
Léčba anakinrou u Schnitzler‑ syndromu – výsledky první retrospektivní multicentrické studie šesti pacientů z České republiky
Anakinra Treatment in Schnitzler Syndrome – Results of the First Retrospective Multi‑center Study in Six Patients from the Czech Republic
Background:
Schnitzler syndrome is a very rare, acquired, autoinflammatory disease of mostly adult onset with characteristic combination of chronic recurrent urticaria and monoclonal immunoglobulin M or G gammopathy predisposing the patients to malignant lymphoproliferation. In this work, we analyzed the results of biological therapy with anakinra on a national level aiming to supply data for effective pharmacoeconomic estimates, lay the grounds of nationwide patient registry, raise awareness among professional public and optimize provided health care.
Patients and Methods:
The retrospective study (10/ 2006– 9/ 2013) included six males with definite Schnitzler syndrome verified by the new Strasbourg criteria. All patients were pretreated with antihistamines, nonsteroidal anti‑inflammatory drugs and glucocorticoids. Four patients underwent two or more treatment lines including intravenous bisphosphonates, 2- chlorodeoxyadenosine (cladribine), interferon‑α, PUVA photochemotherapy, cyclosporine A, thalidomide, bortezomib, chlorambucil, cyclophosphamide, colchicine and methotrexate. Anakinra monotherapy was initiated in standard dosing (100 mg subcutaneously daily).
Results:
Complete and partial remissions were achieved in five (83%) and one patients (17%), respectively. Complete remission was characterized by urticaria and pain regression (within hours), normalization of inflammatory markers (within days) and bone metabolism improvement assessed by the markers of osteoblastic osteoformation and osteoclastic osteoresorption in one case (within weeks). With normalized inflammatory markers (including interleukin‑6 and interleukin‑18), arthralgia and sporadic exacerbations of urticaria and fevers persist in the patient in partial remission with proven Q703K polymorphism in NLRP3 gene. The median treatment follow‑up was 30.5 months (37.2 ± 31.2 (n = 6)). The dosing interval was prolonged in one case of complete remission to 48 hours. No serious adverse reactions occurred during anakinra application.
Conclusion:
In Schnitzler syndrome, anakinra represents an effective, verified and safe medication with potentionally long‑term administration not compromising its original efficacy and subjective tolerance. Anakinra, blocking autonomous inflammatory reaction of the organism via interleukin‑1 pathway, is a generally accepted first line treatment that should be made available in standard dosing for all Schnitzler patients.
Key words:
Schnitzler syndrome – monoclonal gammopathy – interleukins – anakinra – acute‑ phase proteins – osteogenesis
Autori:
P. Szturz 1; A. Šedivá 2; M. Žurek 3; Z. Adam 1; J. Štork 4; Z. Čermáková 5; P. Steyerová 6; A. Vokáčová 7; J. Hrbek 8; M. Sýkora 9; I. Špička 10; Z. Mechl 1; J. Mayer 1
Pôsobisko autorov:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Ústav imunologie 2. LF UK a FN v Motole, Praha
2; III. interní klinika – nefrologická, revmatologická a endokrinologická LF UP a FN Olomouc
3; Dermatovenerologická klinika 1. LF UK a VFN v Praze
4; Oddělení klinické biochemie, FN Brno
5; Radiodiagnostická klinika 1. LF UK a VFN v Praze
6; Ústav nukleární medicíny 1. LF UK a VFN v Praze
7; Radiologická klinika LF UP a FN Olomouc
8; Oddělení klinické hematologie, Nemocnice České Budějovice
9; I. interní klinika – klinika hematologie 1. LF UK a VFN v Praze
10
Vyšlo v časopise:
Klin Onkol 2014; 27(2): 111-126
Kategória:
Original Articles
Súhrn
Východiska:
Schnitzler‑ syndrom představuje velmi vzácné získané autoinflamatorní onemocnění převážně dospělých jedinců vyznačující se typickou kombinací chronicky recidivující kopřivky a monoklonální gamapatie typu imunoglobulinu M nebo G, která predisponuje pacienty k rozvoji maligní lymfoproliferace. V této práci analyzujeme výsledky biologické léčby anakinrou na národní úrovni s cílem poskytnout data pro efektivní farmakoekonomický odhad, položit základy celostátního registru pacientů, zvýšit osvětu odborné veřejnosti a optimalizovat péči o tyto nemocné.
Soubor pacientů a metody:
Do retrospektivní studie (10/ 2006– 9/ 2013) bylo zařazeno šest mužů s jistou diagnózou Schnitzler‑ syndromu ověřenou dle nových štrasburských kritérií. Všichni byli předléčeni antihistaminiky, nesteroidními antiflogistiky a glukokortikoidy. Čtyři pacienti absolvovali dvě nebo více léčebných linií zahrnujících intravenózní bisfosfonáty, 2- chlorodeoxyadenozin (kladribin), interferon-α, PUVA fotochemoterapii, cyklosporin A, thalidomid, bortezomib, chlorambucil, cyklofosfamid, kolchicin a metotrexát. Žádný z těchto preparátů však nevykazoval dostatečnou účinnost při uspokojivé toleranci. Monoterapie anakinrou byla zahájena ve standardním dávkování 100 mg podkožně denně.
