
The Role of Heat Shock Proteins in Leukemia
Úloha bielkovín tepelného šoku v leukémii
Bielkoviny tepelného šoku (heat shock proteins – HSPs) HSP27, HSP70 a HSP90 sú molekulárne šaperóny, ktorých expresia sa zvyšuje ovplyvnením buniek po pôsobení enviromentálneho stresu, akými sú tepelný šok, ťažké kovy, oxidačný stres alebo pri patologických podmienkach ako napr. ischémia, infekcia a zápal. Ich protektívna úloha pomáha bunke vyrovnať sa s letálnymi podmienkami. HSPs sú skupina bielkovín, ktoré v zdravých bunkách zodpovedajú za udržanie homeostázy, za interakciu s rôznymi bielkovinovými substrátmi na zabezpečenie ich správneho zbalenia, zabraňujú zbaľovaniu intermediátorov, ktoré vedú ku tvorbe chybne zbalených alebo poškodených molekúl. Ukázalo sa, že interagujú s rôznymi kľúčovými bielkovinami a zohrávajú úlohu v regulácii apoptózy. Viaceré bielkoviny tepelného šoku preukázali priamu interakciu s rozličnými zložkami úzko regulovanej kaspázovo-závislej programovanej bunkovej smrti. Tieto bielkoviny rovnako ovplyvňujú kaspázovo-nezávislú dráhu apoptózy väzbou s apoptickými faktormi. Bielkoviny tepelného šoku sú odlišne exprimované v hematologických malignitách. Z dôvodu ich asociácie a úlohy v leukémiách, HSPs predstavujú zaujímavý cieľ v antileukemickej terapii. Tento prehľadový článok opisuje rôzne molekuly intaragujúce s antiapoptotickými bielkovinami HSP70 a HSP90, ktoré by mohli byť využité v nádorovej terapii na základe ich inhibície.
Klúčové slová:
bielkoviny tepelného šoku – inhibítory – leukémia – apoptóza
Táto práca bola podporená grantom „Zvýšenie možností kariérneho rastu vo výskume a vývoji v oblasti lekárskych vied“ (IMTS 26110230067), operačný program Vzdelávanie, doc. MUDr. Ján Staško, PhD., mim. prof., 2012–2015.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Obdržané:
7. 8. 2015
Prijaté:
11. 10. 2015
Authors:
K. Kliková; I. Pilchova; A. Stefanikova; J. Hatok; D. Dobrota; P. Racay
Authors‘ workplace:
Department of Medical Biochemistry, Jessenius Faculty of Medicine, Comenius University, Martin, Slovak Republic
Published in:
Klin Onkol 2016; 29(1): 29-38
Category:
Review
doi:
https://doi.org/10.14735/amko201629
Overview
Heat shock proteins (HSPs) HSP27, HSP70 and HSP90 are molecular chaperones; their expression is increased after exposure of cells to conditions of environmental stress, including heat shock, heavy metals, oxidative stress, or pathologic conditions, such as ischemia, infection, and inflammation. Their protective function is to help the cell cope with lethal conditions. The HSPs are a class of proteins which, in normal cells, are responsible for maintaining homeostasis, interacting with diverse protein substrates to assist in their folding, and preventing the appearance of folding intermediates that lead to misfolded or damaged molecules. They have been shown to interact with different key apoptotic proteins and play a crucial role in regulating apoptosis. Several HSPs have been demonstrated to directly interact with various components of tightly regulated caspase-dependent programmed cell death. These proteins also affect caspase-independent apoptosis by interacting with apoptogenic factors. Heat shock proteins are aberrantly expressed in hematological malignancies. Because of their prognostic implications and functional role in leukemias, HSPs represent an interesting target for antileukemic therapy. This review will describe different molecules interacting with anti-apoptotic proteins HSP70 and HSP90, which can be used in cancer therapy based on their inhibition.
Key words:
heat shock proteins – inhibitors – leukemia – apoptosis
Introduction
The group of proteins involved in folding and unfolding of other proteins were originally called stress proteins or heat shock proteins (HSPs). They are found in all living organisms, from bacteria to humans [1]. HSPs were first discovered in 1962 by Ritossa as a set of highly conserved proteins whose expression was induced in salivary gland chromosome puffs of Drosophila melanogaster in response to transient exposures to elevated temperatures. The cellular response to stress is induced at the molecular level by induced synthesis of heat shock proteins [2]. HSPs have been classified into six families according to their molecular sizes: the small HSPs (between 15 – 30 kDa), HSP40, HSP60, HSP70, HSP90 and HSP100 [3]. Within each family, there are members that are constitutively or inducibly regulated and/ or targeted to different compartments. The functions of HSP90 are in the cytosolic and nuclear compartments, whereas the function of GRP94 is an analogue of endoplasmic reticulum. The members of the HSP70 family are located in cytosol; for example, the major inducible HSP70 (called HSP70 or HSP72) or the constitutively expressed HSC70, mtHSP70 is a mitochondrial protein, whereas GRP78 is localized in the endoplasmic reticulum [4]. HSPs with high molecular weight (e. i. HSP70 and HSP90) are ATP dependent chaperones that require co-chaperones to modulate their conformation and binding to ATP. The function of HSP70, ATP dependent molecular chaperone, is to assist in folding of newly synthetized polypeptides, the assembly of multi-protein complexes and the transport of proteins across cellular membranes. The HSP70 co-chaperones are proteins like HSP40, CHIP, HOP, HIP, BAG-1 and BAG-3. Co-chaperones of HSP90 include proteins such as Cdc37, p23, Aha1, PP5, HOP and CHIP. The small HSPs are ATP independent chaperones [5,6]. The pleiotropic activities of HSPs can contribute to tumorigenesis because they play an essential role as molecular chaperones that provide the cancer cells with an apportunity to alter protein activities, components of cell cycle, kinases and other proteins that influence cell growth [7]. The abnormally high activities and/ or expressions of HSP27, HSP70 or HSP90 are increased after different kinds of stimuli including oxidative stress, hyperthermia, radiation, ligation to death receptors or addition of anticancer drugs [8]. The small family of transcription factors called heat shock factors (HSFs) are the regulators of stress-inducible expression of HSP genes. They bind consensus heat shock elements (HSEs) which are located at various distances upstream of the site of transcription initiation [9]. It is known that the stress-induced formation of homotrimer of HSF1 and a number of post-translation modifications convert the factor into an active form that moves toward a nucleus and binds within the 5‘ promoter regions of HSP genes that is critical point to trigger of HSP transcription. In humans, three heat shock factors HSF1, HSF2 and HSF4 have been characterized. The ubiquitously expressed HSF1 has a pivotal role in the stress-induced expression of HSP genes. Activation of HSF2 happens during inhibition of the ubiquitin-dependent proteasome, hemin-induced cell differentiation or in specific stages of development [10,11]. HSF4 constitutively binds to DNA and appears to be preferentially expressed in human tissues, such as heart, pancreas, sceletal muscle, brain and lung. It has been suggested to be an inhibitor of stress-induced gene expression [12,13]. HSF4 consists of two isoforms HSF4a and HSF4b, which are derived by alternative RNA splicing events. HSF4b has the transcriptional potential, whereas HSF4a does not [13 – 16]. HSF4 may have physiological roles during development [17]. It has been shown recently that mutations of HSF4 are associated with dominant inherited cataracts in humans [18]. The group of dr. Fujimoto has found that HSF4 has a major HSE-binding activity specifically in the lens extract. They generated mice in which the HSF4 gene was mutated. The anomalies of the lens revealed novel HSF4 target genes that are essential for cell growth and differentiation [19]. The over-expression of HSFs has been associated with therapeutic resistance and with a poor clinical outcome in acute leukemias and myelodysplasic syndrome [20,21]. In this way, HSPs may have important therapeutic implications and they can be targeted by specific drugs and/ or inhibitors.
