
Inhibitory proteazomu v léčbě mnohočetného myelomu
Proteasome Inhibitors in Treatment of Multiple Myeloma
Multiple myeloma, a plasma cell malignancy, still remains a hard-to-treat hematological disease that desperately needs new therapy targeting plasmocytes but also the bone marrow microenvironment. Clonal plasmocytes are characterized by increased regulation of ubiquitin-proteasome pathway which augments their sensitivity to proteasome inhibitors. Treatment strategies based on proteasome inhibitors belong to the era of new drugs, and they have become increasingly important for treatment of multiple myeloma in recent years. Bortezomib became the first proteasome inhibitor approved for the treatment of multiple myeloma and showed remarkable anti-myeloma activity. However, despite its high efficiency, a large proportion of patients have became bortezomib resistant. The second generation of proteasome inhibitors – carfilzomib, marizomib and MLN9708 – were developed in an effort to overcome bortezomib-resistance and find proteasome inhibitors with a better toxic profile. These drugs brought a chance that multiple myeloma would become a chronic disease.
Key words:
multiple myeloma – proteasome inhibitors – bortezomib – carfilzomib – marizomib – MLN9708
:
L. Kubiczková; J. Matějíková; L. Sedlaříková; F. Kryukov; R. Hájek; S. Ševčíková
:
Babákova myelomová skupina, Ústav patologické fyziologie, LF MU Brno
:
Klin Onkol 2013; 26(1): 11-18
:
Reviews
Mnohočetný myelom, maligní onemocnění plazmatických buněk, zůstává stále velmi obtížně léčitelným hematoonkologickým onemocněním, pro které je nutné hledat nové možnosti terapie ovlivňující jak plazmocyty samotné, tak i mikroprostředí kostní dřeně. Klonální plazmocyty se vyznačují zvýšenou regulací ubikvitin-proteazomové kaskády, což zvyšuje jejich citlivost k působení inhibitorů proteazomu. Léčebné přístupy využívající inhibitory proteazomu patří do éry nových léků a v posledních letech se ukázaly být velice důležitou součástí léčby pacientů s mnohočetným myelomem. Prvním inhibitorem proteazomu schváleným pro léčbu mnohočetného myelomu se stal bortezomib, který vykazoval silné antimyelomové účinky. Bohužel, navzdory jeho vysoké účinnosti se u velkého procenta pacientů s mnohočetným myelomem pacientů po čase objevuje rezistence k jeho působení. Ve snaze překonat rezistenci k bortezomibu a vyvinout inhibitor proteazomu s lepším toxickým profilem byly vyvinuty inhibitory proteazomu druhé generace – carfilzomib, marizomib a MLN9708, které by mohly nadějně změnit průběh mnohočetného myelomu v onemocnění chronické.
Klíčová slova:
mnohočetný myelom – inhibitory proteazomu – bortezomib – carfilzomib – marizomib – MLN9708
Úvod – inhibitory proteazomu
Proteazom jako nová buněčná struktura byl poprvé identifikován na počátku 70. let skupinou doktora Harrise [1]. Mnoho dalších objevů bylo učiněno později, koncem let 70. a počátkem let 80. v laboratoři prof. Avrama Hershka, a bylo zjištěno, že jeho funkce spočívá v ATP-dependentní degradaci intracelulárních proteinů a jeho specifita je dána interakcí pouze s takovými proteiny, které jsou označeny polyubikvitinovým řetězcem anebo obsahují specifickou sekvenci aminokyselin [2,3]. Za tento významný objev získali prof. Ciechanover, Hershko a Rose v roce 2004 Nobelovu cenu za chemii.
Proteazom je útvar cylindrického tvaru. Skládá se ze čtyř homologních prstenců tvořených sedmi podjednotkami α nebo β, které jsou nad sebou uspořádány v pořadí α-β-β-α [4]. V této struktuře lze rozeznat tři různé typy proteolytických podjednotek – β1, β2 a β5. Každá z nich obsahuje aktivní místo na svém N-konci, kde se nachází zbytek aminokyseliny threoninu (Thr1) [5]. Ačkoli proteazom obsahuje více katalytických míst, k inhibici jeho funkce postačuje pouze zablokování podjednotky β5, která vykazuje chymotrypsinovou aktivitu [6]. V hematopoietických buňkách je hlavním typem proteazomu jeho inducibilní izoforma – imunoproteazom, jehož výskyt koreluje s hladinou cytokinů. Je charakterizován nahrazením proteolyticky aktivních podjednotek β1, β2 a β5 jejich ekvivalenty β1i (LMP2), β2i (MELC1) a β5i (LMP7) [7].
Nejčastějšími proteiny určenými k degradaci jsou špatně sbalené proteiny a proteiny s krátkým biologickým poločasem, které mají většinou regulační funkci [8]. Produkty proteolytické reakce jsou pak oligopeptidové řetězce s průměrnou délkou 8–12 aminokyselin [9].
Použití inhibitorů proteazomu (IP) patří v současné době mezi jedny z nejúspěšnějších strategií pro léčbu mnohočetného myelomu (MM), nádorového onemocnění způsobeného maligní transformací B lymfocytů s charakteristickou klonální proliferací a akumulací plazmatických buněk v kostní dřeni [10]. IP jsou zpravidla krátké peptidy, ke kterým je kovalentně připojen farmakofor – skupina atomů, která se váže do katalytických míst proteazomu, a tím zabraňuje jeho správné funkci. Konečným důsledkem inhibice proteazomu v myelomových buňkách je indukce apoptotických drah, překonání rezistence ke konvenční chemoterapii a senzitizace vůči dalším terapeutikům.
V tomto přehledovém článku se zaměříme na mechanizmus účinku a roli IP v léčbě mnohočetného myelomu.
První generace inhibitorů proteazomu
Bortezomib
Bortezomib, známý také pod označením PS-341 a komerčním názvem Velcade (Millenium Pharmaceuticals), je prvním a jediným IP, který byl doposud oficiálně schválen pro klinickou praxi. Po chemické stránce se jedná o dipeptidylový derivát kyseliny borité se sumárním vzorcem C19H25BN4O4 (obr. 1).
