k poruche regulácie proliferácie buniek. Je spojená s nepriaznivou prognózou. U FLT3 TKD D835 dochádza k bodovej mutácii kyseliny asparágovej D835 v exóne 20 v tyrozínovej doméne receptora. To vedie ku konštitutívnej aktivácii nezávislej na naviazaní liganda (obr. 1). Mutácia je asociovaná s dobrou prognózou. Zisťuje sa u 5–10 % detských pacientov s AML [13]. NPM1 = Nucleophosmin (nucleolar phosphoprotein B23, numatrin) je gén lokalizovaný na 5q35 [16]. Fosfoproteín zabezpečuje transport ribozomálnych proteínov cez jadrovú membránu. Mutácie sa zisťujú v exóne 12. Viac ako 95 % mutácií predstavuje inzercia o dĺžke 4bp. Incidencia u detských pacientov s AML predstavuje 2–6 % [14]. NPM1 mutácie korelujú s prítomnosťou FLT3-ITD a FLT3-TKD mutáciami. NPM1 mutácie sú spojené s priaznivou prognózou, pokiaľ sa vyskytujú bez FLT3-ITD. MLL = mixed-lineage leukemia, gén je lokalizovaný v chromozómovom prúžku 11q23. MLL proteín sa uplatňuje ako pozitívny regulátor expresie génov počas skorého embryonálneho vývoja a hematopoézy. Parciálna tandemová duplikácia MLL génu býva lokalizovaná vo vnútornej časti génu MLL a zaberá exóny 2–6 alebo exóny 2–8. Frekvencia výskytu u detí s AML je zhruba 2,5 % [13]. Aberácie génu MLL predstavujú nepriaznivý prognostický faktor. NADEXPRESIA WT1 slúži ako nešpecifický nezávislý faktor. WT1 (Wilms tumor 1) gén je lokalizovaný v chromozómov prúžku 11p13. Kóduje zinkový prst DNA viažuceho proteínu. WT1 gén sa vyznačuje funkčnou dualitou – pôsobí ako tumorsupresorový gén, aj ako onkogén. Nadexpresia v rôznych tkanivách je asociovaná so zvýšenou proliferáciou (testes, ovária, slezina). Za nadexpresiu sa považuje, ak hodnota expresie presiahne 100-násobne jej fyziologickú hodnotu. U detských AML pacientov sa nadexpresia génu WT1 vyskytuje u 78,3 % pacientov [15]. Expresia WT1 sa môže použiť na sledovanie priebehu ochorenia a stanovenia MRD u pacientov s AML. U detských pacientov sa zisťuje v 12 % prípadov [14]. NADEXPRESIA BAALC slúži ako nešpecifický prognostický faktor. Gén BAALC (brain and acute leukemia cytoplasmatic) je lokalizovaný v chromozómovom prúžku 8q22.3. Exprimuje sa hlavne na tkanivách odvodených z neuroektodermu a na prekurzoroch hematopoetických buniek. Naopak, nenachádza sa v zrelej kostnej dreni a cirkulujúcich bielych krvinkách. Za nadexpresiu sa považuje, ak hodnota expresie 100-násobne presiahne jej fyziologickú hodnotu. Expresia BAALC sa môže použiť na monitoring MRD u pacientov s AML.

Cieľom využitia genetických metód v diagnostike AML ochorenia je nájsť genetický marker charakterizujúci klonový vývoj leukémie. V mnohých prípadoch pomáhajú špecifické cytogenetické markery v spresnení diagnózy (stanoveniu podtypu AML na základe špecifických chromozómových translokácií), vyjadreniu sa k prognóze ochorenia, monitorovaniu odpovede pacienta na špecifickú liečbu, sledovaniu minimálnej reziduálnej choroby a potransplantačného priebehu ochorenia. V súčasnosti sa do popredia dostáva snaha identifikovať prognosticky významné molekulové markery, ktoré by umožnili stratifikovať pacientov s normálnym karyotypom bez cytogenetického nálezu do prognosticky užších skupín. Zavedenie diagnostiky genetických markerov môže pomôcť identifikovať u každého pacienta aspoň jeden cytogenetický alebo molekulárny marker na monitorovanie priebehu ochorenia, na predikciu včasného relapsu, alebo na sledovanie efektívnosti terapie. Cieľom predkladanej práce bolo zistiť incidenciu jednotlivých genetických markerov u detských AML pacientov na Slovensku. Ďalej skrínovať mutácie v génoch FLT3, NPM1, MLL metodikami PCR a stanoviť hladinu expresie génov BAALC a WT1 pomocou kvantitatívnej real-time RT-PCR a sledovať celkové prežívanie detských pacientov vo vzťahu ku genetickému nálezu.