HSPs in apoptosis
One of the mechanisms to maintain cancer cell survival are the cytoprotective functions of stress-inducible HSP proteins, such as HSP27, HSP70 and HSP90. The metabolic and signal transduction pathways are extensively rewired in the cancer cells, thereby becoming dependent on these proteins. Their anti-apoptotic effect is associated with key effectors of apoptosis (Fig. 1) [8]. HSP70 has an important role at the death receptors level because it binds to DR4/ 5 (death receptors 4 and 5), thereby inhibiting TRAIL-induced assembly and activity of DISC (death inducing signaling complex) [22]. Antiapoptotic protein HSP70 binds and blocks c-Jun N-terminal Kinase (JNK) activity at the pre-mitochondrial level. The activation of JNK signaling pathway and activation of caspase 3 is induced by deficiency of HSP70 that triggers apoptosis by hyperosmolarity [23]. Inhibition of Bax translocation and its insertion by HSP70 is a consequence of prevention to permeabilize the mitochondrial membrane and to release cytochrome c and apoptosis inducing factor (AIF) [24]. It has been demonstrated that HSP70 and HSP90 bind to the apoptotic protease activating factor 1 (Apaf-1) and inhibit its oligomerization and further recruitment of procaspase-9 to the apoptosome [25]. The lysosomal membrane permeabilization is inhibited by HSP70 thereby preventing proteases cathepsines to release [26,27]. The genes coding for HSPs are induced in hemin-induced differentiation of human K-562 erythroleukemic cells [28]. One of another mechanism of HSPs is the subcellular localization that determines whether a cell is to die or to differentiate. Lanneau with his colleagues have demonstrated that a nuclear HSP70 has the essential role for erythroid differentiation. HSP70 and caspase-3 are accumulated in the nucleus of the erythroblast during formation of red blood cells [29,30]. The erythropoiesis requires transcription factor GATA-1 which directly binds to HSP70. As a result, erythroblasts continue their differentiation process instead of dying by apoptosis [31].
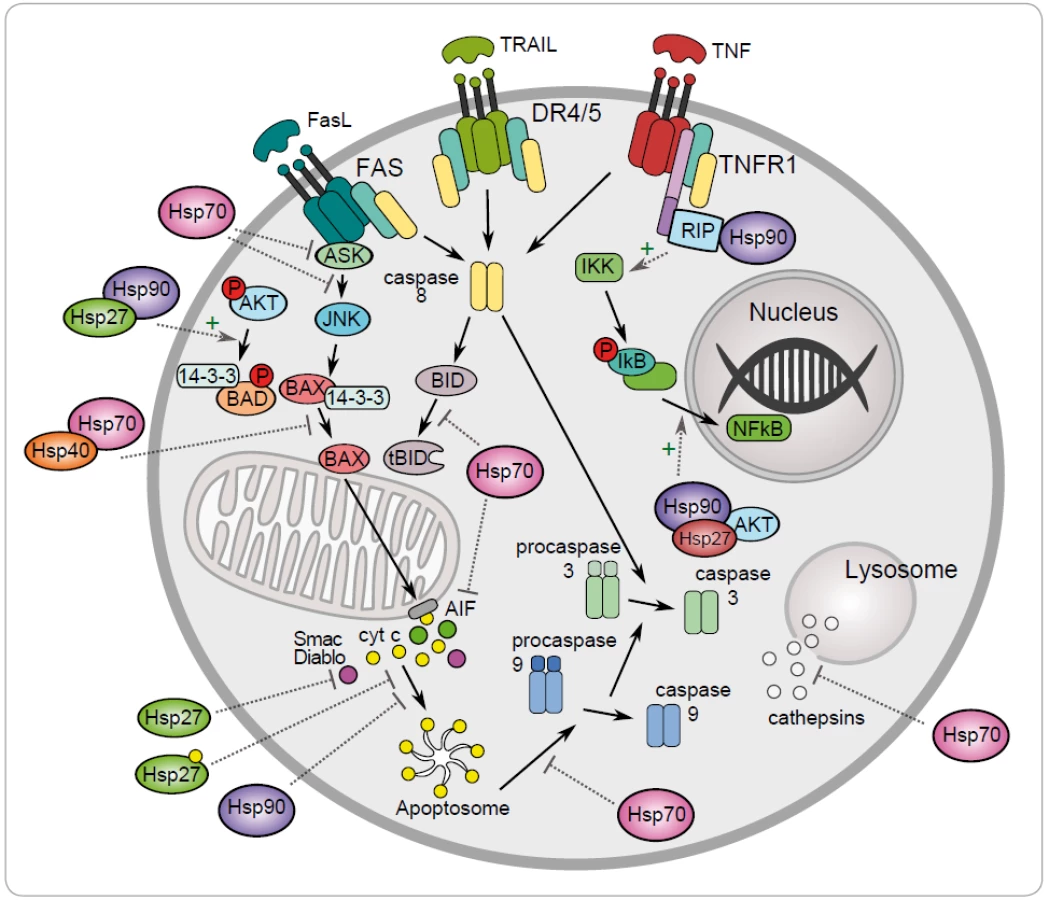
Antiapoptotic function of HSP90 can be explained by its chaperone role assuring the stability and activity of different cellular proteins, kinases and transcription factors, such as NF-κB, p53, Akt, Raf-1 and JNK. It has been demonstrated that HSP90α gene expression could be repressed by p53 in UV-irradiated cells [32]. HSP90 interacts with and stabilizes receptor RIP (receptor interacting protein). Upon ligation of TNFR-1,RIP-1 is recruited to the receptor and promotes the activation of NF-κB and JNK. Degradation of RIP-1 in the absence of HSP90 precludes activation of NF-κB mediated by TNF-α and senzitizes cells to apoptosis [33]. NF-κB signaling pathway is regulated by HSP90 via IKK complex, which is composed of two catalytic p65/ p50 subunits and one regulatory subunit – IκB [34]. The anti-apoptotic action of HSP90 is reflected by its capacity to interact with phosphorylated serine/ threonine kinase Akt/ PKB, a protein that generates a survival signal in response to growth factor stimulation. Interaction HSP90 with phosphorylated Akt leads to phosphorylation of proapoptotic Bcl-2 family protein Bad and caspase-9, leading to their inactivation and to cell survival [35]. Akt has been shown to phosphorylate IκB kinase, which results in promotion of NF-κB mediated inhibition of apoptosis [36].