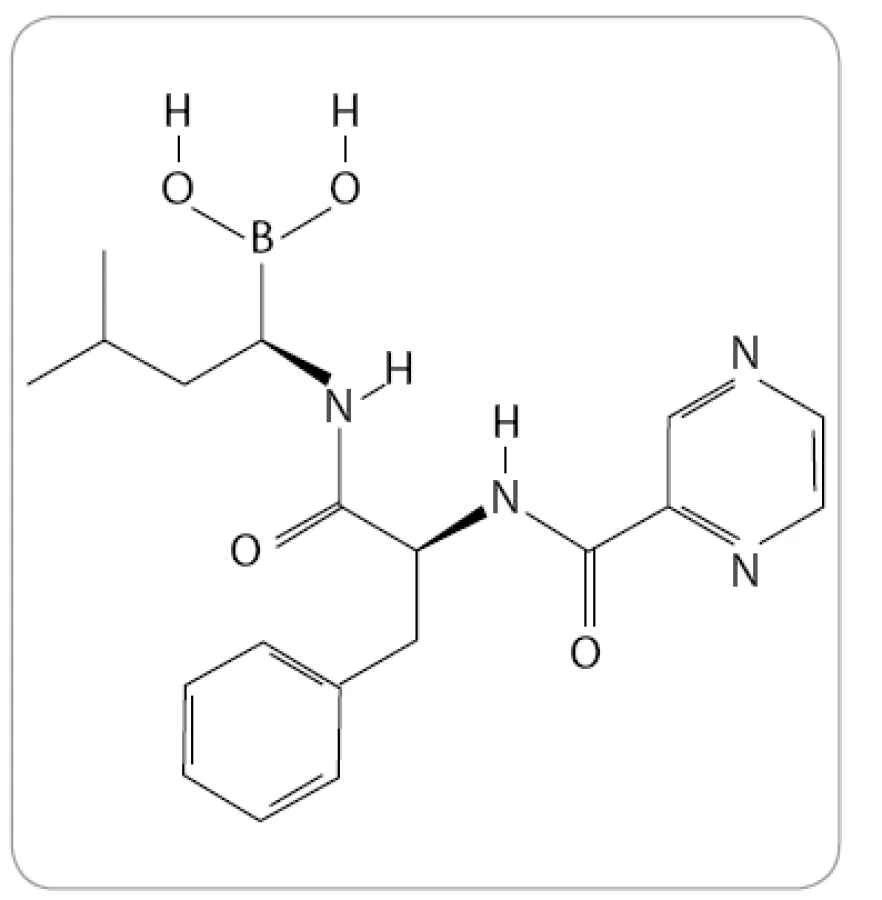
Poprvé byl bortezomib syntetizován v polovině 90. let minulého století společností Myogenics [11]. O jeho vysoké specifitě, účinnosti a oxidační stabilitě vypověděly výsledky studií in vitro u 60 rakovinných linií [12]. První klinická studie s Velcade pro léčbu hematologických malignit byla zahájena v listopadu 1999. Tým pod vedením dr. Orlowského ve studii s nízkými dávkami léku, sloužícími pouze k ověření jeho bezpečnosti, zaznamenal úplné vymizení příznaků (CR) MM u 47leté pacientky. U ostatních osmi pacientů z celkového počtu jedenáct došlo alespoň k minimální léčebné odpovědi (MR) nebo stabilizaci onemocnění [13]. Výsledek to byl natolik převratný, že po ověření v dalších fázích klinických studií vedl k urychlenému schválení bortezomibu pro léčbu relabujícího a refrakterního myelomu v roce 2003 v USA a o rok později také v České republice [14,15].
Mechanizmus účinku
Mechanizmus inhibice proteazomu bortezomibem spočívá v jeho kovalentní vazbě na β5 podjednotku, případně LMP7 podjednotku imunoproteazomu. S nižší afinitou se bortezomib váže také na podjednotky β1 a β2 [16]. Rozdílnost v afinitě je dána odlišnými interakcemi postranních řetězců inhibitoru s jednotlivými podjednotkami [17].
Vazebná konformace je zaujímána v podobě antiparalelního β skládaného listu, který je stabilizován vodíkovými můstky mezi atomy hlavního řetězce agens a konzervovanými zbytky katalytických míst (Gly47N, Thr21N, Thr21O a Ala49O). Samotná inhibice je zprostředkována farmokoforovou skupinou, v tomto případě zbytkem kyseliny borité. Atom boru zde kovalentně interaguje s nukleofilem, kterým je volný elektronový pár kyslíku Thr1O. Vzniklá elektronová mezera je stabilizována vazbou Gly47N na hydroxyl boru. Tetrahedrická struktura proteolyticky inaktivního produktu je dále zpevněna vazbou druhého hydroxylu na aminoskupinu v katalytickém místě [17]. Vzniklý adukt je charakterizován nízkým stupněm disociace, a proto, i když se jedná o reverzibilní reakci, zůstává po několik hodin prakticky vysoce stabilní.
Jelikož je proteazom zapojen do obratu intracelulárních proteinů, patří mezi primární důsledky jeho inhibice hromadění nefunkčních proteinů a chyby v signálních drahách, které vyúsťují v narušení adheze myelomových buněk, potlačení novotvorby cév, zastavení buněčného cyklu, omezení odpovědi na poškození DNA a indukci apoptózy [18].