Výsledky:
U pěti pacientů (83 %) bylo dosaženo kompletní, u jednoho pacienta (17 %) parciální remise. Při kompletní remisi došlo k ústupu kopřivky a bolestí (během několika hodin), normalizaci laboratorních markerů zánětů (během několika dnů) a úpravě kostního metabolizmu měřeného ukazateli osteoblastické osteoformace a osteoklastické osteoresorpce v jednom případu (během několika týdnů). U pacienta v parciální remisi s prokázaným Q703K polymorfizmem v genu NLRP3 přetrvávají artralgie a sporadické exacerbace kopřivky a febrilií při normalizovaných zánětlivých ukazatelích (včetně hladin interleukinu‑ 6 a interleukinu‑ 18). Medián sledování při probíhající léčbě je aktuálně 30,5 měsíce (37,2 ± 31,2; n = 6). V jednom případu kompletní remise jsme prodloužili interval dávkování na 48 hodin. Během podávání anakinry nedošlo k rozvoji žádných závažných nežádoucích projevů.
Závěr:
V indikaci Schnitzler‑ syndromu je anakinra účinným, ověřeným a bezpečným preparátem s možností dlouhodobého podávání při zachování původní efektivity a tolerance. Tento lék blokující cestou interleukinu‑ 1 autonomní zánětlivou reakci organizmu je všeobecně uznávaným lékem první volby a měl by být dostupný ve standardním dávkování všem pacientům se Schnitzler‑ syndromem.
Klíčová slova:
Schnitzler‑ syndrom – monoklonální gamapatie – interleukiny – anakinra – proteiny akutní fáze – osteogeneze
Úvod
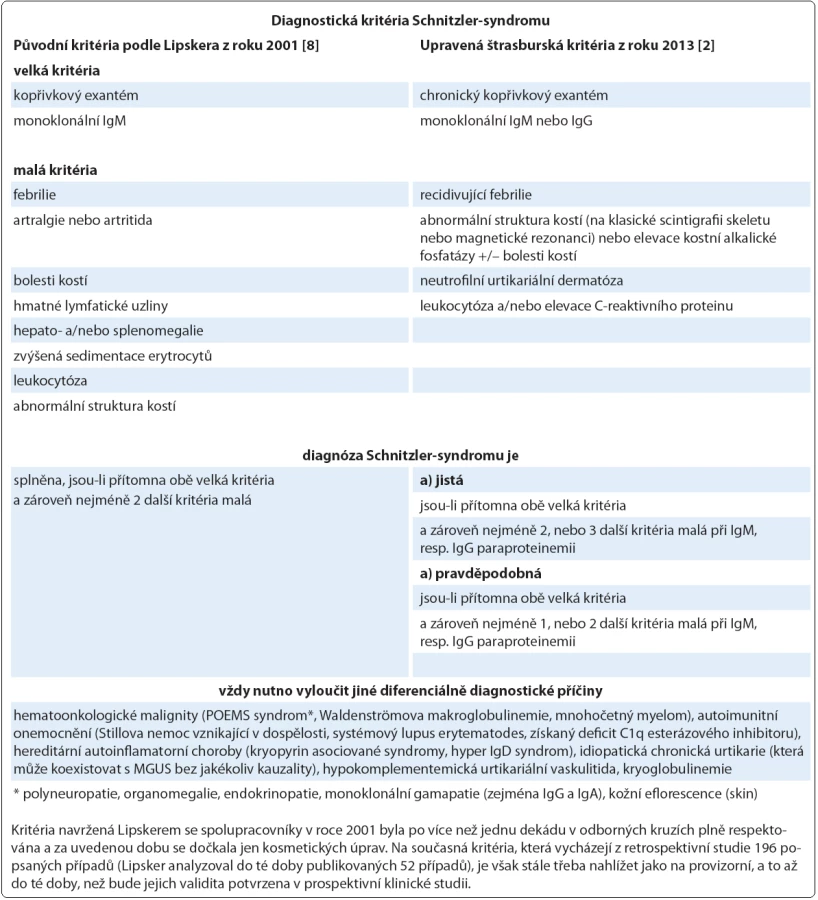
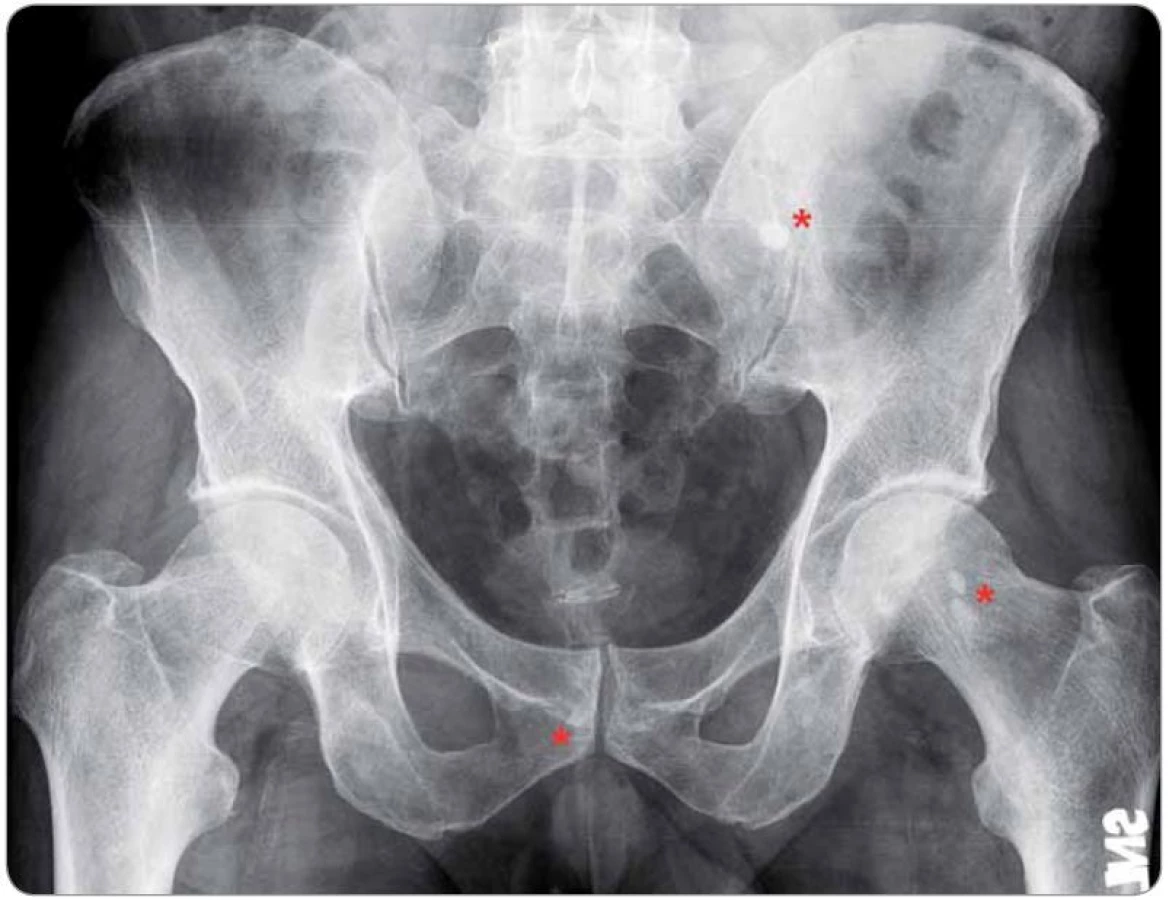
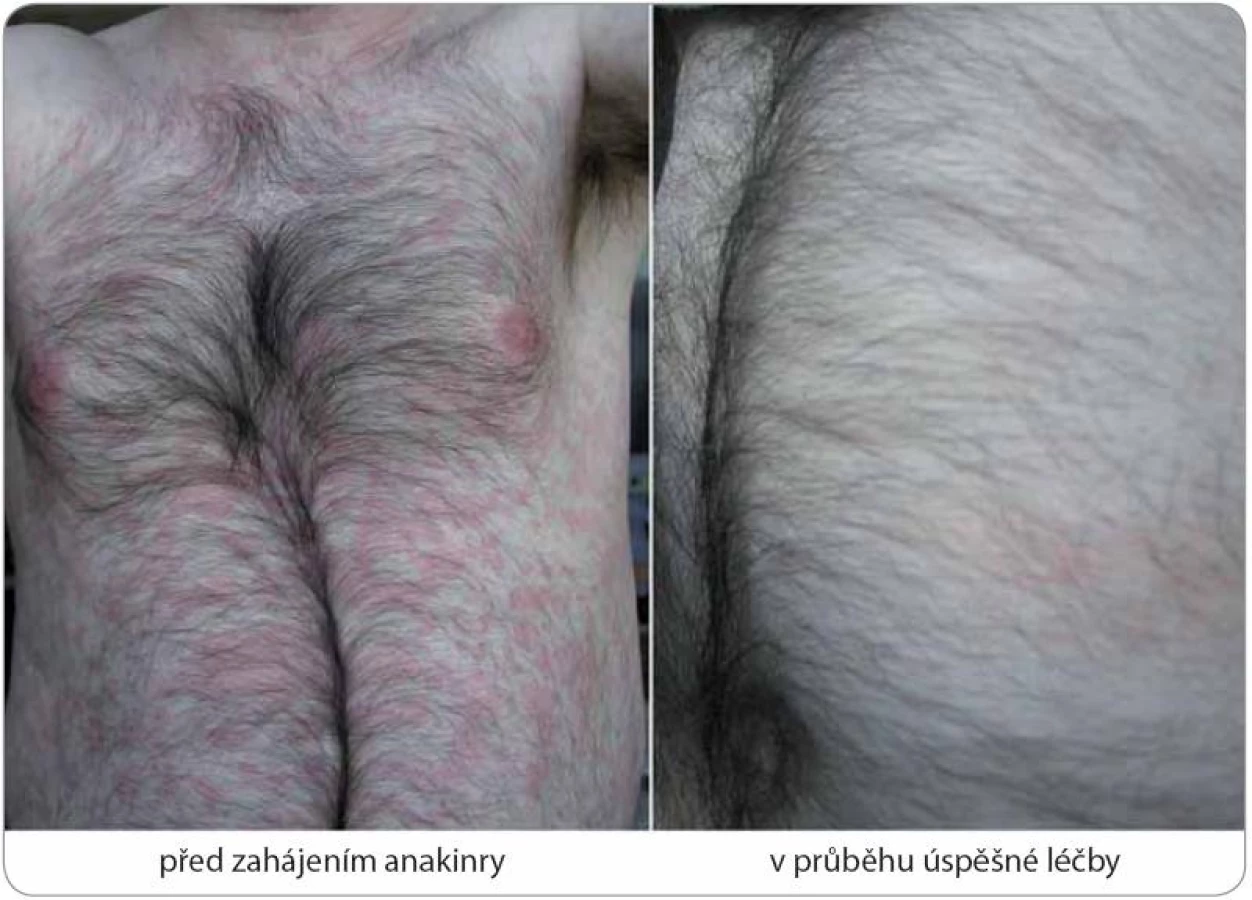
Na Schnitzler‑ syndrom, vzácné multisystémové onemocnění s neznámou patogenezí, se v současnosti pohlíží jako na získanou autoinflamatorní chorobu postihující především dospělé jedince mužského pohlaví (poměr 1,76 : 1) s mediánem věku v době diagnózy 51 let [1,2]. Charakteristický soubor příznaků byl poprvé popsán v roce 1972 a pak uceleně publikován v roce 1974 francouzskou dermatoložkou Liliane Schnitzler [3,4]. Během následujících 40 let se ve světové literatuře objevily, dominantně formou kazuistik, zprávy asi o 200 případech tohoto onemocnění, které byly statisticky shrnuty ve dvou rozsáhlých pracích [2,5]. Lze však předpokládat, že skutečná prevalence této jednotky, vyznačující se typickou kombinací chronické kopřivky (obr. 1) a monoklonální gamapatie typu imunoglobulinu M (IgM), je mnohem vyšší a podle autorské skupiny Jain et al může dosahovat až 1,5 % všech pacientů s IgM paraproteinemií (obr. 2) [6]. Další často se vyskytující příznaky zahrnují recidivující epizody subfebrilií či febrilií, bolestí kostí, kloubů a svalů. Radiograficky, scintigraficky a histopatologicky lze v některých případech prokázat abnormity skeletu a z laboratorního rozboru krve bývá patrná leukocytóza s neutrofilií, zvýšená sedimentace erytrocytů (fähraeus‑ westergren – FW), elevace C‑ reaktivního proteinu (C‑ reactive protein – CRP), ale i dalších reaktantů akutní fáze (feritin, fibrinogen) [7]. V roce 2013 byla na základě konsenzu předních odborníků při setkání ve Štrasburku upravena původní diagnostická kritéria publikovaná v roce 2001 Lipskerem se spolupracovníky, jak ukazuje tab. 1 [2,8].
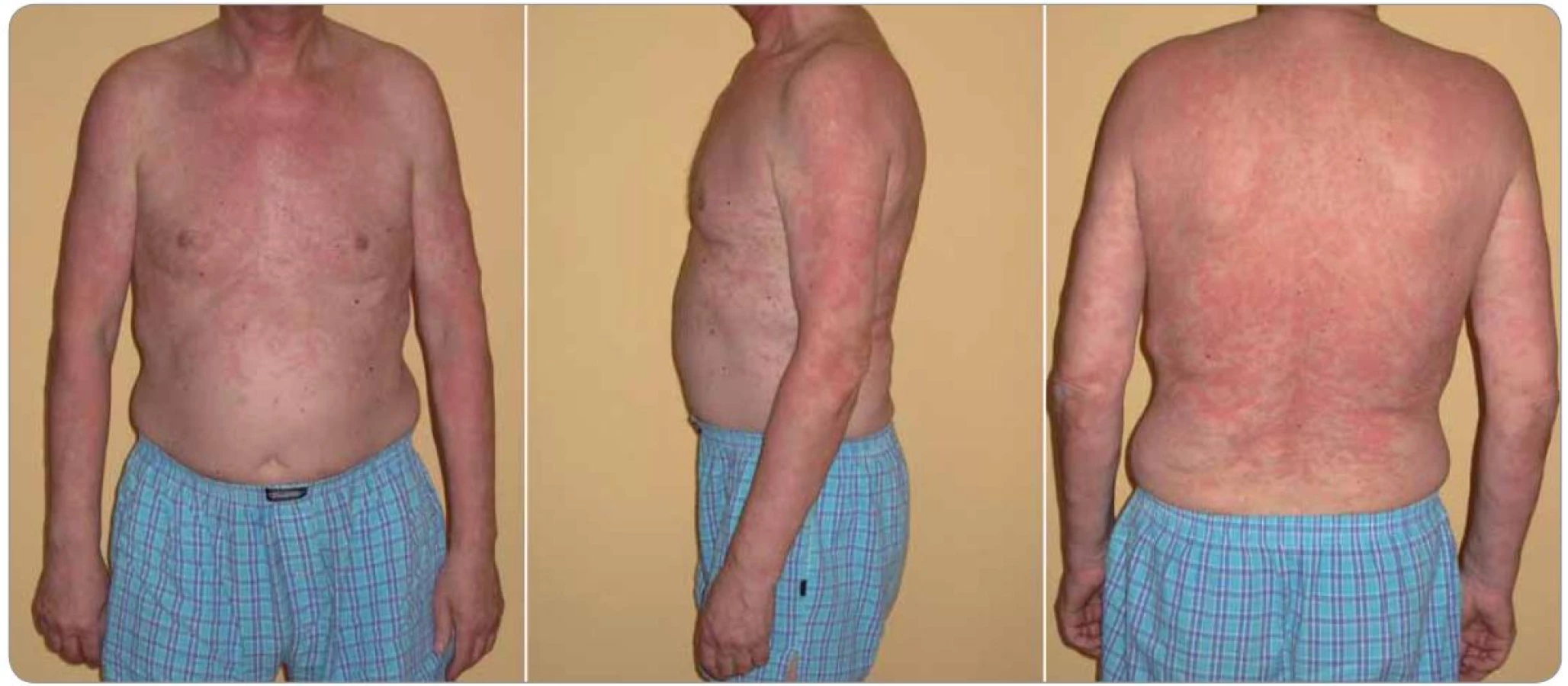

Kožní změny charakteru chronicko‑recidivující až chronicko‑kontinuální kopřivky bývají obvykle hlavním příznakem nemocných. Erytematózní makulopapulózní ložiska mohou splývat do geografických ploch velikosti i více než 10 cm v průměru. K výsevu nových morf dochází v souvislosti s konzumací alkoholu, kořeněných jídel, po tělesném vypětí nebo jiném stresu, ale i bez zjevné příčiny. Postiženy bývají obvykle končetiny a trup, zatímco hlava a krk, stejně tak jako chodidla a ruce, zůstávají ušetřeny. Svědění je nekonstantním příznakem, který může zpočátku chybět a objevit se až s odstupem několika měsíců či roků [5,9]. Histologický nález neutrofilní urtikariální dermatózy (obr. 3) je společný pro Schnitzler‑ syndrom, Stillovu nemoc vzniklou v dospělosti, systémový lupus erythematodes a zřejmě i některá hereditární autoinflamatorní onemocnění (skupina kryopyrin asociovaných syndromů) [10]. Přítomnost monoklonálního IgM (u minoritní skupiny IgG) je nezbytná pro stanovení diagnózy Schnitzler‑ syndromu. U 89 % nemocných se jedná o IgM typu κ. Koncentrace monoklonálního imunoglobulinu je obvykle při stanovení diagnózy nízká (pod 10 g/ l) a zůstává stabilní nebo se pozvolna v průběhu času zvyšuje rychlostí 0,5– 1 g/ l/ rok. Vyšší hodnoty vyvolávají podezření na transformaci do maligní lymfoproliferace (zvláště Waldenströmovy makroglobulinemie) [1]. Detailní analýza jednotlivých příznaků je součástí práce dříve publikované v Klinické onkologii [7].