Metódy
Na Oddelení onkologickej genetiky Národného onkologického ústavu v Bratislave sme v rokoch 2008–2010 u detí s AML vyšetrili vzorky kostnej drene a periférnej krvi metódou konvenčnej a molekulárnej cytogenetiky (FISH) a molekulovou RNA/DNA diagnostiku (RT-PCR). Odobraté vzorky sme za účelom cytogenetického vyšetrenia spracovali 24-hodinovou alebo 72-hodinovou kultiváciou, pri teplote 37 °C v 10 ml MarrowGrow kultivačného média. Množstvo vzorky pridanej do média sme vypočítali podľa vzorca 10/počet leukocytov v 1 ml = množstvo vzorky v ml. Po 24-hodinovej kultivácii sme do kultivačnej nádoby pridali cytostatikum vinkristín alebo po 72 hod roztok kolcemidu a nechali sme pôsobiť v termostate pri 37 °C 10 min. Následne sme ich centrifugovali. Po centrifugácii sme roztok odsali a ponechali iba sediment, ktorý sme resuspendovali v 4 ml hypotonického roztoku (0,075 M KCl) a skúmavku sme týmto roztokom doplnili do výsledného objemu 8 ml. Nasledovala inkubácia 30 min pri 37 °C a opätovná centrifugácia. Po kvapkách a za nepretržitého trepania sme k sedimentu pridávali Carnoyov fixačný roztok. Materiál sme ďalej inkubovali pri laboratórnej teplote 30 min (pri 24-hodinovej kultivácii) alebo 60 min (pri 72-hodinovej kultivácii) a následne sme scentrifugovali. Tento postup sme zopakovali aspoň trikrát, avšak už bez inkubácie. Na záver sme sediment ešte resuspenzovali vo fixačnom roztoku a nakvapkali na suché namrazené podložné sklá (–22 °C), ktoré sme ponechali na vzduchu do vysušenia. Jedno sklo sme využili pre FISH a druhé sa zafarbilo pre potreby konvenčnej cytogenetiky [2]. Pri vyšetrení FISH metódou sme na identifikáciu aberácií použili fluorescenčne značené sondy diagnostikujúce prestavby charakteristické pre AML – MLL, C-MYC, BCR/ABL, RARA, ETV6(TEL), AML1/ETO, CBFB. Preparáty pripravené 24/72-hodinovou kultiváciou sme prezerali pod svetelným mikroskopom pri 100-násobnom zväčšení – kontrolovali sme kvalitu, hustotu a čistotu skiel a ďalšími postupmi pripravili tak, aby ich bolo možné priamo hodnotiť pod fluorescenčným mikroskopom pri 1 000-násobnom zväčšení s vhodnými filtrami podľa použitej sondy. Na identifikáciu molekulovo-genetických markerov charakteristických pre AML ochorenie sme použili nasledujúce gény BCR/ABL, CBFB/MYH, AML1/ETO, PML/RARA-bcr1,2,3, FLT3-ITD a D835, MLL PTD, NPM1, expresia génov BAALC a WT1. Izoláciu celkovej RNA sme realizovali pomocou trizolu, podľa protokolu odporúčaného výrobcom. Koncentráciu a čistotu RNA sme zisťovali spektrofotometrickým meraním pri vlnovej dĺžke 260 nm a 280 nm použitím prístroja eppendorf BioPhotometer. Izolovanú RNA sme uschovali pri –20 °C. Pri syntéze cDNA sme vychádzali z 1 000 µg RNA/reakciu. Vypočítaný objem RNA sme nariedili vodou do výsledného objemu 11 µl. Pridali