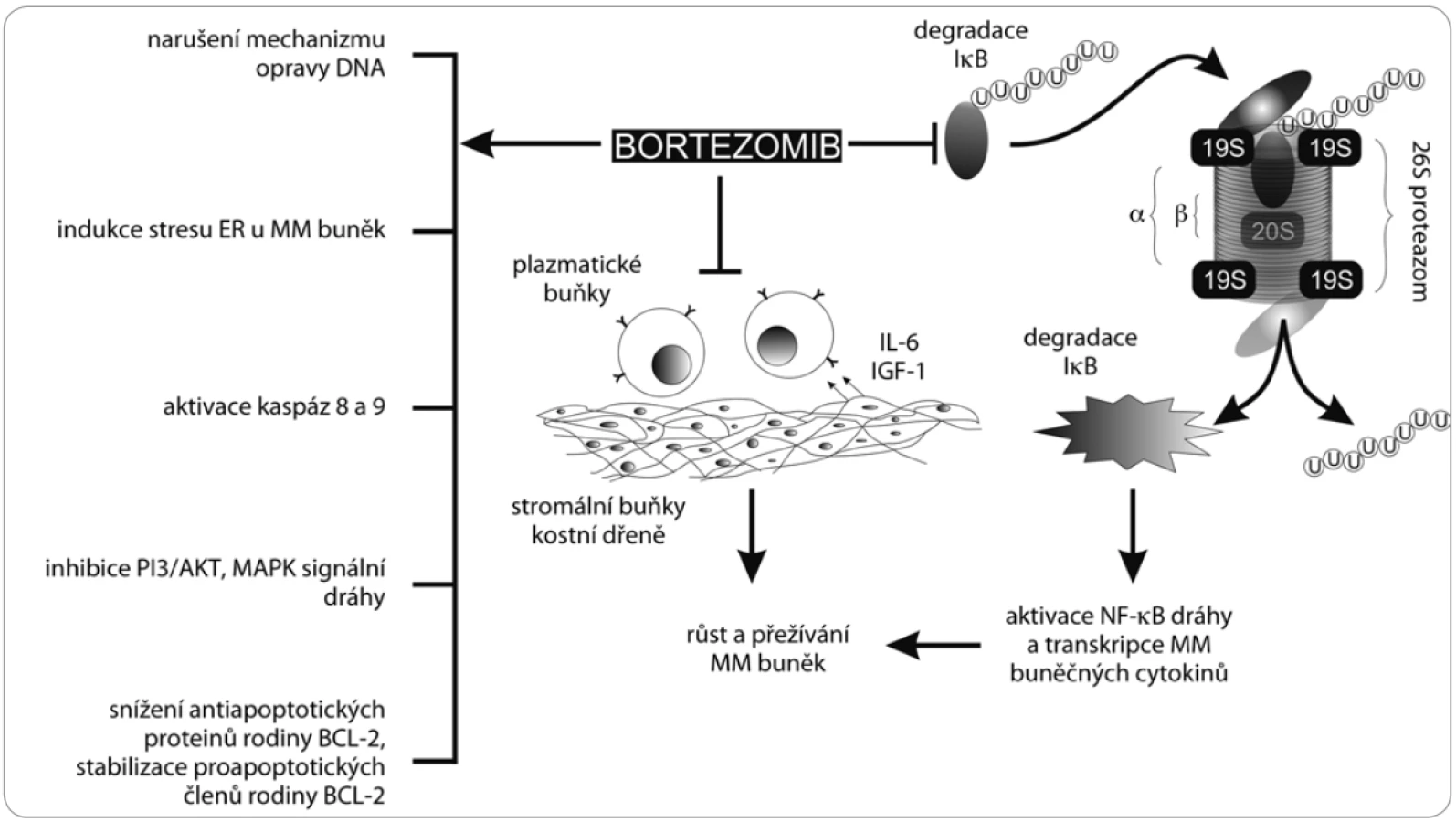
Původní hypotézou hlavního biologického účinku bortezomibu na myelomové buňky byla inhibice transkripčního faktoru NF-κB, a tím zabránění transkripce genů, které blokují apoptózu. Hlavní zástupce NF-κB se nachází v cytoplazmě v podobě inaktivního komplexu s vlastním inhibitorem I-κB, jehož degradace proteazomem je klíčem k aktivaci samotného transkripčního faktoru. Model proteazomové inhibice počítal se zastavením degradace I-κB, a tedy konstitutivní inhibicí NF-κB [19]. Ačkoli se tento předpoklad nepotvrdil, existuje ještě alternativní nekanonická dráha aktivace NF-κB, která by eventuálně mohla být bortezomibem blokována [20].
Klíčovou událostí v navození apoptózy myelomových buněk bortezomibem zůstává aktivace iniciačních kaspáz 8 a 9, které předávají apoptotický signál efektorovým kaspázám štěpícím obsah buňky zevnitř. Iniciační kaspázy mohou být aktivovány nejrůznějšími způsoby. U bortezomibem navozené inhibice proteazomu byla pozorována zvýšená aktivita c-Jun N-terminální kinázy (JNK), která souvisí s apoptózou odehrávající se přes Fas receptor. Fas patří do rodiny TNF receptorů, které po navázání ligandu spouštějí proapoptotický signál, který, pokud není interferován, vyústí v buněčnou smrt. Zablokováním přirozeného obratu intracelulárních proteinů dochází také k disproporcionaci apoptotických proteinů rodiny Bcl-2 ve prospěch jejích proapoptotických členů, což vyústí v permeabilizaci vnější mitochondriální membrány a taktéž v aktivaci efektorových kaspáz. Další cestou je aktivace proteinu p53 s výraznými proapoptotickými účinky a jeho následná stabilizace štěpením příslušného ubikvitin-ligačního enzymu MDM2 [21,22].
Dále bortezomib zabraňuje opravám poškozené DNA, indukuje stres endoplazmatického retikula (ER) v buňkách MM, snižuje adhezi nádorových buněk k buňkám kostní dřeně inhibicí signalizační dráhy MAPK, zabraňuje nádorové angiogenezi a podílí se na apoptóze osteoklastů a diferenciaci osteoblastů [23,24] (obr. 2).
Klinické studie
Klinické studie fáze II, SUMMIT a CREST, potvrdily antimyelomový účinek bortezomibu, který byl pozorován ve studiích preklinických fází a v první fázi klinického výzkumu [25,26]. Do studie SUMMIT bylo zařazeno 202 pacientů, z nichž 193 bylo následně hodnoceno. Léčebné odpovědi dosáhlo 35 % z nich, přičemž kompletní remise byla pozorována v sedmi případech, a to ve velmi krátké době (medián odpovědi 1,3 měsíce). U dalších 24 % pacientů byla pozorována stabilizace nemoci. Navíc, 74 pacientů s nedostatečnou odpovědí na monoterapii bortezomibem dostalo v kombinaci dexametazon a v 13 případech bylo dosaženo následného zlepšení léčebné odpovědi [25].
Mezinárodní randomizovaná klinická studie fáze III APEX srovnávala účinnost bortezomibu v porovnání s dexametazonem. Do studie bylo zapojeno 669 pacientů s relabujícím MM, z nichž bylo nakonec vyhodnoceno 627. Již při prvních výsledcích bylo zřejmé, že bortezomib je signifikantně mnohem účinnější než dexametazon v léčebné odpovědi (38 % vs 18 %), kompletní remisi (6 % vs < 1 %) i v jednoročním přežití (80 % vs 66 %) [18].
Klinická studie fáze III VISTA byla podkladem pro schválení bortezomibu pro primoléčbu. Cílem její pozdější analýzy bylo potvrdit, že kombinace bortezomibu s melfalanem a prednisonem zvyšuje celkové přežití nemocných [27].