Etiopatogeneze Schnitzler‑ syndromu je značně komplexní a doposud není zcela objasněna. Podle některých hypotéz podpořených klinickými i laboratorními daty je klíčovým bodem patogeneze zánět způsobený interleukinem‑ 1 (IL‑1), který je považován za hlavní prozánětlivý cytokin a představuje zřejmě hnací sílu typických příznaků Schnitzler‑ syndromu [2,11]. Jeho ústřední roli dokládá především univerzální léčebná odpověď na aplikaci selektivního receptorového antagonisty IL‑1, anakinry (Kineret™, Sobi), jehož význam v této indikaci bude dále podrobně rozebrán. Zatím ojedinělé nálezy aktivující mutace V198M v genu NLRP3 (nucleotide‑binding domain protein and leucine‑ rich repeat containing gene family, pyrin domain containing 3) vysvětlují predispozici pro nadměrnou sekreci solubilní formy IL‑1 (IL‑1β) u těchto pacientů [12,13]. S ohledem na recidivující charakter zánětlivé odpovědi organizmu, výše zmíněnou roli IL‑1 a jisté podobnosti s kryopyrinopatiemi (tedy přítomnost neutrofilní urtikariální dermatózy, terapeutického efektu inhibitoru IL‑1 a snad i mutace, případně polymorfizmu genu NLRP3) je Schnitzler‑ syndrom stále častěji řazen mezi získaná autoinflamatorní onemocnění [1,2]. Nicméně jeho maligní potenciál, možnost rozvoje systémové amyloidózy a skutečnost, že pacienti jsou často odesíláni na hematoonkologické kliniky k diferenciální diagnostice monoklonální gamapatie, jsou hlavní důvody, proč by měli být onkologové seznámeni s touto raritní jednotkou.
Schnitzler‑ syndrom lze považovat za poddiagnostikované a často nesprávně diagnostikované onemocnění. Literatura uvádí typický časový odstup mezi prvními příznaky a stanovením diagnózy asi pět let [1]. V České republice dosud neexistuje žádný registr, který by sdružoval pacienty s touto diagnózou na národní úrovni, stal se platformou pro odbornou diskuzi mezi zdravotnickými centry a základem pro efektivní farmakoekonomický odhad. Tento článek si klade za cíl existující deficit částečně kompenzovat. Na základě spolupráce mezi třemi klinickými pracovišti zde představujeme první multicentrickou retrospektivní analýzu výsledků biologické terapie anakinrou u šesti případů. Jedinečnost této studie spočívá dále v tom, že k datu odeslání příspěvku do redakce zahrnuje všechny pacienty se Schnitzler‑ syndromem z České republiky na uvedené léčbě. Práce tedy poskytuje cenná referenční data pro diskuzi s financujícími subjekty k zajištění plnohodnotné zdravotní péče pro tuto skupinu nemocných. Připojená obrazová dokumentace pak zachycuje některé klinické a paraklinické nálezy, které dokreslují komplexnost probírané tematiky a dokládají tak potřebu pečlivé diferenciální diagnostiky, často s úzkou mezioborovou spoluprací.
(Pozn. k terminologii: tradované eponymum „Schnitzlerův syndrom“ by správně mělo být dle svého původu nahrazeno označením „syndrom Schnitzlerové“. Abychom však předešli nejasnostem, prosazujeme v našich publikacích neutrální název „Schnitzler‑ syndrom“, kterým se rovněž přibližujeme termínu „Schnitzler syndrome“ užívanému v anglosaské literatuře.)
Soubor pacientů a metody
Pacienti
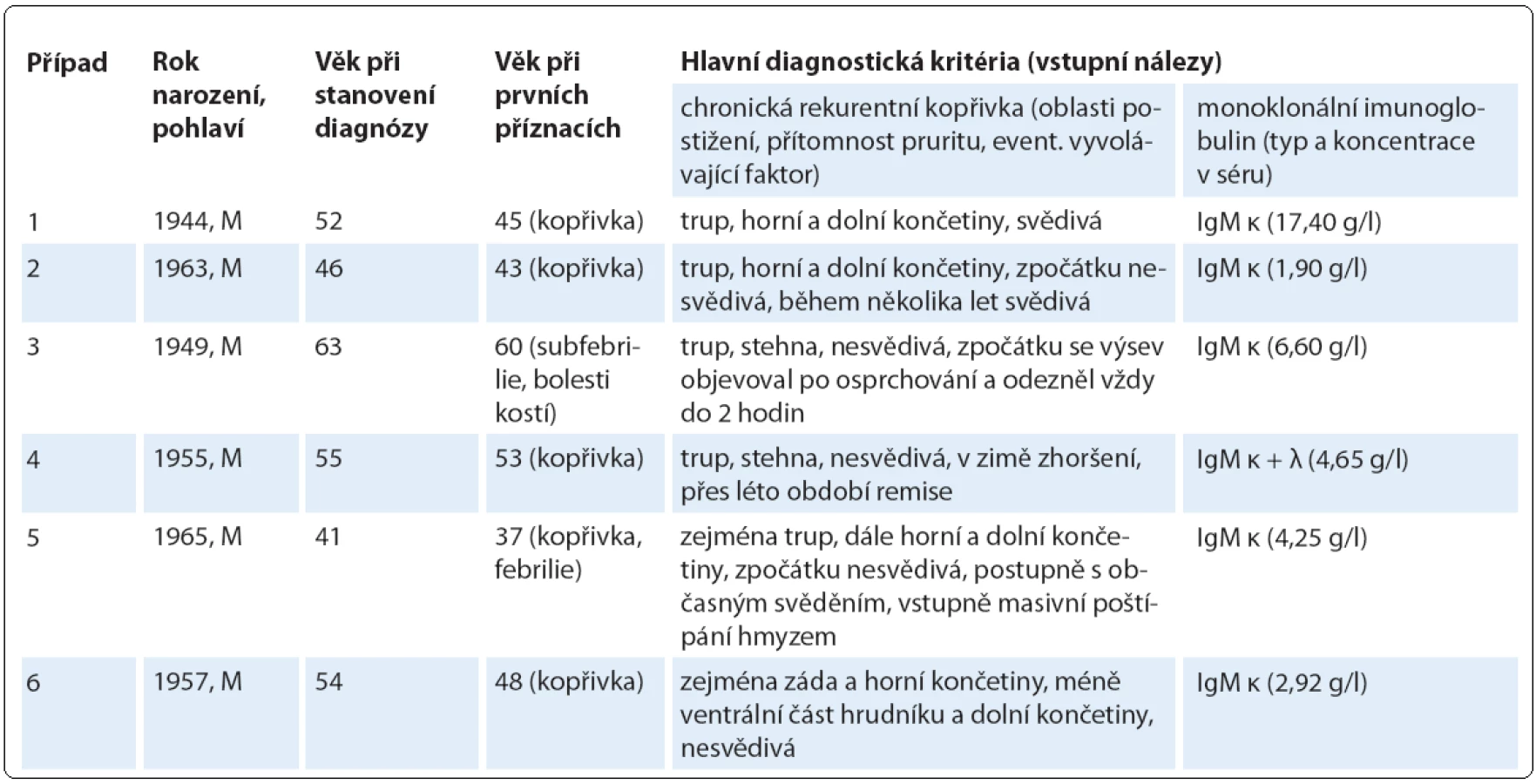
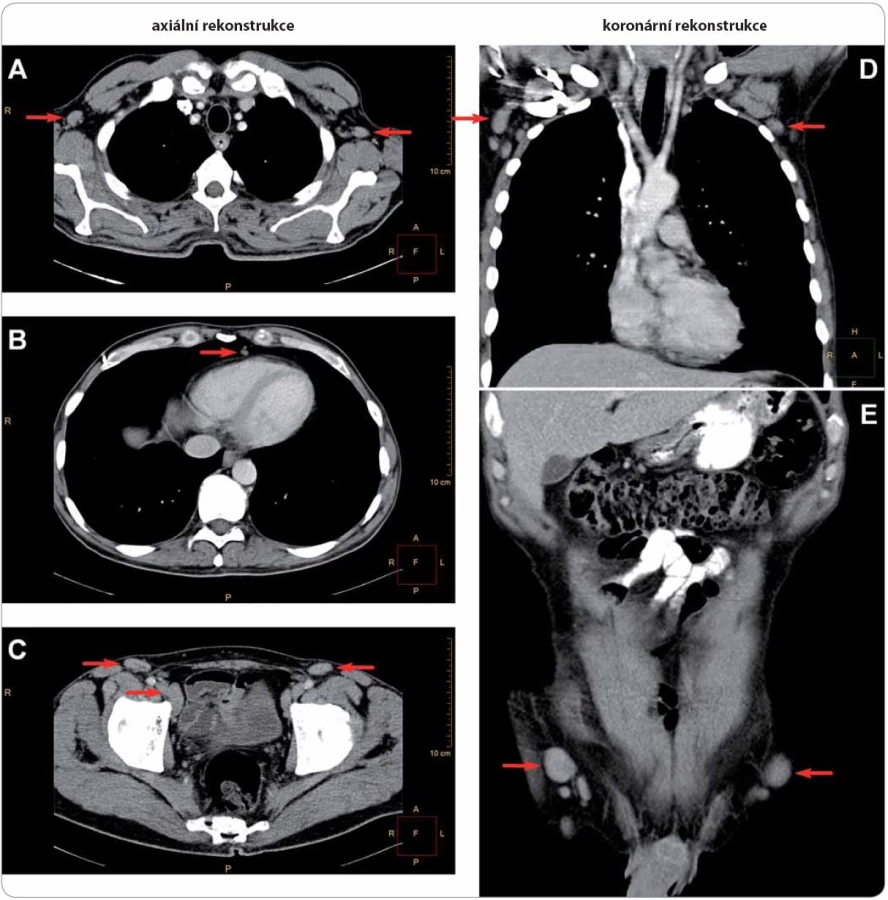
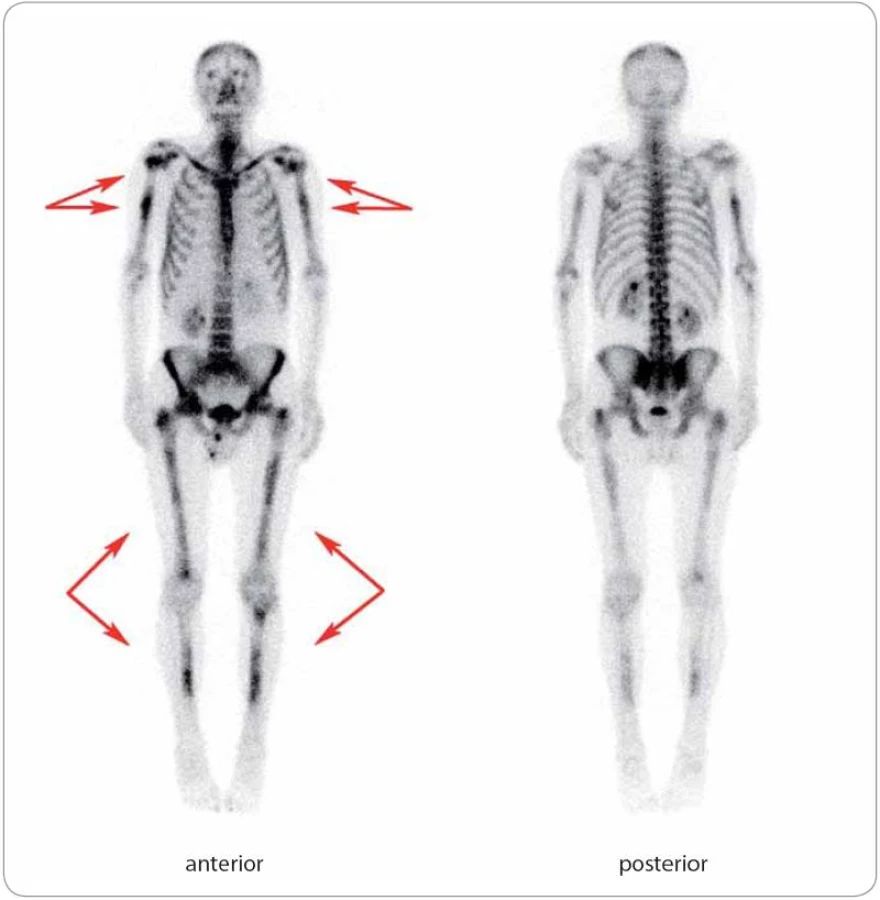