sme 1 µl primerov tzv. náhodných hexamérov. Reakčnú zmes sme inkubovali 5 minút pri 70 °C. Potom sme vzorky 5 min chladili pri 4 °C. Hneď po schladení sme pridali 4 µl 5× reakčného pufru, 2 µl dNTP a 1 µl RNáza inhibítoru. Po krátkom zamiešaní jednotlivých vzoriek sme ich preniesli do prístroja Gene Amp® PCR System 9700 (Applied Biosystems), v ktorom prebiehal prepis RNA do cDNA pri teplotnom profile: 25 °C – 15 min; 42 °C – 60 min; 70 °C – 10 min; 4 °C – podľa potreby. Pri 25 °C sme po 5 minútach prebiehajúceho cyklu pridali 1 µl reverznej transkriptázy. Po ukončení prepisu sme produkt zriedili s vodou v pomere 1 : 1 a uskladnili pri –20 °C. Na identifikáciu jednotlivých molekulovo-genetických markerov sme ďalej použili metódu RT-PCR. Postupovali sme podľa protokolov odporúčaných výrobcom pre dané markery. Podľa týchto protokolov sme pripravili reakčné zmesi modifikované pre každú zisťovanú anomáliu. Každá zmes obsahovala mix enzýmov potrebných pre priebeh PCR reakcie, začiatočné a koncové primery, vodu bez nukleáz a cDNA ako produkt reverznej transkripcie. RT-PCR prebiehali podľa teplotných profilov opäť prispôsobených pre daný marker. Na identifikáciu falošne pozitívnych a falošne negatívnych výsledkov RT-PCR reakcie sme spolu s vyšetrovanými vzorkami analyzovali pozitívnu kontrolu PK (pozitívny pacient) a negatívnu kontrolu NK (voda bez nukleáz). Získané PCR produkty sme analyzovali elektroforézou v agarózovom géli. V niektorých prípadoch, ako napríklad pri detekcii mutácie FLT3 D835, sme museli produkt PCR ďalej identifikovať pomocou restrikčnej analýzy. Reakčné médium tvorili produkt PCR reakcie, Buffer B2, enzým ECOR V a voda bez nukleáz. Následná PCR prebiehala podľa teplotného profilu odporúčaného výrobcom. Produkty restrikčnej analýzy sme hodnotili elektroforézou v agarózovom géli. Elektroforetická separácia produktov PCR prebiehala v 2% alebo 5% agaróze v tlmivom roztoku TBE pri jednosmernom elektrickom prúde s napätím 120 V po dobu 120 min, respektíve 30 min. Spolu s PCR produktmi sme vždy pustili aj pozitívnu kontrolu, negatívnu kontrolu a vodu.
Výsledky
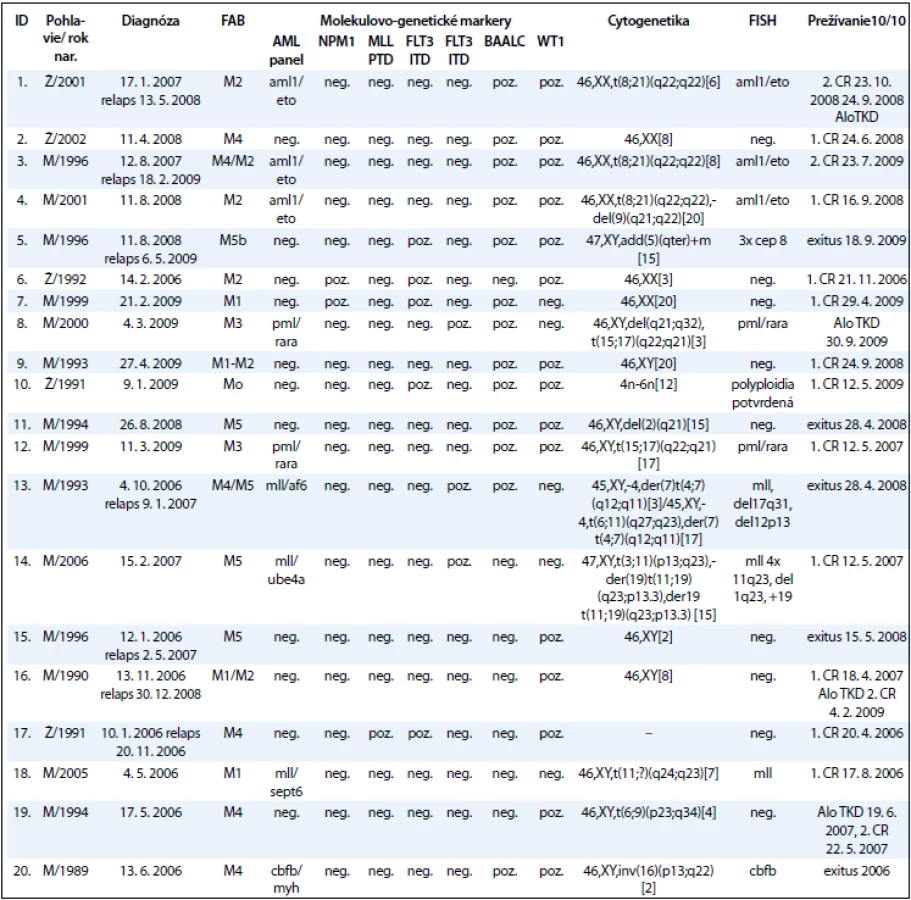
Súbor pacientov tvorí 20 detí s AML (do dovŕšenia 18. roku života), ktoré boli v období rokov 2008–2010 vyšetrení na Oddelení onkologickej genetiky Národného onkologického ústavu (tab. 2). Súbor zahŕňa 5 dievčat a 15 chlapcov. Do súboru neboli zaradení 4 AML pacienti s Downovým syndrómom. Priemerný vek pacientov v čase stanovenia diagnózy bol 11 rokov. Najmladší pacient bol v čase stanovenia diagnózy vo veku 1 roku a najstarší 18 rokov. V čase stanovenia diagnózy boli pacienti zaradení do príslušných FAB skupín. Najviac ich bolo v M4 (6 detí, 30 %) a najmenej v M0 (1 dieťa, 5 %) skupine podľa FAB klasifikácie.