Minulý rok vyšla studie francouzské skupiny, která porovnávala použití bortezomibu subkutánně (s.c.) proti intravenóznímu podávání (i.v.) s překvapivými výsledky. Ve studii bylo hodnoceno celkem 222 nemocných randomizovaných v poměru 2 : 1 (s.c. : i.v.). Z výsledků studie vyplývá, že všechny sledované parametry související s léčebnou účinností léků byly velmi podobné a nezávislé na použití aplikační cesty. Ale četnost nežádoucích účinků při podkožním podání byla pro určité typy toxicit nejméně o 10 % nižší (gastrointestinální, dýchací potíže a potíže související s mediastinem a hrudníkem, průjem), především však byla nižší četnost výskytu periferní polyneuropatie, která při intravenózním podání bortezomibu znamená závažný problém. Periferní neuropatie stupně 2 a výše byla pozorována u 38 % při podkožním podání oproti 53 % u nemocných s intravenózním podáním. Pro polyneuropatie stupně 3–4 byly poměry rovněž ve prospěch podkožního podání (6 % vs 16 %). Při podkožním podání nelze konstatovat, že by poškození nervů přestalo být problémem, ale jde o významnou redukci četnosti nežádoucího účinku výhradně změnou cesty podání [28]. V EU se přechází na podání bortezomibu jednou týdně, lze tedy očekávat – v kombinaci s podkožním podáním – další redukci četnosti polyneuropatie. Evropská komise EMA již odsouhlasila na základě těchto výsledků možnost podkožního podání bortezomibu v běžné praxi.
Toxicita, rezistence a limity
Původní hypotéza, že biologické účinky inhibice proteazomu bortezomibem jsou nezávislé na typu buňky s fatálním dopadem pro lidský organizmus, byla vyvrácena. Faktem však zůstává, že působení bortezomibu na ostatní buňky organizmu se odráží v podobě relativně vysoké míry nežádoucích účinků. Mezi ty hematologické patří trombocytopenie, kdy s každým podáním bortezomibu dochází k poklesu počtu trombocytů. Dále anémie, dyspeptické poruchy a zejména periferní neuropatie, které bývají hlavním důvodem pro ukončení léčby [13,15,29]. Periferní neuropatie se vyskytuje s incidencí 35–55 % po podání bortezomibu a je pravděpodobně způsobena účinkem bortezomibu na proteázu HtrA2/Omi – stresem indukovanou proteázu důležitou pro přežití nervových buněk a formaci neuritů [30].
Navzdory vysoké účinnosti bortezomibu disponuje až 60 % pacientů primární rezistencí nebo se u nich v průběhu léčby vyvine rezistence sekundární [25]. Doposud bylo identifikováno několik málo molekulárních mechanizmů, pomocí kterých tyto rezistence mohou vznikat. Jedním z nich jsou mutace v propeptidovém lokusu pro β5 podjednotku proteazomu a její nadměrná syntéza [31]. Se zvýšenou rezistencí nádorových buněk také koreluje zvýšená hladina antiapoptotických proteinů rodiny Bcl-2 a proteinů tepelného šoku Hsp 27, 70 a 90 [32,33].
Studie Zhang et al (2011) odhalila, že u buněčných linií rezistentních k bortezomibu zůstala enzymatická aktivita proteazomu inhibovaná i po další léčbě bortezomibem stejně jako u buněčné linie citlivé k tomuto léku. Tyto výsledky naznačují, že mechanizmus rezistence se objevuje až později v signalizační kaskádě [34].
V poslední době se navíc ukázalo, že přírodní produkty obsahující vicinální dioly jsou schopné inhibovat účinek bortezomibu vazbou na zbytek kyseliny borité. Mezi tyto produkty patří zelený čaj obsahující epigalokatechin-3-galát a vitamin C [35,36].
Druhá generace inhibitorů proteazomu
Úspěch bortezomibu vzbudil zájem vědecké obce o proteazomové inhibitory. Optimalizace dávek a kombinace bortezomibu s jinými protinádorovými terapeutiky sice omezily jeho vedlejší účinky a částečně potlačily rezistenci, bylo však jasné, že druhá generace inhibitorů proteazomu může přinést daleko lepší výsledky. Carfilzomib, Marizomib a MLN9708 reprezentují druhou generaci IP a nabízejí řadu výhod v podobě zvýšené účinnosti, bezpečnosti lékového profilu a překonání rezistence k bortezomibu díky své odlišné chemické struktuře, biologickým vlastnostem, mechanizmu účinku, i/reverzibilitě inhibice proteazomu a způsobu užívání [37].
Carfilzomib
Carfilzomib (známý též jako PR-171, Kyprolis, Onyx Pharmaceuticals) je tetrapeptidový epoxyketon se sumárním vzorcem C40H57N5O7 (obr. 3). V preklinických výzkumech byl identifikován jako vysoce potentní IP. Bylo prokázáno, že dokáže navodit apoptózu u bortezomib-naivních i u bortezomibem předléčených myelomových buněk bez zvýšené toxicity a je schopen překonat primární i sekundární rezistenci [38,39]. Ačkoli mechanizmus překonání rezistence nebyl doposud zcela objasněn, jedním z možných vysvětlení může být odlišný způsob a typ vazby, který dovoluje obejít důsledky mutací v genech pro proteazomové podjednotky. Jiným vysvětlením může být jeho vyšší selektivita vůči katalytickým podjednotkám imunoproteazomu, jehož význam doposud nebyl dostatečně reflektován [39–41].
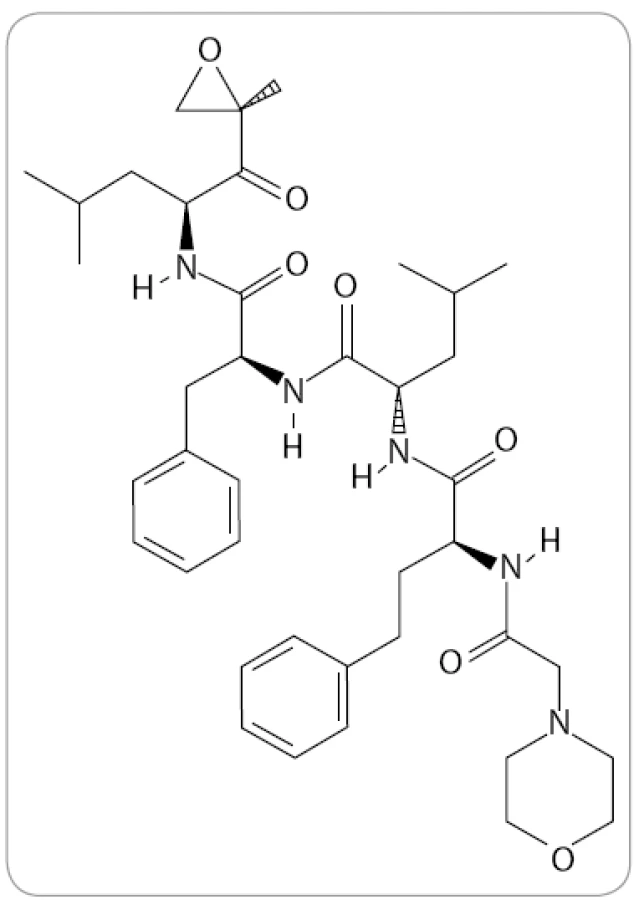
V červenci 2012 schválila FDA carfilzomib pro léčbu pacientů s MM, kteří už mají za sebou více než dvě linie léčby včetně bortezomibu a nějakého imunomodulačního léku a u kterých došlo k progresi onemocnění do 60 dnů od ukončení předchozí léčby (www.onyx.com). Předpokládá se, že nejpozději do dvou let od schválení v USA bude lék schválen i pro Českou republiku.