Do této retrospektivní studie bylo zařazeno šest pacientů s diagnostikovaným Schnitzler‑ syndromem, u nichž byla v průběhu 6leté doby sledování od 10/ 2006 do 9/ 2013 zahájena a podávána biologická léčba anakinrou ve třech klinických centrech České republiky (u čtyř pacientů ve FN Brno, u dalších dvou ve FN v Motole a ve FN Olomouc, tab. 2). U všech zařazených pacientů byla diagnóza Schnitzler‑ syndromu stanovena na základě původních Lipskerových kritérií z roku 2001 [8] a její platnost jsme později ověřili z pohledu nových štrasburských kritérií z roku 2013 [2], podle nichž se vždy jednalo o jistou diagnózu, kdy byla splněna obě velká kritéria a alespoň dvě další kritéria malá (ve třech případech splněna dvě, ve dvou případech tři, v jednom případu všechna čtyři; tab. 3). Zevrubná diferenciální diagnostika zahrnovala dermatologická, imunologická, revmatologická, onkologická a mikrobiologická vyšetření, která doplňovala zobrazovací metody radiologické (ultrasonografie, konvenční radiografie (RTG), výpočetní tomografie (CT), magnetická rezonance (MRI)) i scintigrafické (klasická scintigrafie skeletu pomocí technecia pyrofosfátu (TcS), jednofotonová emisní výpočetní tomografie (SPECT), pozitronová emisní tomografie s 18F‑ fluorodeoxyglukózou (PET nebo hybridní vyšetření PET/ CT)) (obr. 4– 6). Stanovení diagnózy Schnitzler‑ syndromu předcházelo u všech pacientů období různě dlouhého pátrání po příčině chronické kopřivky, recidivujících febrilií, bolestí muskuloskeletálního systému a případně i proinflamatorního stavu organizmu (zejména neinfekční elevace CRP), kdy zásadní obrat správným diagnostickým směrem nastal až po zjištění přítomnosti monoklonálního imunoglobulinu v séru. Na výše uvedená tři klinická pracoviště byli pacienti odesláni s diagnózou Schnitzler‑ syndromu (případ 4), vysokým podezřením na toto onemocnění (případ 3), v rámci diferenciální diagnostiky paraproteinemie (případ 1, 2), blíže nespecifikované imunologické poruchy (případ 5) či systémového onemocnění pojiva (případ 6). Spektrum aplikovaných vyšetřovacích modalit jsme v případech 1, 2 a 5 rozšířili o molekulárně biologickou analýzu NLRP3 genu ze vzorku bioptované kůže, kdy jsme zjišťovali výskyt mutací a polymorfizmů v rozsáhlém exonu 3, který jimi bývá nejčastěji zasažen.
Léčba
Spektrum léčebných modalit použitých ve sledované kohortě tvořila dominantně systémová terapie (cytostatika, monoklonální protilátky, imunomodulancia a další preparáty) podávaná parenterálně i perorálně, přičemž u případu 1 jsme využili i světloléčbu kožních příznaků v podobě metody PUVA (psoralen + UV‑ A záření). Operativa našla uplatnění při diagnostických výkonech (pět diagnostických kožních biopsií a jeden odběr lymfatické uzliny). Před zahájením anakinry absolvoval každý pacient nejméně jednu léčebnou linii (glukokortikoidy), větší předléčenost byla zaznamenána ve čtyřech případech. Aplikované režimy shrnuje tab. 4. Kortikoterapie úplně potlačila projevy Schnitzler‑ syndromu v případu 2, kde se však poměrně vysoká kumulativní dávka metylprednisolonu promítla do obrazu těžké formy Cushingova syndromu. Ve zbylých případech hodnotíme efekt glukokortikoidů jako parciální či minimální remisi. Pouze v případu 1 jsme pozorovali částečný nebo časově omezený účinek rovněž u jiných preparátů, z nichž si však jen bisfosfonáty (v chronologickém pořadí pamidronát, klodronát, ibandronát, vše parenterálně) uchovaly dlouhodobý terapeutický potenciál při zvládání bolestí kostí, nicméně ostatní symptomy neovlivňovaly. Antihistaminika byla zcela neúčinná, nesteroidní antiflogistika měla jen aditivní analgetický, případně antipyretický význam napříč celou kohortou. Přínos žádného z těchto léků tudíž nelze hodnotit jako dostatečný, a to i z důvodu mnohdy nezanedbatelných nežádoucích reakcí.