HSP90 structure
HSP90 is abundantly expressed in the cytoplasm of most human cells and makes up 1 – 2 % of cytosolic proteins [37]. Five isoforms of HSP90 have been identified so far: HSP90AA1 (HSP90α1), HSP90AA2 (HSP90α2), HSP90AB1 (HSP90β), HSP90B1 (GRP94), TRAP1 (mitochondrial HSP90), HSP90N is an artefact [38]. Prominent cytoplasmic isoforms are HSP90α (inducible form) and HSP90β (constitutive form) that are expressed by two distinct genes [39]. All of the mammalian HSP90 isoforms share a similar structure. HSP90 exists as a homodimer, which contains three relevant domains: A: the C-terminal dimerization domain contains EEVD motif that enables the binding to tetratricopeptide repeat-binding (TRP) domain of cochaperones, B: the charged middle linker region with high affinity for co-chaperones and the client proteins, and C: the adenine binding N-terminal domain [40,41], which is absent in HSP90N [42]. The N-terminal dimerization of HSP90 stimulates the intrinsic and essential ATPase activity which is controlled by set of accessory proteins called co-chaperones [43].
HSP90 client proteins
The chaperone function of HSP90 ensures the correct conformation, intracellular localization, activity and proteolytic turnover of a various proteins that are implicated in cell growth, differentiation and survival [44]. HSP90 client proteins play a role upon binding and hydrolysis of ATP and in the conformations changes of HSP90. The essential role of HSP90 is important for stability and function of many oncogenic client proteins that include apoptotic mediators (Bcl-2, Apaf-1), cell cycle regulatory proteins (hTERT, CDK4), tumor supressor genes (p53) [45], transcription factors (HSF-1, HIF-1), signaling molecules (Src, Lck, Akt, Raf-1), steroid hormone receptors, such asestrogen, androgen, progesterone, glucocorticoid receptors and mutant fusion kinase (BCR-ABL) [44]. Inhibition of HSP90 ATPase activity disrupts the “folding cycle”, which involves multiple co-chaperone proteins leading to the destabilization, ubiquitination and degradation of client proteins, such as signaling proteins (the ligand-dependent transcription factors) or signal transducing kinases by the proteasome [39].
HSP70 structure
The stress inducible form of heat shock protein HSP70 is upregulated by HSF. HSP70 protects cells from apoptosis, both upstream and downstream of the effector caspase activation [46]. HSP70 inhibits the apoptotic pathway by binding to Bax and Apaf-1 preventing them from continuing the apoptotic cascade [29]. A novel HSP70 co-chaperone called HSP70 binding protein 1 (HspBP1), an intracellular protein abundant in tissue [47], was found to bind to the ATPase domain of HSP70 and inhibit its ability to refold denatured proteins [48]. The molar ratio of HspBP1 to HSP70 in cells might be expected to be an important determinant of the interaction between these two proteins as well as of the function of the resulting complex [49]. The role of HSP70 as ATP dependent molecular chaperone is to assist in folding of newly synthesized polypeptides, to assemble the multi-protein complexes and to transport the proteins across cellular membranes [50]. HSP70 contains two distinct functional domains: a peptide binding domain (PBD) includes a carboxyl-terminal chaperone EEVD motif which is responsible for substrate binding and refolding, and the amino-terminal ATPase domain (ABD) [37].
HSP70 client proteins
The ABD domain releases the client proteins of HSP70 after ATP hydrolysis. The co-chaperones bind to HSP70 and regulate its chaperone function. There are three classified groups of HSP70 co-chaperones: 1. the co-chaperones with J-domain are large group that binds to the HSP70 ABD and stimulate the low ATPase activity of this chaperone, such as HSP40, 2. the nucleotide exchange factor co-chaperones catalyze the release of ADP which is necessary for completion of HSP ATPase cycle, such as BAG-1, HSP110 and HSPB1, 3. the TPR domain co-chaperones that bind to the C-terminal EEVD motif, such as Hop and CHIP. Both of them are essential co-chaperones of HSP70 and HSP90 complexes and CHIP has an ubiquitin ligase activity that is implicated in the ubiquitination of HSP client proteins [37]. BAG-1 is the apoptosis regulatory protein interacting with HSP70 that simultaneously regulates the activities of proteins, such as Bcl-2 and Raf-1. HSP70/ BAG-1 complex regulates the cell growth in the response of the stress and activity of Raf-1/Erk kinase [51,52].
Overexpression of HSP90 and HSP70 in hematological malignancies
The basal level of HSP protein expression is low or absent in normal, non-transformated cells and tissues. In contrast, the HSPs are abundantly expressed in hematological malignancies, including lymphoid diseases and chronic (CML) or acute myeloid leukemias (AML). Cancer cells need high levels of HSPs for their survival [53]. Over 90% of cases of CML possess a human fusion oncogene Bcr-Abl as a result of reciprocal translocation between chromosome 9 and 22 [54]. Bcr-Abl positive cells are characterized by increased proliferation with high resistance to anti-leukemia drugs [55]. HSP70 and HSP90 bind and stabilize BCR-ABL tyrosine kinase and were found to be overexpressed in CML blast crisis and in cell line K562 established from CML primary cells [56]. The cell proliferation could be stimulated by upregulation of HSP70 and HSP90 through the control of BCR-ABL tyrosine kinase functions, which phosphorylates and activates Akt that, in turn, inactivates Bad and caspase-9 and phosphorylates STAT5. Phosphorylated STAT5 binds DNA and increases the expression of anti-apoptotic protein Bcl-xL. In contrast, when the activity of tyrosine kinase BCR-ABL and PI-3K is inhibited with imatinib or wortmannin, decreased expression of HSP70 and downregulation of STAT5 activity were observed [22]. The conformational stability and activation of mutant oncoproteins, such as c-Kit, FLT3-ITD or BCR-ABL, are strongly dependent on HSPs, leading to poor prognosis in AML [57]. The inhibition of HSP90 using 17-AAG reduces the level of the FLT3-ITD, downstream STAT5 activity [58]. In peripheral blood and bone marrow of patients diagnosed with de novo acute myeloblastic leukemia as well as in leukemia cell lines (K-562, Jurkat and CCRF-CEM), increased expression of HSP90α at the level of mRNA was observed [49]. Similar results were published in the study by Thian et al. where they investigated the expression of HSP90α a in different leukemia cell lines (HL-60, NB4, Molt-4) and human bone marrow mononuclear cells derived from acute leukemia patients. In the untreated patients and in patients in remission, higher expression of HSP90α compared to cells from healthy individuals was observed [59]. In our study, we found significantly higher protein levels of HSP27 (p < 0.001), HSP70 (p < 0.001), HSP90α (p < 0.001), HSP90β (p < 0.001) and GRP75 (p < 0.05) in K-562 cells compared to HL-60 cells while difference in GRP78 protein level between analyzed cell lines was not significant. The highest differences were observed in the levels of HSP27 (30.9 fold higher expression in K-562 cells) and HSP70 (14.5 fold higher expression in K-562) [60].