Výsledky cytogenetického vyšetrenia
Pri cytogenetickom vyšetrení bol normálny karyotyp zistený u 6/20 pacientov. U 13/20 pacientov boli detekované kvalitatívne alebo kvantitatívne zmeny karyotypu. U 1/20 pacienta nebolo možné karyotyp vyšetriť. U 6/13 pacientov (46 %) s abnormálnym karyotypom boli detegované chromozómové aberácie asociované so zlou prognózou, u 4 pacientov (31 %) so strednou prognózou, a len u 3 pacientov (23 %) sme zistili karyotyp s aberáciami s dobrou prognózou (tab. 2).
Výsledky molekulovo-genetického vyšetrenia zobrazuje tab. 3 a graf 2 a poukazujú na to, že jednotlivé molekulovo-genetické markery sa vyskytovali u pacientov s rôznou frekvenciou. Zmeny v AML paneli boli detekované u 9 pacientov z 20 (45 %). Najpočetnejšie boli zastúpené AML/ETO v 3 prípadoch a MLL, taktiež v 3 prípadoch. U 2 pacientov bol potvrdený fúzny gén PML/RARA. U ďalších pacientov sa vyšetrením metódou FISH vyskytol len jeden z nasledovaných genetických markerov: cep8, del17q31, del12p13, del11q23, CBFB a polyploidia. Markery asociované so zlou prognózou, ako mutácia FLT3 ITD a MLL PTD/MLL translokácie, boli zistené u 9/20 pacientov (45 %), pričom 5/20 mali mutáciu FLT3 ITD a 4/20 MLL PTD/MLL translokácie, z toho MLL PTD sme zistili u 1 pacienta. Markery asociované s dobrou prognózou, mutácia FLT3 D835 a NPM1, boli detegované u 5/20 (25 %), z toho mutácia FLT3 D835 u 5/20 pacientov (15 %) a NPM1 u 2/20 pacientov (10 %). Zvýšené expresie WT1 génu a BAALC génu patria medzi prognosticky nezávislé markery. Nárast expresie niektorého z génov bol prítomný u 18/20 pacientov, t. j. u 90 % pacientov. U viac ako polovice týchto pacientov sa vyskytla nadexpresia u oboch génov súčasne. Nadexpresiu sme hodnotili nárast expresie o 100 % a viac oproti fyziologickej hodnote expresie. Za zvýšenú expresiu sme označili nárast expresie o 10 % až 100 % voči fyziologickej úrovni. Nárast expresie do 10 % sa klasifikuje ako mierne zvýšená expresia.