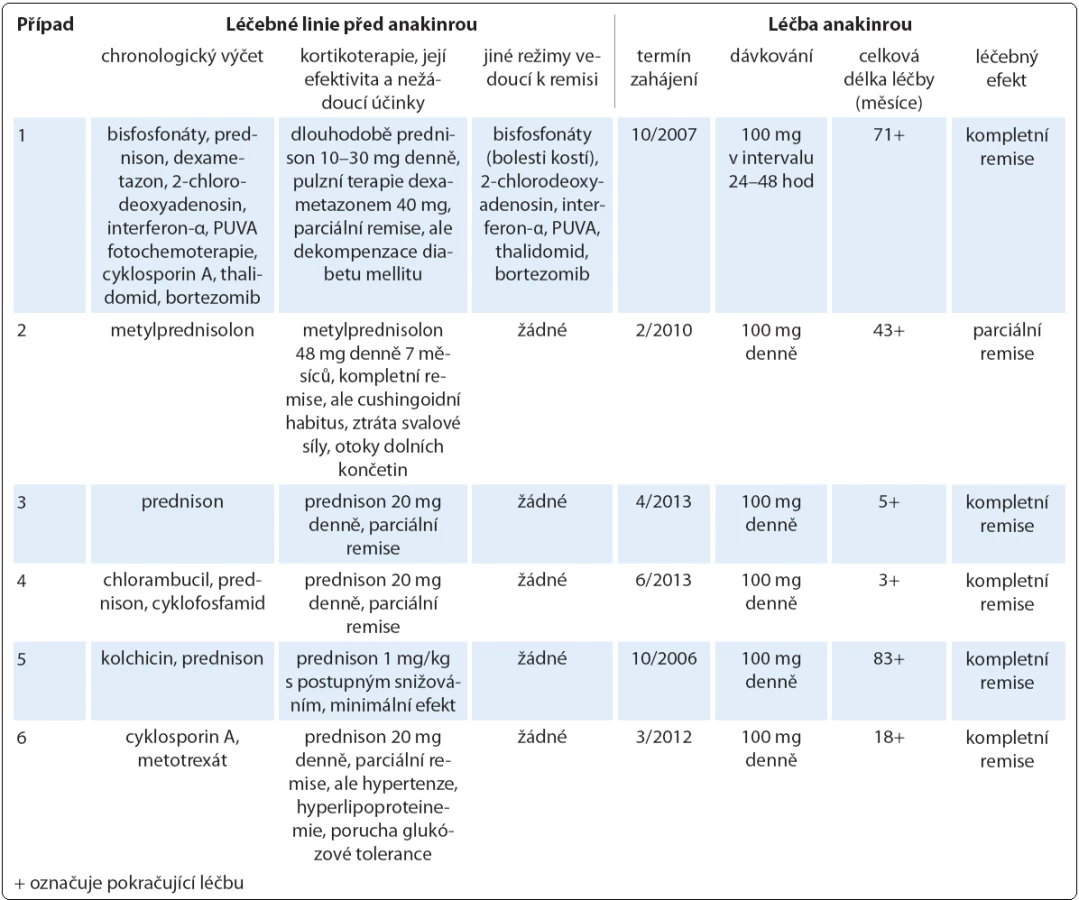
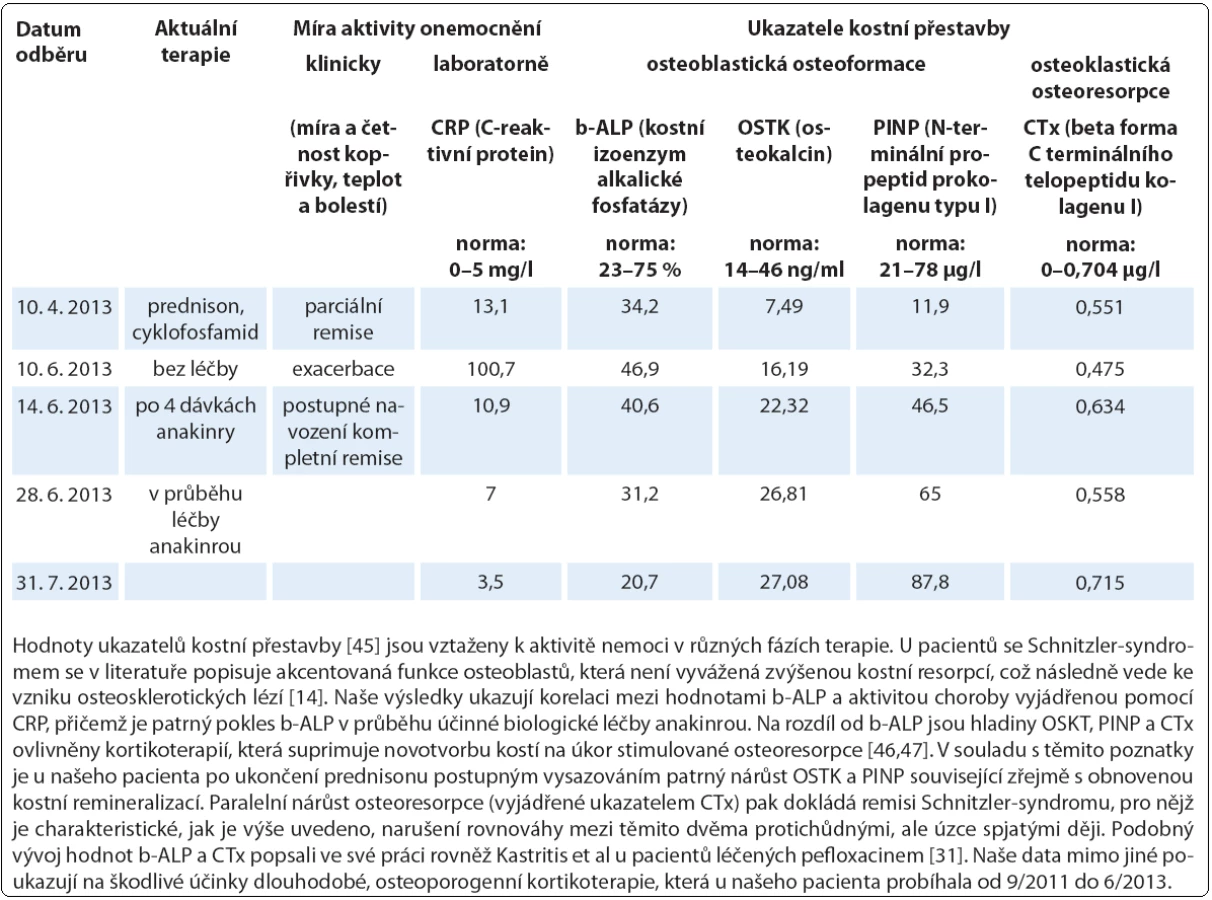
Léčbu biologickým preparátem anakinra jsme u pacientů zahájili ve standardním dávkování 100 mg denně podkožní injekcí po vysvětlení očekávaného přínosu a možných rizik s podepsáním informovaného souhlasu. Po vstupní krátkodobé hospitalizaci probíhala aplikace anakinry v domácím prostředí s plnou soběstačností všech zařazených pacientů. Sledování léčebné odpovědi se opíralo o klinické zhodnocení zdravotního stavu (kožní nález, měření tělesné teploty, anamnestické stanovení míry bolesti na vizuálně analogové škále) a laboratorní měření reaktantů proinflamatorního stavu organizmu ze vzorků periferní krve (množství leukocytů a neutrofilních granulocytů, hodnoty CRP, FW, feritinu, fibrinogenu). Jako doplňkové vyšetření sloužilo u vybraných pacientů monitorování hladin IL‑6 a IL‑18 (případ 2) a dále i ukazatelů kostního metabolizmu (případy 3 a 4). Termínem „kompletní remise“ jsme označili stav charakterizovaný úplným odezněním kožních příznaků, epizod zvýšených tělesných teplot a bolestí pohybového aparátu při normalizaci krevních zánětlivých parametrů. Podmínky kompletní remise tedy nezahrnují vymizení patologických změn na zobrazovacích modalitách (lymfadenopatie, osteoskleróza), i když např. Terpos et al uvádějí u čtyř pacientů se Schnitzler‑ syndromem po mediánu 6 měsíců léčby anakinrou významný pokles metabolické aktivity osteoblastických lézí dle TcS [14]. Scintigrafické nálezy totiž nemusejí vždy korelovat s klinickým obrazem, jak dokládáme případem 4,u něhož sice neukázala kontrolní TcS po dvou letech medikace chlorambucilu, prednisonu a cyklofosfamidu žádné z původních patologických osteoblastických okrsků (obr. 6), celkový klinický prospěch při přetrvávajících bolestech pohybového aparátu byl ale nevýznamný. Podobně není pro hodnocení efektivity léčby směrodatná hladina monoklonálního imunoglobulinu, která zůstává nezměněna či pozvolna narůstá. Monitorování paraproteinemie, stejně tak jako stavu lymfatických uzlin se ovšem doporučuje v rámci sekundární prevence maligní lymfoproliferace [2].
Výsledky
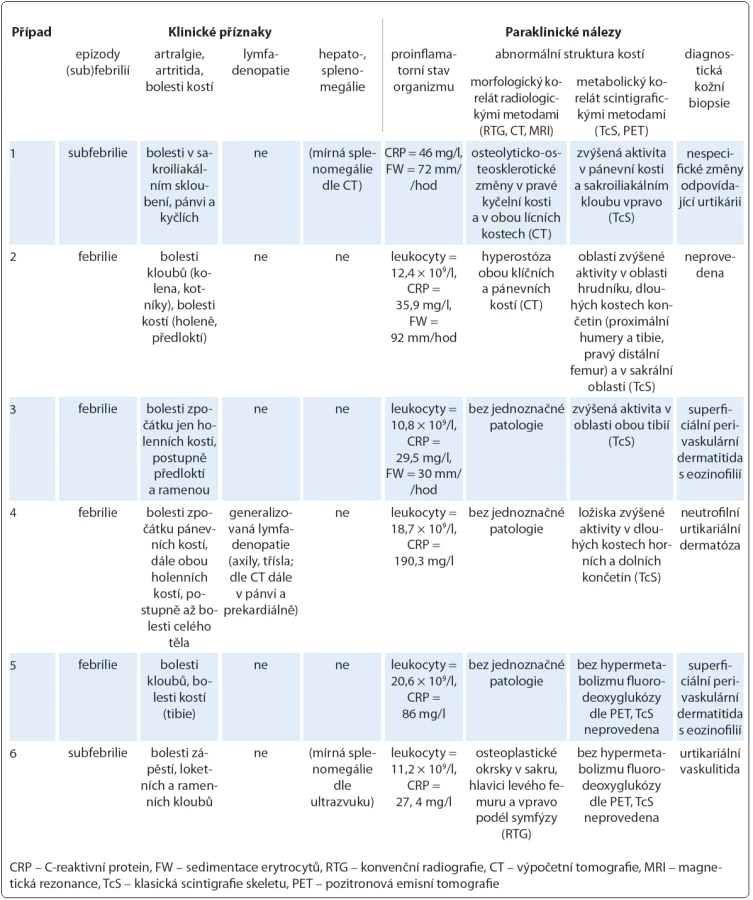
V této retrospektivní multicentrické studii pacientů se Schnitzler‑ syndromem léčených anakinrou jsme hodnotili celkem šest mužů. Věk při prvních příznacích, které zahrnovaly kopřivku (n = 5), zvýšenou tělesnou teplotu (n = 2) a bolesti kostí (n = 1), se pohyboval v rozmezí od 37 do 60 let (47,7 ± 7,3; n = 6), věk v době stanovení diagnózy pak v rozpětí od 41 do 63 let (51,8 ± 7,0; n = 6). Medián latence od prvních příznaků ke stanovení správné diagnózy byl 3,5 roku (4,2 ± 1,8; n = 6). Počátek doby sledování pacientů na uvedených třech klinických pracovištích se ve většině případů překrýval s obdobím definitivního potvrzení diagnózy. Medián celkové doby sledování, který se následně počítal vždy k datu poslední ambulantní kontroly (9/ 2013), je 70 měsíců (71,2 ± 59,5; n = 6). Elektroforéza séra prokázala u pěti pacientů monoklonální IgM κ a v případu 4 biklonální IgM κ, λ s mediánem koncentrace rovným 4,45 g/ l (6,29 ± 5,18; n = 6). Všichni pacienti byli postiženi chronicky recidivující kopřivkou, ve třech případech (50 %) doprovázenou svěděním (tab. 2). Další chorobné příznaky ve sledované kohortě tvořily (tab. 3): zvýšená tělesná teplota (100 %), bolesti kostí a kloubů (100 %), přičemž u čtyř pacientů (67 %) se jednalo o bolesti holenních kostí, dále laboratorní známky zánětu (100 %) a abnormální struktura kostí dle TcS (67 %) a dle CT nebo RTG (50 %). Hmatné zvětšené lymfatické uzliny byly zjištěny jen u jednoho pacienta (případ 4), stejně tak příznačný nález neutrofilní urtikariální dermatózy ukázal jen jeden histopatologický rozbor (případ 4) z celkem pěti diagnostických kožních biopsií. Zajímavá je však vazba na onemocnění prostaty u dvou pacientů (karcinom prostaty u případu 3, prostatitida u případu 5) v počátečních fázích Schnitzler‑ syndromu. Analýza NLRP3 genu potvrdila Q703K polymorfizmus u případu 2.