HSPs inhibitors – pharmacological targets
As molecular chaperons, HSP90 and HSP70 have been implicated in pathogenesis of multiple diseases, such as leukemia [61 – 64], neurodegenerative disorders [65], cancer [66], and infectious disease [67]. HSPs are interesting targets in therapy of hematological malignancies because of their antiapoptotic and tumourigenic properties [68]. Several oncogenic kinases, such asreceptor tyrosine kinase FLT3 involved in AMLs, fusion proteins NPM-ALK and BCR-ABL involved in chronic myeloid leukemia and anaplastic large-cell lymphomas are client proteins for HSP90 [69,70]. Studies in Bcr-Abl human leukemia cells suggest that HSP70 is a promising therapeutic target for reversing drug resistance, probably due to its ability to inhibit caspase-dependent and caspase-independent death pathways both upstream and downstream of the mitochondrial signaling [22,71]. One of the strategies to modulate intracellular regulatory pathways is to develop several direct inhibitors of HSP70 and HSP90 that would be useful alone but also in combination with other drugs in leukemia treatment.
The inhibitors of HSP70
One of the contributing factors of HSP70 is its strong connection to apoptosis; relatively little progress has been made in bringing HSP70 inhibitors to the clinic [72]. The complexity of HSP70 functions (e. g. folding, degradation, trafficking and remodeling) and its ubiquitous expression patterns create numerous challenges in designing safe and effective therapeutics [73]. Specific HSP70 inhibitors are now investigated in preclinical models but only one of hese agents, AG-858, has already reached clinical trials (Tab. 1) [74].
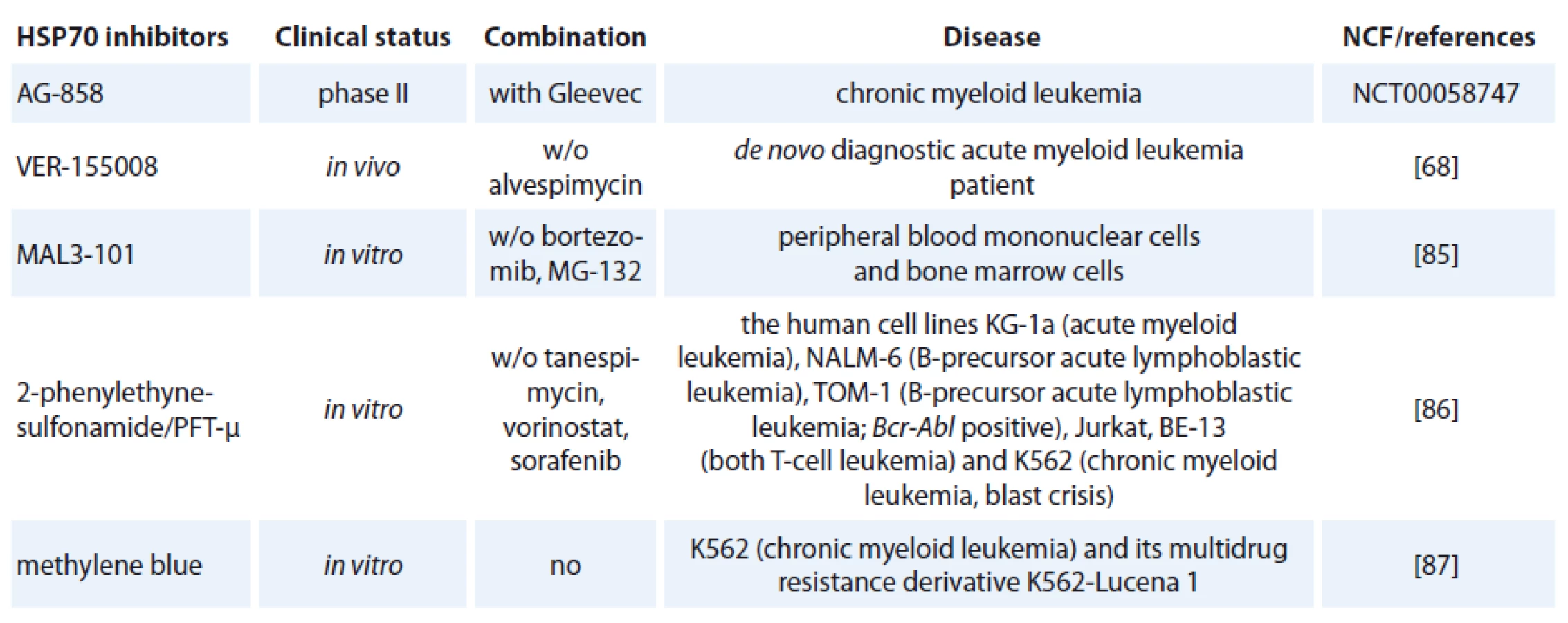
HSP70 inhibitors fall into three basic classes:
- small molecule inhibitors,
- aptamers,
- antibodies [75].
The class of the small molecule inhibiting HSP70 is represented by e. g. pifithrin-µ (PFT-µ), identified as a specific inhibitor of inducible HSP70 (Tab. 1). PFT-µ interferes with the carboxyterminal peptide-binding domain (PBD) of HSP70 and disrupts its association with client proteins [76]. The first study demonstrating significant antileukemic in vitro effects of PFT-µ alone and in combination with different antineoplastic drugs had been evaluated in ALL and AML cell lines as well as in primary AML blasts [77].
The second class of inhibitors are DNA aptamers, such as JH6, JH19, and K19 [78] or RNA aptamers, for example Antisoma [79] that are isolated from a large pool of nucleic acids by selection process called SELEX. Aptamers have the ability to bind to proteins and specifically inhibit their functions with minimal or no harmful side-effects [80]. Only few HSP70-targeting aptamers were developed, one example is the most potent aptamer A17 that binds to the NBD of HSP70 and disrupts the function of HSP70 [81]. The dual testing of aptamers with HS70/ HSP90 inhibitors could be the next step in leukemia treatment. The mechanism of action of HSP70 aptamers in leukemia is still unknown.