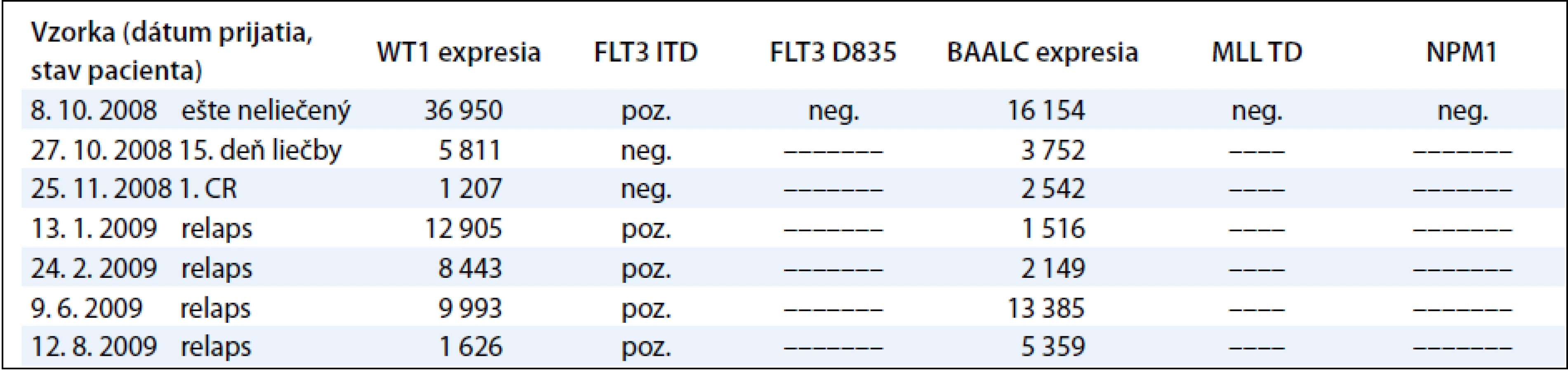
Sledovanie expresie génov WT1 a BAALC u detí s AML počas liečby demonštrujeme na príklade 9-ročného chlapca s diagnózou AML-M3, cytogenetickým nálezom 46,XY,t(15;17)(q22; q32) a FISH nálezom s 83,5 % jadierkami s prestavbou PML/RARA a kvalitatívnym potvrdením fúzneho génu PML/RARA s miestom scelenia bcr3. V čase diagnózy 11. 3. 2009 sme zistili nadexpresiu génov WT1 a BAALC bez mutácie v génoch FLT3 ITD, FLT3 D835, MLL TD a NPM1. Počas špecifickej liečby pre AML pacientov sme po indukcii v 35. deň liečby zistili výrazný pokles expresie génu WT1 a BAALC (tab. 4, graf 3). Po 2 mesiacoch nastala 1. klinická remisia (1. CR) a pokles hladiny expresie WT1 génu na fyziologickú hodnotu a pretrvávanie zvýšenej expresie u génu BAALC. U druhého pacienta, 12-ročného chlapca s AML-M5 a cytogenetickým nálezom 47,XY,add(5)(qter),+m[15], FISH vyšetrením s 35 % vyšetrenými jadrami s trizómiou chromozómu 8 sme metódou molekulovo-genetickej diagnostiky vylúčili prítomnosť translokácií charakteristických pre AML. V čase diagnózy 8. 10. 2008 sme zistili nadexpresiu génov WT1 a BAALC s mutáciou v géne FLT3 ITD. Mutácie v génoch, FLT3 D835, MLL TD a NPM1 sme vylúčili. Počas špecifickej liečby pre AML pacientov sme po indukcii v 15. deň liečby zistili výrazný pokles expresie génu WT1 a BAALC a elimináciu nádorových buniek s FLT3 ITD (tab. 5 a graf 4). Po 35 dňoch nastala 1. klinická remisia (1. CR) a výrazný pokles hladiny expresie WT1 a BAALC génov bez dosiahnutia fyziologickej hodnoty. Po 2 mesiacoch narástla hladina expresie oboch génov WT1 na 10-násobnú hodnotu, pričom bol zaznamenaný relaps ochorenia na hematologickej úrovni. Hladina expresie génu BAALC stúpla 2-násobne a bol potvrdený klon s mutáciou FLT3 ITD. Napriek špecifickej liečbe hladina expresie oboch génov narastala a pacient po stanovení relapsu ochorenia po 8 mesiacoch ochoreniu podľahol. Výskyt molekulových markerov v súbore poukazuje na to, že u viac ako polovice pacientov (11/20, 55 %) bola zistená prítomnosť až 3 molekulovo-genetických markerov. U 6 pacientov (30 %) to boli 2 markery a u 3 pacientov (15 %) 1 marker. Pri porovnaní prítomnosti molekulových markerov u pacientov bez prítomnosti BCR/ABL, CBFB/MYH, AML1/ETO, PML/RARA-bcr1,2,3 sme zistili súčasný výskyt 3 ďalších molekulových markerov typu FLT3-ITD a D835, MLL PTD, NPM1, expresie génov BAALC a WT1 u 5/20 pacientov (45 %). U rovnakého počtu pacientov (5/11) sme zistili súčasný výskyt 2 zo sady týchto ďalších markerov. V jednom prípade bol prítomný len jeden z týchto markerov.