Zahájení léčby anakinrou představovalo zlomové řešení vedoucí během několika hodin k ústupu kopřivky (obr. 7) a bolestí a v řádu několika dnů k normalizaci laboratorních markerů zánětu (graf 1, 2), stejně tak jako hladin IL‑6 a IL‑18 v případu 2. Kompletní (83 %) nebo parciální (17 %) remise onemocnění bylo dosaženo u všech sledovaných subjektů s mediánem délky podávání anakinry 30,5 měsíce (37,2 ± 31,2; n = 6) (tab. 4). Podrobný popis léčebného úspěchu v případech 1 a 2 jsme zveřejnili v dřívějších publikacích [15,16]. Interval mezi jednotlivými 100 mg dávkami, který standardně činí 24 hod, jsme v případu 1 prodloužili až na 48 hod při uspokojivé toleranci, kdy je pacient schopen sám si jej regulovat dle aktuálního zdravotního stavu. Příznivý vliv blokády signalizační dráhy IL‑1 jsme zaznamenali i při laboratorním hodnocení kostní remodelace (tab. 5). Přes rychlou a jednoznačnou léčebnou odpověď na anakinru u případu 2 nedošlo k úplnému vymizení všech projevů onemocnění a u pacienta nadále přetrvávají artralgie, k nimž se asi po roce a půl podávání anakinry připojily námahou vyvolané ataky kopřivkových výsevů a febrilií s frekvencí přibližně jednou za dva měsíce při normalizovaných zánětlivých ukazatelích. Ačkoliv nebyla etiologie kloubních příznaků zpočátku zcela jednoznačná, vzhledem k dalšímu vývoji je lze přiřadit k symptomatice Schnitzler‑ syndromu a stav nyní hodnotíme jako parciální remisi. S ohledem na omezenou dostupnost biologika nebyla intenzita dávkovacího schématu zatím stupňována. S relapsem původní symptomatiky jsme se setkali dále v případu 5 při exacerbaci chronické cholecystitidy, jednalo se jen o krátkodobé zhoršení stavu. Během podávání anakinry nedošlo ve sledované kohortě k rozvoji žádných závažných nežádoucích projevů (tedy stupně III a IV dle CTCAE, Common Terminology Criteria for Adverse Events, verze 4.0) či jiných interkurencí vynucujících si přerušení terapie, případně úpravu dávkování. Neutrofilní granulocytopenie se v naší kohortě nevyskytly, nebo byly přechodné, jak přehledně znázorňuje graf 1.

![Hodnoty CRP [mg/l] u třech pacientů v průběhu zahájení léčby anakinrou.](https://pl-master.mdcdn.cz/media/image/77fa7e7ac5465feaa40e64eb91c34d37.png?version=1537794699)
Diskuze
Někteří autoři pohlížejí na Schnitzler‑ syndrom jako na premaligní stav, a to s ohledem na možný přechod do lymfoproliferativního onemocnění (lymfom marginální zóny, Waldenströmova makroglobulinemie, mnohočetný myelom). Riziko transformace se tradičně odhaduje asi na 15– 20 %, což přibližně odpovídá výskytu tohoto jevu u početnější skupiny pacientů s monoklonální gamapatií nejasného významu (monoclonal gammopathy of undetermined significance – MGUS) [1]. Skutečnosti však bude zřejmě více odpovídat nedávno publikovaný statistický rozbor databáze z Mayo Clinic, který není zatížen pozitivní selekcí těžkých forem choroby, jako popisy jednotlivých případů či malých skupin pacientů, a míru pravděpodobnosti stanovuje nižší, asi kolem 8 % [6]. K malignizaci pak dochází nejdříve po 10– 20 letech od prvních příznaků nemoci. Pro předpověď přechodu do maligní lymfoproliferace nebyly dosud popsány žádné prognostické faktory, varovným signálem může být narůstající hodnota paraproteinu. Další potenciálně závažné komplikace zahrnují těžký průběh anémie chronických chorob a ojediněle popisovaný rozvoj sekundární (AA) amyloidózy u neléčených nemocných. Z hlediska mortality je však průběh příznivý, 91 % nemocných žije déle než 15 let [8,17]. Pacient, který byl původně popsán Liliane Schnitzler, zemřel 23 let od prvních příznaků nemoci na difuzní lymfoplazmocytární infiltraci jater a kostní dřeně [18].
Ačkoliv se Schnitzler‑ syndrom vyznačuje kolísavým průběhem s různě dlouhými klidovými pauzami, dlouhou dobu byla choroba považována za chronicky invalidizující, bez možnosti uzdravení. Jistý názorový obrat nastal po zveřejnění dvou případů spontánní remise, které byly publikovány v roce 2010 a 2011. U prvního pacienta došlo po osmi letech k samovolnému útlumu aktivity nemoci trvajícímu již více než pět roků, během nichž se chladem či stresem indukovaná exacerbace vyskytla jednou, maximálně dvakrát do roka při neměnné hladině monoklonálního imunoglobulinu (30 g/ l) [1]. Druhým byl případ muže předléčeného antihistaminiky, kortikoidy a nesteroidními antiflogistiky, u něhož se po 3letém průběhu Schnitzler‑ syndromu přesmykla produkce IgM κ na třídu IgG κ s následným pozvolným a úplným snižováním hladiny paraproteinu doprovázeným odezněním všech původních příznaků nemoci (kopřivka, únava, bolesti kloubů a kostí dolních končetin). Tento pozoruhodný průběh nemoci upozornil na potenciální kauzalitu mezi monoklonální gamapatií a typickou klinickou manifestací tohoto syndromu [19].
Do roku 2005, kdy se objevila první zpráva o úspěšné biologické terapii preparátem anakinra [20], byla léčba této choroby svízelná a frustrující a lékem první volby se na dlouhou dobu staly nesteroidní antiflogistika a kortikosteroidy. Blokátory cyklooxygenázy sice účinkovaly při symptomatické léčbě teploty a bolestí kostí a kloubů, neměly však většinou žádný vliv na kožní projevy [21]. Obdobně glukokortikoidy na jedné straně snižují intenzitu kožních projevů a povšechné zánětlivé reakce, ale pro dosažení léčebného efektu je zapotřebí takových dávek (obvykle minimálně 20 mg prednisonu), při jejichž dlouhodobém užívání vznikají velmi nepříjemné nežádoucí účinky v podobě iatrogenního Cushingova syndromu [15]. Vyzkoušeny byly také kolchicin (Colchicum-Dispert™) [8,22], dapson [23], hydroxychlorochin (Plaquenil™) a chlorochin [24], dále plazmaferéza [23,25] a nitrožilní imunoglobuliny [26], cytostatika (metotrexát, cyklofosfamid, chlorambucil) a imunosupresiva (azathioprin, cyklosporin) [8], rituximab [27], inhibitory tumor nekrotizujícího faktoru α (etanercept, adalimumab) [28], s významnějším léčebným efektem i interferon α [20], thalidomid [29], fototerapie metodou PUVA [8] a pefloxacin (Abaktal™), antibiotikum fluorochinolonové řady, který kromě svých antimikrobiálních účinků vykazuje rovněž imunomodulační a antiinflamatorní vlastnosti [30,31]. Antihistaminika jsou u Schnitzler‑ syndromu na kožní projevy neúčinná [24,32]. Bolesti kostí však mohou mírnit, či zcela odstraňovat bisfosfonáty [16].
Zásadní změnu přinesl až preparát anakinra, antagonista lidského receptoru pro IL‑1 získávaný rekombinantní technologií z buněk Escherichia coli, tedy látka selektivně zaměřená na tlumení zánětu kompetitivní inhibicí hlavního prozánětlivého cytokinu. Již první injekce (100 mg subkutánně) navodí během několika hodin kompletní remisi nemoci [11]. Anakinra odstraní zcela svědění kůže, kopřivkovité projevy, zvýšenou tělesnou teplotu, stejně tak jako bolesti pohybového systému a únavu. Léčebný účinek anakinry se projeví také v laboratorních nálezech, kde je patrný pokles zánětlivé odpovědi organizmu, včetně hladin proinflamatorních cytokinů IL‑6 a IL‑18 [15]. IL‑6 hraje důležitou roli při proliferaci plazmocytů a je spojován s chronicky zvýšenou hodnotou CRP, neutrofilií a s anémií chronických chorob přítomnou u těchto pacientů [33]. IL‑18, označovaný jako interferon γ indukující faktor, pak zastupuje proinflamatorní cytokin z IL‑1 rodiny štěpený kaspázou‑ 1 [34]. Naproti tomu plazmatické hladiny IL‑1β (výhradně solubilní forma IL‑1) a IL‑1α (převážně membránová forma IL‑1) jsou u pacientů se Schnitzler‑ syndromem pod limitem detekce ELISA (< 1 pg/ ml) [12,35]. Pro jejich stanovení v CD14+ buňkách (monocytech) lze použít stimulaci pomocí bakteriálního endotoxinu (lipopolysacharidu), která zvýší nejen sekreci IL‑1, ale dále i IL‑6 a tumor nekrotizujícího faktoru‑α (TNF‑α). Ryan et al v ex vivo podmínkách ukázali, že IL‑1 inhibitor tlumí stimulovanou hypersekreci jak samotného IL‑1, tak i IL‑6 a TNF‑α, čímž poskytli důkaz podporující klinickou odpověď po podání anakinry [36]. Přes evidentní a zásadní příznivý vliv na zánět nemá však léčba blokádou IL‑1 žádný vliv na monoklonální komponentu [11]. Vzhledem ke krátkému biologickému poločasu anakinry je její aplikace každodenní a trvalá a po vysazení se příznaky dostavují do 35– 45 hodin [1].