Mechanizmus působení
Inhibice chymotrypsinové podjednotky proteazomu carfilzomibem je z mechanického hlediska ireverzibilní reakcí snižující aktivitu proteazomu na méně než 20 %. Znovunastolení proteazomové aktivity v buňce je možné pouze nasyntetizováním jednotlivých podjednotek a jejich sestavením do nových proteazomů [38].
Carfilzomib se primárně navazuje na β5 katalytickou podjednotku proteazomu a LMP7 podjednotku imunoproteazomu s vyšší selektivitou než bortezomib [7]. Navázáním epoxybutanového farmakoforu, který vykazuje vysokou specifitu vůči hydroxylové a především aminové skupině N-terminálního Thr1, vzniká šestičlenný morfolinový kruh. Tato intermolekulární cyklizace probíhá dvoustupňovým mechanizmem, kdy tedy kyslík hydroxylové skupiny Thr1 nukleofilně atakuje uhlík epoxyketonu za vzniku hemiacetalu. Druhým krokem je nukleofilní atak α-amino dusíku Thr1 na C2 uhlík epoxidového kruhu, což má za následek vytvoření morfolinového aduktu [42,43].
V buňkách MM vystavených působení carfilzomibu byla pozorována indukce vnější i vnitřní apoptotické kaskády s výrazným zvýšením hladiny kaspázy 3, 7, 8 a 9. Programovaná buněčná smrt byla asociována s aktivací JNK, depolarizací mitochondriální membrány, vylitím cytochromu c, počátečním poklesem fosforylovaného eIF2 v souvislosti se stresem endoplazmatického retikula navozeným akumulací nefunkčních proteinů a zvýšením hladiny proapoptotického proteinu Noxa, který je členem rodiny proteinů Bcl-2 [7,38].
Klinické studie
První klinická studie s carfilzomibem pro léčbu hematologických malignit byla zahájena v září 2005. Studie 29 pacientů prokázala snášenlivost a klinickou aktivitu terapeutika. Objektivní léčebné odpovědi bylo dosaženo u dvou z deseti pacientů s MM [44]. Pokračováním fáze I bylo vyhodnocení bezpečnosti a účinnosti carfilzomibu v kombinaci s lenalidomidem a dexametazonem, standardními léky pro pacienty s relabujícím myelomem. Ačkoli se jedná teprve o první zhodnocení studie, Niesvizky et al (2009) zaznamenali klinický přínos v 78 % případů [45]. U šesti pacientů došlo ke kompletní remisi a nebyly pozorovány závažné vedlejší účinky. Výsledky by měly být potvrzeny v rámci fáze III začínající klinické studie ASPIRE. Nedávno byly publikovány výsledky otevřené multicentrické studie fáze II s carfilzomibem v monoterapii pro relabující/refrakterní myelom. Do studie bylo zařazeno 129 pacientů, z nichž 47,6 % dosáhlo léčebné odpovědi. Ve třech případech došlo ke kompletní remisi [46].
V klinické studii fáze I/II u nově diagnostikovaných pacientů s MM léčených kombinací carfilzomibu, lenalidomidu a nízko dávkovaného dexametazonu dosáhlo 62 % pacientů téměř kompletní remise [47]. Odpověď na léčbu se zlepšovala v průběhu času a tento léčebný režim byl vyhodnocen jako vysoce účinný a dobře tolerovatelný.
Toxický profil carfilzomibu není zdaleka tak bohatý jako u bortezomibu. Periferní neuropatie byly pozorovány u relativně nízkého procenta pacientů a nejčastějšími vedlejšími účinky byly únava, anémie, trombocytopenie a nauzea [46].
Marizomib
Marizomib, známý také jako NPI-0052 nebo Salinosporamid A, je prvním přírodním proteazomovým inhibitorem, který byl zařazen do klinického výzkumu MM. Jedná se o produkt obligátní mořské bakterie, aktinomycéty Salinispora tropica [48]. Po chemické stránce je marizomib bicyklo γ-laktam-β-lakton se sumárním vzorcem C15H20ClNO4 (obr. 4). Na rozdíl od předešlých IP ve své struktuře neobsahuje peptidový řetězec. V preklinických výzkumech s buněčnými liniemi MM bylo prokázáno překonání rezistence na konveční terapie a léčbu bortezomibem spolu s vyšší účinností. Kombinace bortezomibu s marizomibem by mohla umožnit používat takovou koncentraci jednotlivých léků, která nepůsobí na pacienty toxicky a zlepšuje společný antimyelomový účinek jednotlivých léků [49].
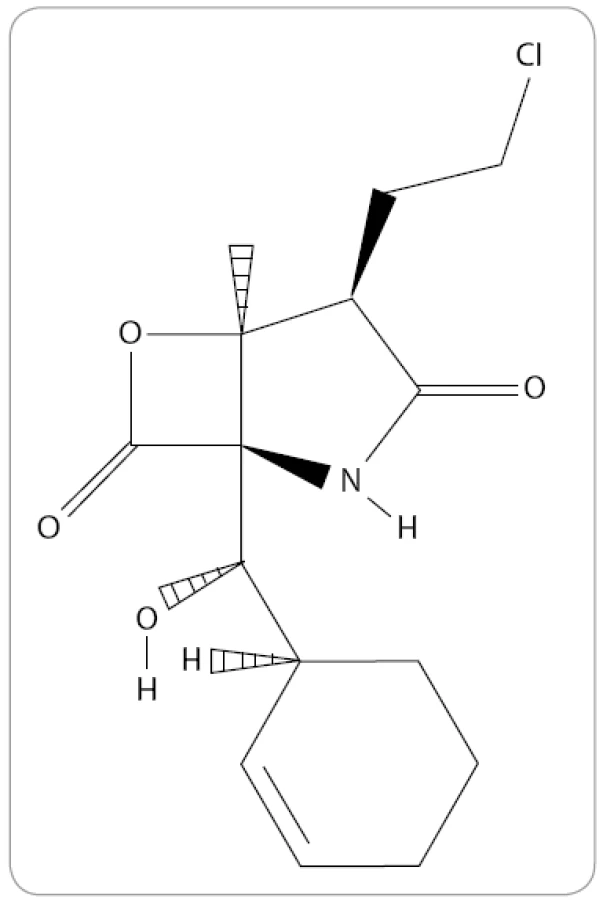
Mechanizmus působení
Marizomib se přednostně navazuje do β5 a β1 katalytického místa proteazomu a s nižší afinitou také na β2 podjednotku. Za ireverzibilnost vazby zodpovídá chloretylová skupina substituující β-lakton. Skupina se navazuje do S2 vazebné kapsy aktivního místa a chlor se chová jako odstupující atom, čímž umožní vytvořit stabilní komplex konečného produktu po acylaci Thr1O β-laktonem inhibitoru [49].