Diskusia
U AML boli doposiaľ identifikované mnohonásobné chromozómové a génové aberácie, korelujúce s biologickými a klinickými vlastnosťami ochorenia [13]. Z klinického hľadiska predstavujú jednotlivé chromozómové aberácie dôležitý prognostický a terapeutický faktor, napomáhajúci stanoviť optimálnu terapiu a efektívne ju modifikovať v ďalšom priebehu liečby. V tejto štúdii sme sa zaoberali zostavením súboru detských pacientov s AML a sledovaním výskytu genetických aberácií. V našom súbore bola najpočetnejšou skupinou skupina pacientov s AML–M4, ktorá tvorila až 30 % všetkých detských pacientov s AML. V literatúre sa uvádza najviac zastúpená skupina AML M0–M2, ktorá tvorí viac ako 40 %. Aj v prezentovanom súbore sa nachádza 45 % pacientov s morfológiou AML M0–M2, čo sa zhoduje s literárnym údajom [18]. Metódou konvenčnej cytogenetiky sme u 65 % detských pacientov s AML vyšetrili chromozómové aberácie a normálny karyotyp u 35 % detí s AML. V literatúre sa často uvádza výskyt približne 50 % pacientov s normálnym karyotypom bez chromozómových aberácií. V našom súbore sa podarilo vyšetriť väčší počet, čo možno pripísať výbornej kvalite štandardného vyšetrovacieho postupu pri kultivácii a spracovanie materiálu. Podľa genetického nálezu sme mohli AML pacientov zaradiť do prognostických skupín. Do prvej skupiny patrilo 23 % pacientov s dobrou prognózou, 31 % so strednou a 46 % so zlou prognózou. Teda u 54 % pacientov predpokladáme priaznivý priebeh ochorenia pri špecifickej liečbe. Ako v literatúre, tak aj v našom súbore sme zistili, že väčšina pacientov umiera rýchlo po stanovení diagnózy (do 2–3 rokov). Preto sa vyvíja snaha u pacientov bez nálezu chromozómových aberácií alebo u tých s priaznivou a strednou prognózou hľadať ďalšie prognostické genetické ukazovatele, ktoré by pomohli stratifikovať takýchto pacientov do užších prognostických skupín. Z tohto dôvodu sa zvyšuje záujem o molekulovo-genetické markery, ktoré by mohli napomôcť v tejto stratifikácii. Do dnešných dní boli v cytogeneticky normálnych myeloblastových bunkách identifikované mutácie viacerých génov. K takýmto mutáciám patria interná tandemová duplikácia a bodová mutácia v tyrozín kinázovej doméne FLT3 génu, parciálna tandemová duplikácia MLL génu, mutácie v génoch CEPBA, NMP1, NRAS, ako aj zvýšená expresia BAALC a WT1 génu. Na Oddelení onkologickej genetiky NOÚ bolo v poslednom období zavedené vyšetrovanie mutácií FLT3 ITD a D835, MLL PTD, NPM1 a expresie génov BAALC a WT1. Jednotlivé molekulovo-genetické markery sa vyskytovali u našich pacientov s rôznou frekvenciou. Metódami molekulárnej genetiky sme zachytili molekulovo-genetické markery asociované so zlou prognózou, t. j. mutácia FLT3 ITD a MLL PTD, 6/20 pacientov (30 %). Prítomnosť mutácie FLT3 ITD sme zistili u 5 detí (25 %) – troch dievčat a troch chlapcov. V literatúre sa incidencia mutácie FLT3 ITD uvádza u asi 15 % detských pacientov s AML [15]. V našom súbore bola incidencia vyššia u dievčat ako u chlapcov (3 : 2/dievčatá : chlapci). U 2 FLT3 ITD pozitívnych pacientov sme zistili normálny karyotyp, u dvoch sme zistili prítomné chromozómové aberácie. U jedného pacienta nebolo možné karyotyp vyšetriť. V 1 prípade chlapec s touto mutáciou exitoval. U tohto pacienta sme detegovali chromozómové aberácie asociované so zlou prognózou. FLT3 ITD mutácie sa nachádzajú často spolu s mutáciou v géne NPM1. V našom súbore sme túto koreláciu zachytili u 2/5 pacientov. O výskyte mutácie MLL PTD sa veľa nevie. V našom súbore sme túto aberáciu zachytili len u jednej pacientky (5 %). V literatúre sa výskyt