The third strategy for developing HSP70 inhibitors utilizes the immune system. Exciting new strategies that might affect future anti-leukemia immunotherapy include treatment using monoclonal antibodies [82]. The role of HSP70 antibody on virus production was investigated. Incubation of two rabbit transformed T-cell lines; RH/ K30 (asymptomatic) and RH/ K34 (leukemogenic) with rabbit anti-HSP70 antibodies prevented the production of human T-cell-lymphoptropic virus type I (HTLV-I) specifically in the leukemogenic cell line. The results indicated a relationship between HSP70 and virus production [83]. Recently developed monoclonal antibody, cmHSP70.1, was used in treatment of colon cancer mouse model (CT26) that led to a significantly decreased tumor weight and volume [84]. It is the only HSP70-targeted therapy currently in clinical trials (clinicaltrials.gov).
An adenosine-derived compound binding to amino-terminal ATPase domain of HSP70 is called VER-155008. It inhibits the chaperone activity of proteins of the HSP70 family [37]. VER-155008 caused a dose-dependent inhibition of cytokine-dependent AML cell proliferation both in suspension cultures and in a colony forming assay. HSP70 inhibition has both anti-leukemic and pro-apoptotic effects when tested alone, and the combination of VER-155008 and 17-DMAG (Tab. 1) inhibitors of HSP90 seems to have additive anti-leukemic effects for primary human AML cells in vitro [67]. Methylene blue (MB) is a phenothiazine with radio and photosensitizing properties and anti-tumor activity. The group of dr. Kirszberg has shown that MB (Tab. 1) was capable of inhibiting the in vitro growth of erythroleukemic cells with multidrug resistance (MDR). Lymphocytes and erythroleukemic cells were much more sensitive to the effects of MB than melanoma cells and melanocytes [87]. In our study, we have documented that the effect of methylene blue was different on the relative viability of both leukemic cell lines K-562 and HL-60. MB at concentration 1 µmol/ l and higher, decreased cell viability of HL-60 already after 24 hours. Impact of methylene blue on the viability of K-562 was slow and potent decrease of relative viability was observed after 72 hours of incubation (not published).
Inhibitors of HSP90
Inhibition of HSP90 function has been shown to cause degradation of client proteins via the ubiquitin-proteasome pathway [88], which results in depletion of multiple oncoproteins, in down-regulation of signals propagated through oncogenic signaling pathways and modulation of the malignant phenotype [89].
Inhibitors of HSP90 are classified into four groups:
- benzoquinone ansamycines and their derivatives,
- radicicol and its derivates,
- small synthetic inhibitors,
- other inhibitors [90].
Geldanamycin (GM) is a benzoquinone ansamycin, which inhibits the ATPase activity of HSP90 by competing with ATP for binding to the N-terminal nucleotide binding site, leading in ubiquitin mediated proteosomal degradation of HSP90 client proteins [91]. GM was never evaluated in clinical trials because of its poor solubility, limited in vivo stability and significant hepatotoxicity in animals [92,93]. Geldanamycine analogues have been developed that maintain similar anticancer activities but with an improved toxicity profile. 17-AAG (17-allylamino-17-desmethoxygeldanamycine) entered Phase I trials in 1999 and first therapeutic activity has been seen in melanoma, breast cancer, prostate cancer and multiple myeloma [94]. It is now in clinical trials [95], which mainly focus on specific HSP90 chaperoning targets, such as leukemia expressing Bcr-Abl and Her-2 positive breast cancer [96,97]. 17-AAG has low water-solubility, instability in solution and a lack of oral bioavailability (Tab. 2) [37]. The N,N-dimethylethylamino analogue of 17-AAG (17-DMAG), more water-soluble, has entered Phase I clinical trials in various types of leukemia (Tab. 2) and displayed higher oral bioavailability, tolarable toxicity, increased stability compared with 17-AAG [98]. 17-DMAG with arsenic trioxide has emerged as a promising therapeutic combination since they synergize to induce apoptosis and mitotic arrest in leukemic cells [99].
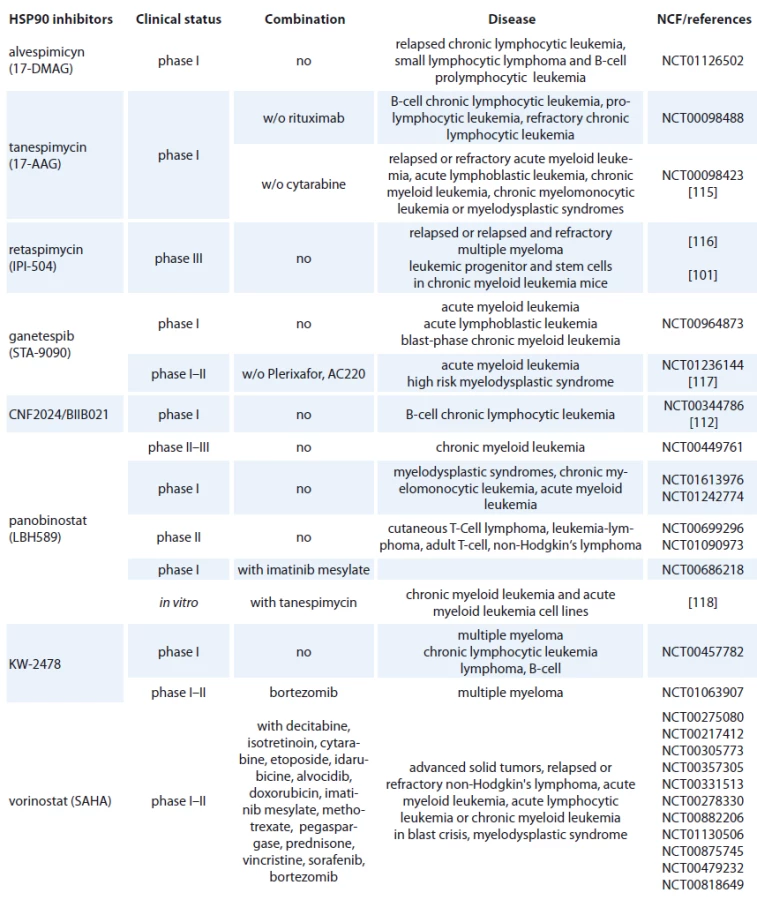
Another hydroquinone hydrochloride analogue of 17-AAG is IPI-504 (Retaspimycin) that has improved water solubility properties (Tab. 2) [100]. IPI-504 has entered the Phase III clinical trials to evaluate its potential for treating Ph-chromosome positive CML that has become resistant to therapy with tyrosine kinase inhibitors [101].
Geldanamycin-derived product, such as 17-AAG, is being clinically tested in combination with chemotherapeutic imatinib (STI-571). This agent is an effective therapy for CML characterized by the expression of the oncoprotein BCR-ABL [102]. Since BCR-ABL is a client HSP90 protein, a combination with 17-AAG is being tested in phase I clinical tials in Bcr-Abl positive leukemia with encouraging results [103].