Z těchto a mnoha dalších pozorování upevňujících postavení anakinry v léčbě pacientů se Schnitzler‑ syndromem čerpali účastníci mezinárodního expertního setkání ve francouzském Štrasburku v květnu 2012 při přípravě oficiálních terapeutických doporučení, která podle závažnosti chorobných projevů dělí nemocné do dvou skupin a zahrnují rovněž problematiku dispenzarizace (tab. 6) [2]. U lehkých forem se kromě standardních režimových opatření (vystříhání se faktorů vyvolávajících vzplanutí příznaků) připouští i možnost bedlivého sledování bez medikamentózních zásahů. Při nedostatečné toleranci pak lze vyzkoušet některý z následujících preparátů – kolchicin (účinný asi jen ve čtvrtině případů, ale příznivý bezpečnostní profil a běžně dostupný na lékařský předpis), nesteroidní antiflogistika (v analgetické indikaci při sporadických exacerbacích), pefloxacin (zřejmě největší potenciál z této skupiny, použití však limitováno nežádoucími účinky, zejména tendinopatií), hydroxychlorochin (především u bolestivých kloubů, výborný bezpečnostní profil, ale jen ojediněle dostatečný efekt). Naopak v podmnožině závažnějších forem Schnitzler‑ syndromu má anakinra dominantní pozici (stupeň doporučení C podle zásad medicíny založené na důkazech). Léčebné alternativy uváděné štrasburskými doporučeními v případě mírného průběhu onemocnění se ale vztahují jen k minoritní skupině pacientů. S ohledem na všeobecně udávaný dlouhý interval od příznaků k diagnóze se totiž u většiny nemocných rozvine torpidní forma vyžadující cílenou biologickou léčbu. Navíc ostatní preparáty, jak je shrnuje tab. 6, vykazují podstatně nižší účinek při potenciálně vyšší toxicitě a jejich upřednostnění v klinické praxi může znamenat jen další oddálení inhibitorů IL‑1 se zbytečným prodlužováním expozice pacientů škodlivému působení prozánětlivého stavu organizmu. Z těchto důvodů naše pracovní skupina jednoznačně podporuje podání anakinry v rámci léčby první linie u všech pacientů s jistou diagnózou Schnitzler‑ syndromu.

Ačkoliv ve většině publikovaných případů navodila první injekce během několika hodin kompletní remisi nemoci, nově se v literatuře objevily sporadické případy rezistence vůči IL‑1 blokádě anakinrou a rilonaceptem (viz níže), které příznivě zareagovaly na antagonistu humánního receptoru pro IL‑6, tocilizumab (RoActemra™, Roche) [37]. Bude to zřejmě otázka několika následujících let, zdali se skutečně potvrdí členění Schnitzler‑ syndromu na pravděpodobně převažující okruh nemocných odpovídajících na anti‑IL‑1 léčbu a na minoritní skupinu určenou pro anti‑IL‑6 preparáty. Štrasburský konsenzus nicméně pro případy nedostatečné responzivity doporučuje v první řadě navýšit dávkování anakinry na 200– 300 mg denně. Na druhé straně u pacientů dobře kompenzovaných anakinrou by měla být vždy snaha nastavit nejnižší účinné dávkování (prodloužením intervalu na 36– 48 hodin, podobně jako u našeho případu 1, nebo aplikací jen části ampulky). Pozornost si v této souvislosti zasluhuje nález Q703K polymorfizmu v genu NLRP3, který jsme odhalili u případu 2, nikoliv ale v případech 1 a 5. Podle nedávno publikované studie totiž může tato varianta predisponovat její nositele k rozvoji klinických projevů podobných těm, které doprovázejí hereditární autoinflamatorní syndromy [38]. Můžeme tak spekulovat o podílu této abnormity potencující proinflamatorní stav organizmu na nedostatečné léčebné odpovědi na standardní dávkování anakinry u případu 2. Zde by řešení mohl přinést intenzifikovaný režim, podání inhibitoru IL‑6 anebo jiného blokátoru IL‑1 (viz níže).
S ohledem na popsané dva případy spontánní remise doporučuje štrasburské usnesení dále přerušení léčby anakinrou po dvou letech kompletní remise ke zhodnocení, zda-li dojde k rozvoji původních symptomů. Toto pozastavení by mělo trvat alespoň dva týdny, neboť díky kompenzatorním mechanizmům může dojít ke vzplanutí příznaků ihned po přerušení aplikace. Někteří experti navrhují před ukončením anakinry překlenovací terapii kolchicinem (zpočátku tři měsíce, při trvání remise následné prodloužení na dalších 3– 6 měsíců) [4]. U našich případů 1, 2, 5 léčených více než dva roky jsme však k těmto krokům nepřistoupili (v případu 2 nebyla dosažena kompletní remise, případ 1 absolvoval během téměř 20 let 10 linií léčby, hodnotíme jej tedy jako těžkou, špatně kompenzovatelnou formu, podobně jako případ 6,u něhož byl s neúspěchem zkoušen i kolchicin). Délka terapie u ostatních pacientů (případy 3, 4, 6) dosud uvedenou hranici nepřesáhla.
Anakinra má výborný bezpečnostní profil, nežádoucí účinky jsou mírné a vyskytují se s nízkou frekvencí (dyspepsie, infekční komplikace, neutropenie), častější kožní reakce bývají jen přechodné [39]. V průběhu podávání by laboratorní odběry měly zahrnovat počet neutrofilních granulocytů, jaterní testy, cholesterol a triglyceridy. Možnou kontraindikaci představuje léková hypersenzitivita a pokles glomerulární filtrace pod 30 ml/ hod [4]. V našem souboru nebyla anakinra spojována s žádnými závažnými nežádoucími projevy. Naše výsledky dále prokazují velmi dobrou toleranci léku při dlouhodobém užívání (medián 30,5 měsíce).
Novinku v léčbě Schnitzler‑ syndromu představují další dva přípravky používané v indikaci kryopyrinopatií, které blokují IL‑1 s delším biologickým poločasem, než má anakinra. I přes jejich nesporný efekt a komfort pro pacienty je však zatím nelze pro nízkou nákladovou efektivnost a nedostatek relevantních dat ze studií zahrnout do běžných terapeutických schémat. Rilonacept (Arcalyst™, Regeneron Pharmaceuticals, Inc., aktuálně není v České republice registrován) je dimerický fúzní protein, který se skládá z extracelulární části receptoru pro IL‑1 (IL‑1R1) a z jeho akcesorního proteinu (IL‑1R‑ AcP), které jsou oba navázány na Fc fragment IgG protilátky. Vzniklý komplex účinně blokuje IL‑1 [40]. Výhodou je možnost aplikovat 160 mg podkožně v intervalu jednoho týdne, přičemž iniciální nasycovací dávka odpovídá 2násobku, tedy 320 mg. Zatím jediná publikace hodnotí bezpečnost a efektivitu u osmi pacientů v rámci prospektivní, jednocentrové, otevřené studie. Při dosažení čtyř kompletních a tří parciálních remisí (jeden případ rezistence) byla tolerance rilonaceptu vynikající, žádné závažné nežádoucí účinky hlášeny nebyly [41].
Druhou účinnou molekulu zastupuje kanakinumab (Ilaris™, Novartis), humánní monoklonální protilátka proti lidskému IL‑1β, která po navázání na tento ligand brání jeho interakci s receptory pro IL‑1 a následnému spuštění zánětlivé kaskády [42]. Aplikuje se 150 mg podkožní injekcí a doporučený dávkovací interval je osm týdnů. Dle medicínské databáze MEDLINE vedl kanakinumab ve všech čtyřech zdokumentovaných případech ke kompletní remisi Schnitzler‑ syndromu, a to při absenci jakýchkoliv závažných nežádoucích účinků. Tento terapeutický úspěch má navíc jednu neméně důležitou patofyziologickou implikaci, podle níž právě IL‑1β zodpovídá za typické příznaky Schnitzler‑ syndromu. Zatímco anakinra blokuje jak IL‑1α, tak IL‑1β, kanakinumab selektivně atakuje toliko IL‑1β za plného léčebnéhoúčinku [43,44].
Závěr
U Schnitzler‑ syndromu bývá chronická kopřivka, někdy provázená intenzivním pruritem, obvykle prvním příznakem nemocných, který je přivede k lékaři. Jelikož je diferenciální diagnostika urtikariálních morf značně rozsáhlá, bývá při sporadických zprávách o Schnitzler‑ syndromu v odborném tisku toto onemocnění často diagnostikováno opožděně. Bez znalosti správné diagnózy je však léčba frustrující a její výsledky neuspokojivé. Dosud nejúčinnější terapie spočívá v blokádě IL‑1 pravidelnou aplikací anakinry, která má nejen ústřední postavení v nejnovějších, štrasburských doporučeních, ale prokázala rovněž jednoznačný efekt v námi prezentované kohortě šesti pacientů ze tří klinických pracovišť v rámci České republiky.
Anakinra je účinný, ověřený a bezpečný preparát s možností dlouholetého podávání při zachování původní efektivity a tolerance. Užívání anakinry zcela odstraní všechny nepříjemné, často invalidizující příznaky Schnitzler‑ syndromu tím, že potlačí autonomní zánětlivou reakci v organizmu. Tento lék by měl být v rámci první linie dostupný ve standardním dávkování 100 mg denně všem pacientům s prokázanou diagnózou Schnitzler‑ syndromu. Věříme, že zde prezentovaná data se stanou nejen podkladem pro efektivní farmakoekonomický odhad v rámci skupiny velmi vzácných diagnóz na národní úrovni, ale že rovněž poskytnou klinickým lékařům ucelené argumenty při jednání s plátci zdravotní péče. Zároveň je však třeba vyzdvihnout úlohu vysoce specializovaných centralizovaných zdravotnických služeb úzce spjatých s existencí celostátního registru, jehož vznik bychom chtěli touto cestou podnítit a podpořit tak mezioborovou a mezicentrovou spolupráci zacílenou na optimalizaci péče o pacienty se Schnitzler‑ syndromem.
Práce byla podpořena granty IGA MZ ČR č. NT12215, NT12130, NT13190, grantem MUNI/A/0723/2012, podpořeno MZ ČR – MZ RVO (FNBr, 65269705).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Petr Szturz, Ph.D.