pohybuje okolo 2,5 % [14]. U našej pacientky bola zároveň prítomná mutácia FLT3 ITD a zvýšená expresia WT1 génu, pričom karyotyp nebolo možné vyšetriť. Molekulovo-genetické markery asociované s dobrou prognózou, t. j. bodová mutácia FLT3 D835 a NPM1, sme detegovali v 5/20 prípadoch (25 %). Bodovú mutáciu FLT3 D835 sme zistili u 3 detí (15 %). Je to o niečo málo viac, ako sa uvádza v literatúre (5–10 %) [12]. V nami sledovanom súbore všetci FLT3 D835 pozitívni pacienti boli chlapci. U 2 pacientov bol normálny cytogenetický nález a nadexpresia WT1 génu. Jeden z týchto chlapcov exitoval. U tretieho chlapca sme zistili chromozómovú aberáciu spojenú so strednou prognózou. Mutáciu NPM1 sme zachytili u 2/20 pacientov (10 %). V literatúre sa uvádza výskyt u detských pacientov 2–6 % [13]. Aj keď je mutácia NPM1 asociovaná s dobrou prognózou, u oboch pacientov sme súčasne zistili aj mutáciu FLT3 ITD, ktorá je naopak spojená so zlou prognózou. Obaja pacienti mali normálny cytogenetický nález. U jedného chlapca sme zistili nadexpresiu BAALC génu a druhého nadexpresiu WT1 génu. U všetkých AML detských pacientov sme mohli po dosiahnutí cytogenetickej remisie sledovať minimálne jeden molekulový marker. Týmto sme dosiahli precízne sledovanie minimálnej reziduálnej choroby a možnosť zistenia včasného relapsu. Zvýšená expresia WT1 génu a BAALC génu patria medzi prognosticky nezávislé markery. Nárast expresie niektorého z génov bol prítomný u 18/20 pacientov, t. j. u 90 % pacientov. Pri korelácii hladiny expresie génov WT1 a BAALC s klinickým priebehom počas liečby sme zistili, že hladina expresie génu WT1 úzko korelovala s dosiahnutím kompletnej remisie ochorenia, čo sa prejavilo poklesom hladiny expresie na úroveň fyziologickej hodnoty. Táto zhoda sa prejavila rovnako aj u pacienta s relapsom, pričom pri klinickom stanovení relapsu, hladina expresie bola zistená so 100 % nárastom. Sledovaním korelácie génu BAALC s klinickým priebehom u oboch pacientov sme zistili diskrepanciu, pri stanovení kompletnej remisie bola hladina expresie BAALC génu zvýšená. Rovnako u druhého sledovaného pacienta sme pri relapse zaznamenali mierne zvýšenú expresiu génu BAALC, pričom expresia génu WT1 bola 100% zvýšená. Z uvedeného vyvodzujeme záver, že sledovanie expresie génu WT1 je citlivejšie pre stanovenie kompletnej remisie, ako aj včasného relapsu. Odporúčame ho využívať v klinickej onkologickej praxi. Nadexpresiu WT1 génu sme zistili u 11/20 pacientov (55 %). V literatúre sa táto hodnota pohybuje okolo 78 % pacientov [18]. U 15 % pacientov bola zvýšená expresia a u 5 % mierne zvýšená expresia. Ostatní pacienti nemali zvýšené hodnoty expresie WT1 génu. Nadexpresiu BAALC génu sme zachytili u 4/20 pacientov (20 %) a zvýšenú expresiu u 45 %. U ostatných pacientov sme namerali fyziologické hladiny expresie BAALC génu. 4 pacienti z vyšetrovaného súboru exitovali. Časový interval od stanovenia diagnózy po exitus bol v rozmedzí 0–2 roky. U jedného z týchto pacientov bol prítomný normálny cytogenetický nález a mutácia FLT3 D835, ktorá je asociovaná s dobrou prognózou. Tento prípad poukazuje na to, že je potrebné hľadať nové markery, ktoré by nám pomohli ešte presnejšie stratifikovať pacientov. Markery spájané s dobrou prognózou ešte nezaručujú prognosticky lepší priebeh ochorenia. Podarilo sa nám identifikovať prídatné molekulárne zmeny, ktoré v skupine pacientov s normálnym karyotypom umožnia ich presnejšiu stratifikáciu do prognostických podskupín a umožnia sledovať hladinu MRD. U každého jedného pacienta v súbore sme identifikovali aspoň jeden genetický marker.