A new approach to targeting HSP90 began with the observation that the antibiotic novobiocin binds with low affinity to a C-terminal region of HSP90 resulting in disruption of HSP90 chaperone activity with subsequent depletion of several client proteins, including Her-2, Raf-1 and mutant p53 [104,105]. More potent analogs of novobiocin have been developed, designated KU135, binds directly to HSP90 and suppresses cell proliferation of Jurkat T-lymphocytes. KU135 was found to be a potent inducer of mitochondria-mediated apoptosis and caused G2/ M arrest of leukemic cells. Indeed, KU135 was found to exert more potent antiproliferative effects than 17-AAG [106].
The second group of HSP90 inhibitors are radicicol and its derivates. Radicicol, a natural macrocyclic antifungal antiobiotic isolated from the fungus Monocillium nordinii, binds to the N-terminal ATP pocket of HSP90 leading to destabilization of HSP90-client proteins, such as Raf-1 [107]. Radicicol displays strong anti-tumor properties in vitro but not in vivo probably because of its chemical instability in tumor xenografts models [108]. This scaffold has led to generate radicicol derivates with improved solubility and stability. Several oxime derivatives and cycloproparadicicol have been developed, such as KF58333 that induced apoptosis in chronic myeloid cell line K-562 [109].
The class of synthetic inhibitors of HSP90 is composed of inhibitors based on purine scaffold. The first synthetic class of such scaffold was the PU series, such as PU-H71 and PU-DZ8. They were developed based on the binding of ADP and geldanamycin inside HSP90 ATP-binding site in combination with crystallographic and docking-based data. These PU mimic the conformation of ADP in the pocket and have a higher afinity for HSP90 than ADP [110]. CNF 2024/ BIIB021 is a purine scaffold oraly bioavailable that induces Hodgkin‘s lymphoma cell death through inhibition of NF-κB signaling pathway [111]. This agent has been evaluated in phase I clinical trial in patients with B-cell CML (Tab. 2) [112].
The other types of inhibitors of HSP90 are the molecules that block the interaction between HSP90 and its client protein or co-chaperone. Shepherdin, a peptidomimetic, is an antagonist of the interaction between HSP90 and its anti-apoptotic client protein survivin that is a key regulator of tumor cell viability [113]. It can interact with the ATP pocket of HSP90 as well as to affect a range of HSP90 client proteins suggesting it has a different mode of action. Shepherdin has anti-leukemia activity in animal models [114]. Most details of the client/ chaperone interactions are currently unknown; therefore, the strategy of targeting these associations is a challenge for further investigation.
Conclusions
The possibility to compare effects of inhibitors of the HSPs in leukemic cells offers the unique opportunity to analyze the biochemistry of these malignancies and their role in apoptosis of this disease. Given the overall poor prognosis of leukemia, great interest surrounds the development of novel and less toxic targeted therapies against signaling pathways that are aberrantly activated in leukemic patients and sustain leukemic cell survival and proliferation.
This work was supported by grant „Career growth opportunities in research and development in the field of medical sciences“ (IMTS 26110230067), operation program Education, assoc. prof. Stasko, 2012–2015.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
RNDr. Katarina Klikova, PhD.
BIOMED
Department of Medical Biochemistry
Jessenius Faculty of Medicine Comenius University
Mala Hora 11161/4D
036 01 Martin
Slovak Republic
e-mail: klikova@jfmed.uniba.sk
Submitted: 7. 8. 2015
Accepted: 11. 10. 2015
Sources
1. De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock 1999; 11 : 1 – 12.
2. Ritossa F. A new puffing pattern induced by heat shock and DNP in drosophila. Experientia 1962; 18 : 571 – 573.
3. Khalil AA, Kabapy NF, Deraz SF et al. Heat shock proteins in oncology: diagnostic biomarkers or therapeutic targets? Biochim Biophys Acta 2011; 1816 : 89 – 104. doi: 10.1016/ j.bbcan.2011.05.001.
4. Jolly C, Morimoto RI. Role of the teat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst 2000; 92(19): 1564 – 1572.
5. Parcellier A, Gurbuxani S, Schmitt E et al. Heat shock proteins, cellular chaperones that modulate mitochondrial cell death pathways. Biochem Biophysic Res Commun 2003; 304(3): 505 – 512.
6. Thomas X, Campos L, Le QH et al. Heat shock proteins and acute leukemias. Hematology 2005; 10(3): 225 – 235.
7. Schmitt E, Gehrmann M, Brunet M et al. Intracellular and extrecellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol 2007; 81(1): 15 – 27.
8. Garrido C, Brunet M, Didelot Y et al. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006; 5(22): 2592 – 2601.
9. Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem 2005; 280(39): 33097 – 33100.
10. Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol 1995; 11 : 441 – 469.
11. Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 1998; 12(24): 3788 – 3796.
12. Nakai A, Tanabe M, Kawazoe Y et al. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol 1997; 17(1): 469 – 481.
13. Tanabe M, Sasai N, Nagata K et al. The mammalian HSF4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J Biol Chem 1999; 274(39): 27845 – 27856.
14. Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005; 10(2): 86 – 103.
15. Frejtag W, Zhang Y, Dai R et al. Heat shock factor-4 (HSF-4a) represses basal transcription through interaction with TFIIF. J Biol Chem 2001; 276(18): 14685 – 14694.
16. Zhang Y, Frejtag W, Dai R et al. Heat shock factor-4 (HSF-4a) is a repressor of HSF-1 mediated transcription. J Cell Biochem 2001; 82(4): 692 – 703.
17. Nakai A. New aspects in the vertebrate heat shock factor system: Hsf3 and Hsf4. Cell Stress Chaperones 1999; 4(2): 86 – 93.
18. Bu L, Jin Y, Shi Y et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 2002; 31(3): 276 – 278.
19. Fujimoto M, Izu H, Seki K et al. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J 2004; 23(21): 4297 – 4306.
20. Thomas X, Campos L, Mounier C et al. Expression of heat shock proteins is assiciated with major adverse prognostic factors in acute myeloid leukemia. Leuk Res 2005; 29(9): 1049 – 1458.
21. Duval A, Olaru D, Campos L et al. Expression and prognostic significance of heat shock proteins in myelodysplastic syndromes. Haematologica 2006; 91(5): 713 – 714.
22. Guo F, Sigua C, Bali P et al. Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005; 105(3): 1246 – 1255.
23. Lee JS, Lee JJ, Seo JS. HSP70 deficiency results in activation of c-Jun N-terminal kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J Biol Chem 2005; 280(8): 6634 – 6641.
24. Stankiewicz AR, Lachapelle G, Foo CP et al. HSP70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 2005; 280(46): 38729 – 38739.
25. Beere HM, Wolf BB, Cain K et al. Heat shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000; 2(28): 469 – 475.
26. Gyrd-Hansen M, Nylandsted J, Jaattela M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 2004; 3(12): 1484 – 1485.