Na rozdíl od bortezomibu, který aktivuje kaspázu 8 i 9, je apoptotický účinek marizomibu zprostředkován především kaspázou 8, v menší míře pak kaspázou 9, ale s odlišným mechanizmem aktivace než u bortezomibu, čímž překonává rezistenci myelomových buněk s mutacemi v genech pro proteiny rodiny Bcl-2. Dochází tedy k uvolnění cytochromu c a proteinu Smac z mitochondrií do cytosolu, generaci kyslíkových radikálů a aktivaci výše zmíněných kaspáz. Studie Chauhana et al (2005) prokázala, že marizomib je schopen indukovat apoptózu u myleomových buněk dokonce i v přítomnosti myelomových růstových faktorů, IL-6 a IGF-1 [32]. Navíc se podílí na zablokování sekrece IL-6 stromálními buňkami kostní dřeně, aniž by došlo k ovlivnění jejich životaschopnosti. Marizomib významně blokuje migraci buněk MM indukovanou VEGF, a potvrzuje tak i své antiangiogenní účinky. U marizomibu byla pozorována jako u jednoho z mála IP i inhibice kanonické NF-κB dráhy a následné sekrece cytokinů [33].
Klinické studie
V současné době se marizomib nachází v doposud nevyhodnocené I. fázi klinické studie [50]. Další klinické studie I právě nabírají pacienty. Jedná se o studii pacientů s relabujícím nebo refrakterním myelomem léčených marizomibem (NPI-0052-101) a studii pacientů trpících některou z pokročilých malignit (NPI-0052-102). Mezi zatím nejčastěji pozorované nepříznivé účinky tohoto léku patří únava, nespavost, nevolnost, průjem, zácpa, bolesti hlavy, halucinace, změny v kognitivních funkcích, ztráta rovnováhy nebo horečky. Významný výskyt periferní neuropatie však nebyl pozorován. Tato data naznačují, že marizomib by mohl být bezpečným lékem bez zkřížené rezistence s ostatními IP a aktivním u pacientů refrakterních k bortezomibu [51,52].
MLN9708
MLN9708 je analog kyseliny borité a první orálně podávaný IP druhé generace, který v preklinických studiích prokázal větší potenciál účinku proti buňkám MM in vivo než bortezomib [53]. Jde o reverzibilní typ IP, který ve vodných roztocích či plazmě okamžitě hydrolyzuje na MLN2238, jeho biologicky aktivní formu (obr. 5) [54]. Je tedy schopen rozsáhlejší distribuce do krve ve stabilní formě a má vyšší farmakodynamický účinek v tkáních [51].
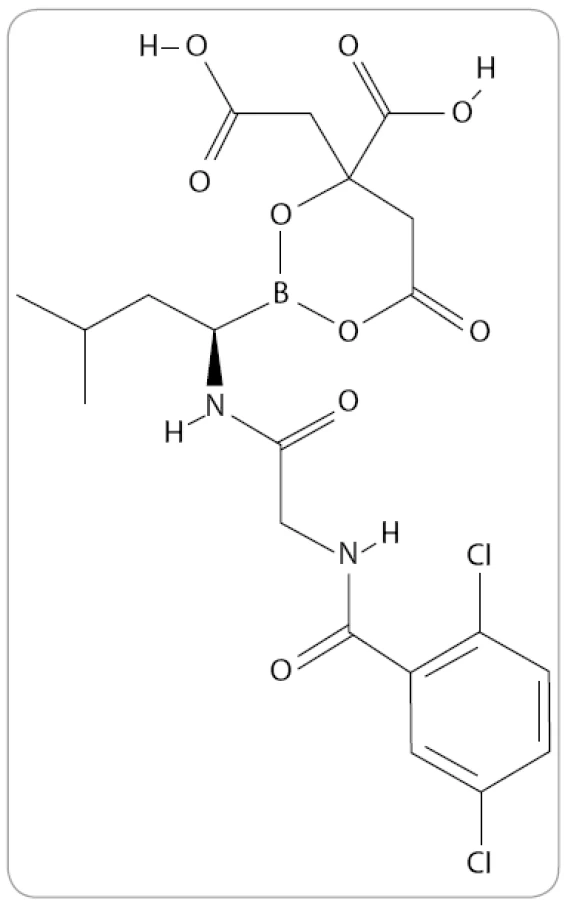
Mechanizmus působení
MLN9708 (MLN2238), stejně jako bortezomib, inhibuje především chymotrypsinovou proteolytickou (β5) podjednotku 20S proteazomu. Navíc je schopen ve vyšších koncentracích inhibovat kaspázovou (β1) a trypsinovou (β2) proteolytickou podjednotku a indukovat akumulaci ubikvitinovaných proteinů [53,55]. K rozpadu 20S podjednotky proteazomu po léčbě MLN9708 dochází šestkrát rychleji než po léčbě bortezomibem.
MLN9708 je zodpovědný za kaspázově dependentní indukci apoptózy myelomových buněk. Po podání dochází k aktivaci kaspáz 8, 9 a 3; dále k navýšení hladiny proteinů p53, p21, NOXA, PUMA, E2F a naopak ke snížení hladiny cyklinu D1 a CDK6. Léčba pomocí MLN9708 vedla také k indukci exprese Bip a CHOP, proteinů stresové odpovědi ER a k účinné inhibici kanonické i nekanonické dráhy NF-κB ovlivňujíce tak sekreci cytokinů důležitých pro růst a přežívání myelomových buněk stromálními buňkami kostní dřeně. Takto jsou narušeny cytoprotektivní účinky mikroprostředí kostní dřeně. Chauhan et al (2011) dále pozorovali snížení počtu buněk nesoucích VEGFR2 a PECAM, což naznačuje inhibici nádorem indukované angiogeneze [53].