Záver
Kombináciou metód klasickej cytogenetiky, FISH a špecifických PCR, RT-PCR metód je možné zistiť široké spektrum genetických aberácií u detských pacientov s AML. Zavádzanie ďalších molekulových markerov v diagnostike AML ochorenia umožňuje rozšíriť spektrum genetických markerov charakterizujúcich klonový vývoj leukémie. V mnohých prípadoch umožňujú ďalšie molekulové markery FLT3-ITD a D835, MLL PTD, NPM1 lepšie vyjadrenie sa k prognóze ochorenia, monitorovanie odpovede pacienta na špecifickú liečbu, sledovanie minimálnej reziduálnej choroby a potransplantačného priebehu ochorenia. V súčasnosti sa do popredia dostáva snaha identifikovať tieto prognosticky významné molekulové markery za účelom lepšej stratifikácie pacientov s normálnym karyotypom bez cytogenetického nálezu do prognosticky užších skupín. Rovnako zavedenie diagnostiky genetických markerov môže pomôcť identifikovať u každého pacienta aspoň jeden cytogenetický alebo molekulárny marker na monitorovanie priebehu ochorenia, na predikciu včasného relapsu, alebo na sledovanie efektívnosti terapie.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
doc. MUDr. Denisa Ilenčíková, PhD.
2. Detská klinika LF UK a DFNsP Bratislava
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: denisa.ilencikova@dfnsp.sk
Obdrženo: 10. 5. 2011
Přijato: 22. 11. 2011
Sources
1. Kubisz P et al. Hematológia a transfuziológia. 1. vyd. Bratislava, Praha: Grada 2006.
2. Nováková P, Ilenčíková D. Genetické metódy v onkológii. Onkologia 2010; 5(2): 59–63.
3. Vardiman JW, Thiele J, Arber DA et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114(5): 937–951.
4. Kaušitz J et al. Onkológia. 1. vyd. Bratislava: VEDA 2003.
5. Sršeň Š, Sršňová K. Základy klinickej genetiky a jej molekulárna podstata. 4. vyd. Martin: Osveta 2005.
6. Vojtašák K et al. Vybrané kapitoly z lekárskej biológie a humánnej genetiky. 1. vyd. Bratislava: Univerzita Komenského 2003.
7. Remick DG, Kunkel SL, Holbrook EA et al. Theory and applications of the polymerase chain reaction. Am J Clin Pathol 1990; 93 (4 Suppl 1): S49–S54.
8. Templeton NS. The polymerase chain reaction. History, methods, and applications. Diagn Mol Pathol 1992; 1(1): 58–72.
9. Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ 2005; 29(3): 151–159.
10. Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006; 368(9550): 1894–1907.
11. Krauter J, Wagner K, Schäfer I et al. Prognostic factors in adult patients up to 60 years with AML and translocations of chromosome band 11q23: individual patient data-based meta-analysis of the German AML Intergroup. J Clin Oncol 2009; 27(18): 3000–3006.
12. Meshinchi S, Alonzo TA, Stirewalt DL et al. Clinical implications of FLT3 mutations in pediatric AML. Blood 2006; 108(12): 3654–3661.
13. Reneville A, Roumier C, Biggio V et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 2008; 22(5): 915–931.
14. Balgobind BV, Hollink IH, Reinhardt D et al. Low frequency of MLL-partial tandem duplications in paediatric acute myeloid leukaemia using MLPA as a novel DNA screenings technique. Eur J Cancer 2010; 46(10): 1892–1899.
15. Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood 2009; 113(23): 5951–5960.
16. Hollink IH, Zwaan CM, Zimmermann M et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 2009; 23(2): 262–270.
17. Blaise A et al. WT1 Overexpression after Induction Therapy in Children AML is Associated with Higher Risk of Relapse. Blood ASH Annual Meeting 2004; 104: Abstract 2998.
18. Leukaemia § lymphoma research. Available from: http://www.llresearch.org.uk/en/1/infdispataml.html.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2012 Issue 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Žilní vstupy v onkologii
- Využitie elektroimpedančnej tomografie v diagnostike karcinómu prsníka
- Identifikácia molekulárnych markerov u detí s akútnou myeloblastovou leukémiou (AML)
- Šestileté sledování pacienta s mnohočetnou angiomatózou postihující skelet, břišní i hrudní dutinu a stěnu trávicí trubice