27. Bivik C, Rosdahl I, Ollinger K. HSP70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome C in human melanocytes. Carcinogenesis 2007; 28(3): 537 – 544.
28. Trinklein ND, Chen WC, Kingston RE et al. Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 2004; 9(1): 21 – 28.
29. Lanneau D, de Thonel A, Maurel S et al. Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion 2007; 1(1): 53 – 60.
30. Zermati Y, Garrido C, Amsellem S et al. Caspase activation is required for terminal erythroid differentiation. J Exp Med 2001; 193(2): 247 – 254.
31. Ribeil JA, Zermati Y, Vandekerckhove J et al. HSP70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007; 445(7123): 102 – 105.
32. Zhang Y, Wang JS, Chen Ll et al. Repression of HSP90 beta gene by p53 in UV irradiation-induced apoptosis of Jurkat cells. J Biol Chem 2004; 279(41): 42545 – 42551.
33. Lewis J, Devin A, Miller A et al. Disruption of HSP90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced factor-kappaB activation. J Biol Chem 2000; 275(14): 10519 – 10526.
34. Lanneau D, Brunet M, Frisan E et al. Heat shock proteins: essencial proteins for apoptosis regulation. J Cell Mol Med 2008; 3(12): 743 – 761. doi: 10.1111/ j.1582-4934.2008.00273.x.
35. Cardone MH, Roy N, Stennicke HR et al. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998; 282(5392): 1318 – 1321.
36. Ozes O, Mayo L, Gustin JA et al. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999; 401(6748): 82 – 85.
37. Jego G, Hazoumé A, Seigneuric R et al. Targeting heat shock proteins in cancer. Cancer Lett 2013; 332(2): 275 – 285. doi: 10.1016/ j.canlet.2010.10.014.
38. Kampinga HH, Hageman J, Vos MJ et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009; 14(1): 105 – 111. doi: 10.1007/ s12192-008-0068-7.
39. Sredhaar AS, Kalmar E, Csermely P et al. HSP90 isoforms: functions, expression and clinical importance. FEBS Lett 2004; 562(1 – 3): 11 – 15.
40. Pearl LH, Prodromou C. Structure, function, and mechanism of the HSP90 molecular chaperone. Adv Protein Chem 2001; 59 : 157 – 186.
41. Onuoha SC, Coulstock ET, Grossmann JG et al. Structural studies on the co-chaperone Hop and its complexes with HSP90. J Mol Biol 2008; 379(4): 732 – 744. doi: 10.1016/ j.jmb.2008.02.013.
42. Schweinfest CW, Graber MW, Henderson KW et al. Cloning and sequence analysis of Hsp89alpha deltaN, a new member of theHsp90 gene family. Biochim Biophys Acta 1998; 1398(1): 18 – 24.
43. Prodromou C, Panaretou B, Chohan S et al. The ATPase cycle of Hsp90 drives a molecular ‚clamp‘ via transient dimerization of the N-terminal domains. EMBO J 2000; 19(16): 4383 – 4392.
44. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5(10): 761 – 772.
45. Eustace BK, Sakurai T, Stewart JK et al. Functional proteomic screens reveal an essential extracellular role for HSP90 alpha in cancer cell invasiveness. Nat Cell Biol 2004; 6(6): 507 – 514.
46. Jaattela M, Wissing K, Kokholm T et al. HSP70 exerts its anti-apoptotic function downstream of caspase-3 like proteases. EMBO J 1998; 17(21): 6124 – 6134.
47. Raynes D, Guerriero V Jr. Inhibition of HSP70 ATPase activity and protein renaturation by a novel HSP70-binding protein. J Biol Chem 1998; 273(49): 32883 – 32888.
48. Kabani M, McLellan C, Raynes DA et al. HspBP1, a homologue of the yeast Fes1 and Sls1proteins, is an Hsc70 nucleotide exchange factor. FEBS Lett 2003; 531(2): 339 – 342.
49. Sedlackova L, Spacek M, Holler E et al. Heat-shock protein expression in leukemia. Tumor Biol 2011; 32(1): 33 – 44. doi: 10.1007/ s13277-010-0088-7.
50. Shi Y, Thomas JO. The transport of proteins into the nucleus requires the 70-kilodalton heta shock protein or its cytosolic cognate. Mol Cell Biol 1992; 12(5): 2186 – 2192.
51. Song J, Takeda M, Morimoto RI. Bag1-HSP70 mediates a physiological stress signalling pathway that regulates Raf-1/ ERK and cell growth. Nat Cell Biol 2001; 3(3): 276 – 282.
52. Gotz R, Kramer BW, Camarero G et al. BAG-1 haplo-insufficiency impairs lung tumorigenesis. BMC Cancer 2004; 4 : 85 – 91.
53. Mjahed H, Girodon F, Fontenay M et al. Heat shock proteins in hematopoietic malignancies. Exp Cell Res 2012; 318(5): 1946 – 1958. doi: 10.1016/ j.yexcr.2012.05.012.
54. Cortes JE, Talpaz M, Beran M et al. Philadelphia chromosome-negative chronic myelogenous leukemia with rearrangement of the breakpoint cluster region. Long-term follow-up results. Cancer 1995; 75(2): 464 – 470.
55. Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood 2000; 96(10): 3343 – 3356.
56. Žáčková M, Moučková D, Lopotová T et al. HSP90 – a potencial prognostic marker in CML. Blood Cells Mol Dis 2013; 50(3): 184 – 189. doi: 10.1016/ j.bcmd.2012.11.002.
57. Reikvam H, Hatfield KJ, Ersvaer E et al. Expression profile of heat shock proteins in acute myeloid leukaemia patients reveals a distinct signature strongly associated with FLT3 mutation status – consequences and potentials for pharmacological intervention. Br J Haematol 2011; 156(4): 468 – 480. doi: 10.1111/ j.1365-2141.2011.08960.x.
58. Yao Q, Nishiuchi R, Kitamura T et al. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and HSP90 inhibitors: the key role of the STAT5 signal transduction pathway. Leukemia 2005; 19(9): 1605 – 1612.
59. Tian WL, He F, Fu X et al. High expression of heat shock protein 90 alpha and its significance in human acute leukemia cells. Gene 2014; 542(2): 122 – 128. doi: 10.1016/ j.gene.2014.03.046.
60. Klikova K, Stefanikova A, Pilchova I et al. Differential impact of bortezomib on HL-60 and K562 cells. Gen Phys Biophys 2015; 34(1): 33 – 42. doi: 10.4149/ gpb_2014026.
61. Gamer J, Bujard H, Bukau B. Physical interaction between heat shock proteins DnaK, DnaJ, and GrpE and the bacterial heat shock transcription factor sigma 32. Cell 1992; 69(5): 833 – 842.