Lee et al (2011) ve své studii na myších modelech prokázali, že na rozdíl od bortezomibu, MLN9708 pravděpodobně také zmírňuje osteolýzu kostí, nejčastější příznak MM [54]. Profilování miRNA v buňkách MM léčených MLN9708 prokázalo zvýšenou expresi miR-33b. Zvýšená exprese této miRNA je asociovaná se sníženou schopností migrace a životaschopností buněk MM stejně jako se zvýšenou apoptózou a citlivostí myelomových buněk k léčbě MLN9708. Navíc zvýšená exprese miR-33b vede k negativní regulaci onkogenu PIM-1. Ve studii Tiana et al (2012) bylo tedy naznačeno, že miR-33b funguje jako nádorový supresor, který se podílí na apoptóze myelomových buněk vyvolané léčbou MLN9708, což vede k inhibici růstu nádoru a prodloužení přežití lidských myelomových xenoimplantátových modelů [56].
Klinické studie
V současné době hodnotí několik klinických studií fáze I bezpečnost MLN9708 u různých populací pacientů léčených různými dávkami tohoto léku. Dvě probíhající studie (C16004 a C16003) hodnotí účinek MLN9708 v monoterapii u pacientů s relabujícím či refrakterním myelomem, kteří byli dříve léčeni některým z IP. Mezi nejčastější nepříznivé účinky léčby MLN9708 patří únava, trombocytopenie, nevolnost, průjmy, zvracení a méně často neutropenie. Je však důležité, že po léčbě MLN9708 trpí pouze 10 % pacientů periferní neuropatií.
Dále probíhají studie účinnosti tohoto léku v různých kombinacích u nově diagnostikovaných pacientů. Jde např. o studii účinku MLN9708 v kombinaci s melfalanem a prednisonem (C16006) a v kombinaci s lenalidomidem a nízkými dávkami dexametazonu (C16005 a C16008) [46]. Bylo rovněž zjištěno, že MLN9708 v kombinaci s lenalidomidem vykazuje synergistickou antimyelomovou aktivitu a tato kombinace léků má potenciál pro klinické studie, neboť jde o vysoce účinný perorální léčebný režim bez známek periferní neuropatie [53].
Budoucnost inhibitorů proteazomu
Doposud byly identifikovány čtyři významné mediátory přímé antimyelomové aktivity IP, a to transkripční faktor NF-κB, pro - a antiapoptické faktory, protein p53 a proteiny stresové odpovědi ER. Účinek IP by však měl být chápán jako komplexní děj zahrnující na mnoha místech všechny tyto hlavní mechanizmy, neboť ani samotná inhibice NF-κB ani samotná mutace zajišťující inaktivitu proteinu p53 nezastaví apoptózu myelomových buněk indukovanou IP. Identifikace podrobných mechanizmů působení IP má velký potenciál pro odhalení možných molekulárních cílů pro budoucí léčiva. Příkladem mohou být dnes známé specifické deubikvitinační enzymy, které mohou zastavit degradaci určitých molekul bez nutné inhibice proteazomu.
Na otázku, jak zajistit vyšší selektivitu, účinnost a výrazně omezit vedlejší účinky dnešních IP, mohou nabídnout odpovědi specifické inhibitory imunoproteazomu.
S ohledem na heterogenní podstatu MM lze do budoucna počítat se zavedením genetického screeningu jednotlivých genových sad kódujících klíčové molekuly, na jehož základě by byla zahájena optimální léčba s predikcí stupně účinnosti IP u jednotlivých nemocných.
Závěr
Důležitou a vysoce efektivní strategií při hledání léčebných přístupů k MM je poznání důležité role, kterou v léčbě tohoto heterogenního onemocnění hraje inhibice proteazomu bortezomibem. Toto poznání vedlo k vývoji IP druhé generace, které se liší ve svých chemických strukturách (boronáty, epoxyketony, salinosporamidy), účinnosti a toxických profilech, a poskytují tak nové možnosti pacientům, kteří se stali rezistentními k bortezomibu. Hlavní cesta dalšího výzkumu ve vývoji nových léčiv by se tedy měla ubírat směrem lepšího porozumění mechanizmům účinku IP a především mechanizmu, kterým buňky získávají rezistenci k těmto lékům.
Práce byla podpořena výzkumnými projekty: Ministerstva školství, mládeže a tělovýchovy MSM0021622434, Gran tové agentury ČR GAP304/10/1395 a Interní grantové agentury Ministerstva zdravotnictví NT13190 a NT11154.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
RNDr. Sabina Ševčíková, Ph.D.
Babákova myelomová skupina
Ústav patologické fyziologie
LF MU
Kamenice 5, A3
625 00 Brno
e-mail: sevcik@med.muni.cz
Obdrženo: 2. 10. 2012
Přijato: 15. 10. 2012
Sources
1. Harris JR. The proteins released from intact erythrocyte ghosts‘ at low ionic strength. Biochem J 1971; 122(5): 38P–40P.
2. Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun 1978; 81(4): 1100–1105.
3. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 1998; 17(24): 7151–7160.
4. Unno M, Mizushima T, Morimoto Y et al. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 2002; 10(5): 609–618.
5. Jäger S, Groll M, Huber R et al. Proteasome beta-type subunits: unequal roles of propeptides in core particle maturation and a hierarchy of active site function. J Mol Biol 1999; 291(4): 997–1013.
6. Arendt CS, Hochstrasser M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required foractive-siteformation. Proc Natl Acad Sci U S A 1997; 94(14): 7156–7161.
7. Parlati F, Lee S, Aujay M et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009; 114(16): 3439–3447.
8. Carvalho P, Goder V, Rapoport TA. Distinct Ubiquitin-Ligase Complexes Define Convergent Pathways for the Degradation of ER Proteins. Cell 2006; 126(2): 361–373.
9. Luciani F, Keşmir C, Mishto M et al. A mathematical model of protein degradation by the proteasome. Biophys J 2005; 88(4): 2422–2432.
10. Adam Z, Ščudla V, Neubauer J. Mnohočetný myelom. In: Adam Z, Vorlíček J, Adamová Z et al. Hematologie II: Přehled maligních hematologických nemocí. Praha: Grada 2001 : 461–498.
11. Goldberg AL. Introduction to the proteasome and its inhibitors. In: Adams J. Proteasome inhibtors in cancer therapy. New Jersey: Humana Press 2004 : 17–39.