Interní hematologická a onkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: petr.szturz@fnbrno.cz
Obdrženo: 15. 9. 2013
Přijato: 26. 9. 2013
Zdroje
1. Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis 2010; 5: 38.
2. Simon A, Asli B, Braun‑ Falco M et al. Schnitzler‘s syndrome: diagnosis, treatment, and follow‑up. Allergy 2013; 68(5): 562– 568.
3. Schnitzler L. Lésions urticariennes chroniques permanentes (érytheme pétaloıde?) Cas cliniques, n° 46 B. Journée Dermatologique d’Angers, 28 octobre 1972.
4. Schnitzler L, Schubert B, Boasson M et al. Urticaire chronique lesions osseuses macroglobulinémie IgM: Maladie de Waldenström? Bull Soc Fr Dermatol Syph 1974; 81: 363– 366.
5. de Koning HD, Bodar EJ, van der Meer JW et al. Schnitzler syndrome: beyond the case reports: review and follow‑up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum 2007; 37(3): 137– 148.
6. Jain T, Offord CP, Kyle R et al. Schnitzler syndrome: an under diagnosed clinical entity. Haematologica 2013; 98(10): 1581– 1585.
7. Szturz P, Adam Z, Šedivá A et al. Schnitzler‑ syndrom: diagnostika a léčba. Klin Onkol 2011; 24(4): 271– 277.
8. Lipsker D, Veran Y, Grunenberger F et al. The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore) 2001; 80(1): 37– 44.
9. SanMartín O, Febrer I, Botella R et al. Urticarial lesions and monoclonal IgM gammopathy. Schnitzler’s syndrome. Arch Dermatol 1994; 130(9): 1195– 1198.
10. Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore) 2009; 88(1): 23– 31.
11. Besada E, Nossent H. Dramatic response to IL1- RA treatment in longstanding multidrug resistant Schnitzler‘s syndrome: a case report and literature review. Clin Rheumatol 2010; 29(5): 567– 571.
12. Loock J, Lamprecht P, Timmann C et al. Genetic predisposition (NLRP3 V198M mutation) for IL‑1- mediated inflammation in a patient with Schnitzler syndrome. J Allergy Clin Immunol 2010; 125(2): 500– 502.
13. Rowczenio DM, Trojer H, Russell T et al. Clinical characteristics in subjects with NLRP3 V198M diagnosed at a single UK center and a review of the literature. Arthritis Res Ther 2013; 15(1): R30.
14. Terpos E, Asli B, Christoulas D et al. Increased angiogenesis and enhanced bone formation in patients with IgM monoclonal gammopathy and urticarial skin rash: new insight into the biology of Schnitzler syndrome. Haematologica 2012; 97(11): 1699– 1703.
15. Szturz P, Adam Z, Klabusay M et al. Schnitzler‑ syndrom: popis případu, zkušenosti s léčbou glukokortikoidy a preparátem anakinra (KineretTM) a sledování cytokinové odpovědi organizmu. Vnitř Lék 2011; 57(1): 97– 112.
16. Adam Z, Krejčí M, Pour L et al. Zhodnocení dvouleté léčby Schnitzlerova syndromu (kopřivkové velkoplošné morfy, monoklonální IgM gamapatie a osteolyticko‑osteosklerotické změny skeletu) preparátem anakinra (Kineret). Vnitř Lék 2009; 55(12): 1196– 1197.
17. Claes K, Bammens B, Delforge M et al. Another devastating complication of the Schnitzler’s syndrome: AA amyloidosis. Br J Dermatol 2008; 158(1): 182– 184.
18. Verret JL, Leclech C, Rousselet MC et al. Schnitzler syndrome and Waldenström disease. Fatal outcome of the original case. Ann Dermatol Venereol 1993; 120(6– 7): 459– 460.
19. Asli B, Brouet JC, Fermand JP. Spontaneous remission of Schnitzler syndrome. Ann Allergy Asthma Immunol 2011; 107(1): 87– 88.
20. Martinez‑ Taboada VM, Fontalba A, Blanco R et al. Successful treatment of refractory Schnizler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum 2005; 52(7): 2226– 2227.
21. Flórez AF, Gallardo Agromayor E, García‑Barredo R et al. Radiological aid to clinical diagnosis of Schnizler’s syndrome: multimodality imaging approach. Clin Rheumatol 2008; 27(1): 107– 110.
22. Sedivá A, Poloučková A, Podrazil M et al. Characterization of the B‑ cell compartment in a patient with Schnitzler syndrome. Scand J Rheumatol 2011; 40(2): 158– 160.
23. Borradori L, Rybojad M, Puissant A et al. Urticarial vasculitis associated with monoclonal IgM gammopathy, Schnitzler’s syndrome. Brit J Dermatol 1990; 123(1): 113– 118.
24. Berdy SS, Bloch KJ. Schnitzler’s syndrome: A broader clinical spectrum. J Allergy Clin Immunol 1991; 87(4): 849– 854.
25. Olsen E, Forre O, Lea T et al. Unique antigenic determinants used as markers in a patient with macroglobulinemia urticaria. Similar idiotypes demonstrated in the skin and on peripheral blood lymphocytes. Acta Med Scand 1980; 207(5): 379– 384.
26. Lebbe C, Rybojad M, Klein F et al. Schnitzler’s syndrome with sensomotor neuropaty. J Amer Acad Dermatol 1994; 30(2 Pt 2): 316– 318.
27. Eiling E, Möller M, Kreiselmaier I et al. Schnizler syndrome: treatment failure to rituximab but response to anakinra. J Am Acad Dermatol 2007; 57(2): 361– 364.
28. Aikawa NE, Silva CA, Bonfá E et al. Schnitzler‘s syndrome improvement after anti‑TNF‑alpha therapy. Joint Bone Spine 2010; 77(5): 491.
29. de Koning HD, Bodar EJ, Simon A et al. Beneficial response to anakinra and thalidomide in Schnitzler‘s syndrome. Ann Rheum Dis 2006; 65(4): 542– 544.
30. Asli B, Bienvenu B, Cordoliani F et al. Chronic urticaria and monoclonal IgM gammopathy (Schnitzler syndrome): report of 11 cases treated with pefloxacin. Arch Dermatol 2007; 143(8): 1046– 1050.
31. Kastritis E, Katoulis A, Terpos E et al. Schnitzler‘s syndrome: increased levels of bone formation and angiogenesis factors are reduced after successful pefloxacin treatment. Clin Lymphoma Myeloma 2008; 8(6): 359– 362.
32. Janier M, Bonvalet D, Blanc MF et al. Chronic urtica and macroglobulinemia (Schnitzler’s syndrome): report of two cases. J Amer Acad Dermatol 1989; 20(2 Pt 1): 206– 211.
33. Asahina A, Sakurai N, Suzuki Y et al. Schnitzler‘s syndrome with prominent neutrophil infiltration misdiagnosed as Sweet‘s syndrome: a typical example of urticarial neutrophilic dermatosis. Clin Exp Dermatol 2010; 35(4): e123– e126.
34. Migliorini P, Del Corso I, Tommasi C et al. Free circulating interleukin‑18 is increased in Schnitzler syndrome: a new autoinflammatory disease? Eur Cytokine Netw 2009; 20(3): 108– 111.
35. Pizzirani C, Falzoni S, Govoni M et al. Dysfunctional inflammasome in Schnitzler‘s syndrome. Rheumatology (Oxford) 2009; 48(10): 1304– 1308.
36. Ryan JG, de Koning HD, Beck LA et al. IL‑1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol 2008; 121(1): 260– 262.
37. Krause K, Feist E, Fiene M et al. Complete remission in 3 of 3 anti‑IL‑6‑treated patients with Schnitzler syndrome. J Allergy Clin Immunol 2012; 129(3): 848– 850.
38. Vitale A, Lucherini OM, Galeazzi M et al. Long‑term clinical course of patients carrying the Q703K mutation in the NLRP3 gene: a case series. Clin Exp Rheumatol 2012; 30(6): 943– 946.
39. Nuki G, Bresnihan B, Bear MB et al. Long‑term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin‑1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double‑blind, placebo‑ controlled trial. Arthritis Rheum 2002; 46(11): 2838– 2846.
40. Stahl N, Radin A, Mellis S. Rilonacept‑ CAPS and beyond. Ann N Y Acad Sci 2009; 1182: 124– 134.
41. Krause K, Weller K, Stefaniak R et al. Efficacy and safety of the interleukin‑1 antagonist rilonacept in Schnitzler syndrome: an open‑ label study. Allergy 2012; 67(7): 943– 950.
42. Church LD, McDermott MF. Canakinumab: a human anti‑IL‑1β monoclonal antibody for the treatment of cryopyrin‑associated periodic syndromes. Expert Rev Clin Immunol 2010; 6(6): 831– 841.
43. de Koning HD, Schalkwijk J, van der Meer JW et al. Successful canakinumab treatment identifies IL‑1β as a pivotal mediator in Schnitzler syndrome. J Allergy Clin Immunol 2011; 128(6): 1352– 1354.
44. Vanderschueren S, Knockaert D. Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum 2013; 42(4): 413– 416.
45. Ščudla V, Budíková M, Petrová P et al. Analýza sérových hladin vybraných biologických ukazatelů u monoklonální gamapatie nejistého významu a mnohočetného myelomu. Klin Onkol 2010; 23(3): 171– 181.
46. Paglia F, Dionisi S, De Geronimo S et al. Biomarkers of bone turnover after a short period of steroid therapy in elderly men. Clin Chem 2001; 47(7): 1314– 1316.
47. Engvall IL, Svensson B, Tengstrand B et al. Impact of low‑dose prednisolone on bone synthesis and resorption in early rheumatoid arthritis: experiences from a two‑year randomized study. Arthritis Res Ther 2008; 10(6): R128.
Štítky
Paediatric clinical oncology Surgery Clinical oncologyČlánok vyšiel v časopise
Clinical Oncology

2014 Číslo 2
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
- Obstacle Called Vasospasm: Which Solution Is Most Effective in Microsurgery and How to Pharmacologically Assist It?
Najčítanejšie v tomto čísle
- Renálny onkocytóm s histologickými črtami invázie – kazuistika
- Aplikace poznatků psychoneuroimunologie v kontextu komplexní onkologické léčby karcinomu prsu
- Lobulární karcinom prsu u muže – kazuistika a přehled literatury
- Další postupný ústup od axilární disekce u časného karcinomu prsu