12. Adams J, Palombella VJ, Sausville EA et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59(11): 2615–2622.
13. Orlowski RZ, Stinchcombe TE, Mitchell BS et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol 2002; 20(22): 4420–4427.
14. Kane RC, Bross PF, Farrell AT et al. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003; 8(6): 508–513.
15. Špička I, Kleibl Z, Hájek R. Bortezomibum. Remedia 2005; 15(3): 193–203.
16. Berkers CR, Verdoes M, Lichtman E et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods 2005; 2(5): 357–362.
17. Groll M, Berkers CR, Ploegh HL et al. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006; 14(3): 451–456.
18. Richardson PG, Sonneveld P, Schuster MW et al. Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352(24): 2487–2498.
19. Hideshima T, Chauhan D, Richardson P et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem 2002; 277(19): 16639–16647.
20. Hideshima T, Ikeda H, Chauhan D et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009; 114(5): 1046–1052.
21. Mitsiades N, Mitsiades CS, Poulaki V et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A 2002; 99(22): 14374–14379.
22. Hideshima T, Mitsiades C, Akiyama M et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003; 101(4):1530–1534.
23. Hideshima T, Richardson P, Chauhan D et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 2001; 61(7): 3071–3076.
24. Mukherjee S, Raje N, Schoonmaker JA et al. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J Clin Invest 2008; 118(2): 491–504.
25. Richardson PG, Barlogie B, Berenson J et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003; 348(26): 2609–2617.
26. Jagannath S, Barlogie B, Berenson J et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol 2004; 127(2): 165–172.
27. Mateos MV, Richardson PG, Schlag R et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated follow-up and impact of subsequent therapy in the phase III VISTA trial. J ClinOncol 2010; 28(13): 2259–2266.
28. Moreau P, Pylypenko H, Grosicki S et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol 2011; 12(5): 431–440.
29. Hájek R, Adam Z, Maisnar V et al. Diagnostika a léčba mnohočetného myelomu. Transfuze Hematol dnes 2012; 18 (Suppl 1): 1–80.
30. Arastu-Kapur S, Anderl JL, Kraus M et al. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res 2011; 17(9): 2734–2743.
31. Oerlemans R, Franke NE, Assaraf YG et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008; 112(6): 2489–2499.
32. Smith AJ, Dai H, Correia C et al. Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem 2011; 286(20): 17682–17692.
33. Chauhan D, Catley L, Li G et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005; 8(5): 407–419.
34. Zhang L, Littlejohn JE, Cui Y et al. Characterization of bortezomib-adapted I-45 mesothelioma cells. Mol Cancer 2010; 9 : 110.
35. Zou W, Yue P, Lin N et al. Vitamin C inactivates the proteasome inhibitor PS-341 in human cancer cells. Clin Cancer Res 2006; 12(1): 273–280.
36. Golden EB, Lam PY, Kardosh A et al. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood 2009; 113(23): 5927–5937.
37. Lonial S, Boise LH. Current Advances in Novel Proteasome Inhibitor–Based Approaches to the Treatment of Relapsed/Refractory Multiple Myeloma. Oncology 2011; 25(2).
38. Kuhn DJ, Chen Q, Voorhees PM et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin--proteasome pathway, against preclinical models of multiple myeloma. Blood 2007; 110(9): 3281–3290.
39. Demo SD, Kirk CJ, Aujay MA et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007; 67(13): 6383–6391.
40. Suzuki E, Demo S, Arastu-Kapur S et al. Bortezomib resistant cell lines have increased proteasome levels but remain sensitive to carfilzomib. Blood 2009; 114: Abstr 2852.
41. Wang L, Kumar S, Fridley B et al. Proteasome beta subunit pharmacogenomics: gene resequencing and functional genomics. Clin Cancer Res 2008; 14(11): 3503–3513.
42. Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol 2001; 8(8): 739–758.
43. Ruschak AM, Slassi M, Kay LE et al. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst 2011; 103(13): 1007–1017.
44. O‘Connor OA, Stewart AK, Vallone M et al. A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin Cancer Res 2009; 15(22): 7085–7091.
45. Niesvizky R, Bensinger W, Vallone M et al. PX-171–006: Phase Ib multicenter dose escalation study of carfilzomib (CFZ) plus lenalidomide (LEN) and low-dose dexamethasone (loDex) in relapsed and refractory multiple myeloma (MM): Preliminary results (Abstract). J Clin Oncol 2009; 27 (15 Suppl); Abstr 8541.
46. Vij R, Wang M, Kaufman JL et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012; 119(24): 5661–5670.
47. Jakubowiak AJ, Dytfeld D, Griffith KA et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012; 120(9): 1801–1089.
48. Fenical W, Jensen PR. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol 2006; 2(12): 666–673.
49. Miller CP, Manton CA, Hale R et al. Specific and prolonged proteasome inhibition dictates apoptosis induction by marizomib and its analogs. Chem Biol Int 2011; 194(1): 58–68.
50. Hofmeister CC, Richardson P, Zimmerman T et al. Clinical trial of the novel structure proteasome inhibitor NPI-0052 in patients with relapsed and relapsed/refractory multiple myeloma (r/r MM). J Clin Oncol 2009; 27 (15 Suppl): Abstr 8505.
51. Ocio EM, Mateos MV, San-Miguel JF. Novel agents derived from the currently approved treatments for MM: novel proteasome inhibitors and novel IMIDs. Expert Opin Investig Drugs 2012; 21(8): 1075–1087.
52. Moreau P. The future of therapy for relapsed/refractory multiple myeloma: emerging agents and novel treatment strategies. Semin Hematol 2012; 49 (Suppl 1): S33–S46.
53. Chauhan D, Tian Z, Zhou B et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res 2011; 17(16): 5311–5321.
54. Lee EC, Fitzgerald M, Bannerman B et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res 2011; 17(23): 7313–7323.
55. Kupperman E, Lee EC, Cao Y et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 2010; 70(5): 1970–1980.
56. Tian Z, Zhao JJ, Tai YT et al. Investigational agent MLN9708/2238 targets tumor suppressor microRNA-33b in MM cells. Blood 2012; 120(19): 3958–3967.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2013 Issue 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Pineal Germ Cell Tumors: Review
- Proteasome Inhibitors in Treatment of Multiple Myeloma
- Malignant Melanoma Treated with Radical Chemotherapy, Resemblance Histology of Melanoma to Soft Tissue Sarcomas, Case Report
- Surgery of the Pulmonary Metastases