62. Rodriguez F, Arsene-Ploetze F, Rist W et al. Molecular basis for regulation of the heat shock transcription factor sigma 32 by the DnaK and DnaJ chaperones. Mol Cell 2008; 32(3): 347 – 358. doi: 10.1016/ j.molcel.2008.09.016.
63. Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010; 11(11): 777 – 788. doi: 10.1038/ nrm2993.
64. Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 2007; 581(19): 3702 – 3710.
65. Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett 2009; 583(16): 2647 – 2653. doi: 10.1016/ j.febslet.2009.04.029.
66. Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene 2004; 23(16): 2907 – 2918.
67. Otvos L, Rogers ME, Consolvo PJ et al. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000; 39(46): 14150 – 14159.
68. Reikvam H, Nepstad I, Sulen A et al. Increased antileukemic effects in human acute myeloid leukemia by combining HSP70 and HSP90 inhibitors. Expert Opin Investig Drugs 2013; 22(5): 551 – 563. doi: 10.1517/ 13543784.2013.791280.
69. Yao Q, Nishiuchi R, Li Q et al. FLT3 expressing leukemias are selectively sensitive to inhibitors of the molecular chaperone heat shock protein 90 through destabilization of signal transduction-associated kinases. Clin Cancer Res 2003; 9(12): 4483 – 4493.
70. Nimmanapalli R, O’Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res 2001; 61(5): 1799 – 1804.
71. Ray S, Lu Y, Kaufmann SH et al. Genomic mechanisms of p210BCR-ABL signaling: induction of heat shock protein 70 through the GATA response element confers resistance to paclitaxel-induced apoptosis. J Biol Chem 2004; 279(34): 35604 – 35615.
72. Assimon V, Gillies AT, Rauch JN et al. Hsp70 protein complexes as drug targets. Curr Pharm Des 2013; 19(3): 404 – 417.
73. Brodsky JL, Chiosis G. Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr Top Med Chem 2006; 6(11): 1215 – 1225.
74. Reikvam H, Brenner AK, Nepstad I et al. Heat shock protein 70 – the next chaperone to target in the treatment of human acute myelogenous leukemia? Expert Opin Ther Targets 2014; 18(8): 929 – 944. doi: 10.1517/ 14728222.2014.924925.
75. McConnell RJ, McAlpine SR. Heat shock proteins 27, 40 and 70 as combinational and dual therapeutic cancer targets. Bioorg Med Chem Lett 2013; 23(7): 1923 – 1928. doi: 10.1016/ j.bmcl.2013.02.014.
76. Leu JI, Pimkina J, Frank A et al. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell 2009; 36(1): 15 – 27. doi: 10.1016/ j.molcel.2009.09.023.
77. Kaiser M, Kuhnl A, Reins J et al. Antileukemic activity of the HSP70 inhibitor pifithrin-μ in acute leukemia. Blood Cancer J 2011; 1(7): 1 – 8. doi: 10.1038/ bcj.2011.28.
78. Yang M, Jiang G, Li W et al. Developing aptamer probes for acute myelogenous leukemia detection and surface protein biomarker discovery. J Hematol Oncol 2014; 7 : 5. doi: 10.1186/ 1756-8722-7-5.
79. Stuart RK, Wei A, Lewis ID et al. A multicenter dose-finding randomized controlled phase IIb study of the aptamer AS1411 in patients with primary refractory or relapsed AML. J Clin Oncol 2010; 28 (Suppl 15): abstr. TPS279.
80. Sundaram P, Kurniawan H, Byrne EM et al. Therapeutic RNA aptamers in clinical trials. Eur J Pharm Sci 2013; 48(1 – 2): 259 – 271. doi: 10.1016/ j.ejps.2012.10.014.
81. Rerole AL, Gobbo J, De Thonel A et al. Peptides and aptamers targeting HSP70: a novel approach for anticancer chemotherapy. Cancer Res 2011; 71(2): 484 – 495. doi: 10.1158/ 0008-5472.CAN-10-1443.
82. Andersen MH. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia 2014; 28(9): 1784 – 1792. doi: 10.1038/ leu.2014.108.
83. Fallouh H, Mahana W. Antibody to heat shock protein 70 (HSP70) inhibits human T-cell lymphoptropic virus type I (HTLV-I) production by transformed rabbit T-cell lines. Toxins 2012; 4(10): 768 – 777. doi: 10.3390/ toxins4100768.
84. Stangl S, Themelis G, Friedrich L et al. Detection of irradiation-induced, membrane heat shock protein 70 (Hsp70) in mouse tumors using Hsp70 Fab fragment. Radiother Oncol 2011; 99(3): 313 – 316. doi: 10.1016/ j.radonc.2011.05.051.
85. Braunstein MJ, Scott SS, Scott CM et al. Antimyeloma effects of the heat shock protein 70 molecular chaperone inhibitor MAL3-101. J Oncol 2011; 2011 : 232037. doi: 10.1155/ 2011/ 232037.
86. Gaudio E, Paduano F, Ngankeu A et al. Heat shock protein 70 regulates Tcl1 expression in leukemia and lymphomas. Blood 2013; 121(2): 351 – 359. doi: 10.1182/ blood-2012-09-457374.
87. Kirszberg C, Rumjanek VM, Capella MA. Methylene blue is more toxic to erythroleukemic cells than to normal peripheral blood mononuclear cells: a possible use in chemotherapy. Cancer Chemother Pharmacol 2005; 56(6): 659 – 665.
88. Demand J, Alberti S, Patterson C et al. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/ proteasome coupling. Curr Biol 2001; 11(20): 1569 – 1577.
89. Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2002; 2(1): 3 – 24.
90. Reikvam H, Ersvaer E, Bruserud O et al. Heat shock protein 90 – a potential target in the treatment of human acute myelogenous leukemia. Curr Cancer Drug Targets 2009; 9(6): 761 – 776.
91. Jhaveri K, Taldone T, Modi S et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 2012; 1823(3): 742 – 755. doi: 10.1016/ j.bbamcr.2011.10.008.
92. Neckers L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb Exp Pharmacol 2006; 172 : 259 – 277.
93. Supko JG, Hickman RL, Grever MR et al. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 1995; 36(4): 305 – 315.
94. Li Y, Zhang T, Schwartz SJ et al. New developments in Hsp90 inhibitors as anti-cancer therapeutics: mechanisms, clinical perspective and more potential. Drug Resist Updat 2009; 12(1 – 2): 17 – 27. doi: 10.1016/ j.drup.2008.12.002.
95. Ronnen EA, Kondagunta GV, Ishill N et al. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs 2006; 24(6): 543 – 546.
96. Dai C, Whitesell L. HSP90: a rising star on the horizon of anticancer targets. Future Oncol 2005; 1(4): 529 – 540.
97. Pacey S, Banerji U, Judson I et al. Hsp90 inhibitors in the clinic. Handb Exp Pharmacol 2006; 172 : 331 – 358.
98. Lancet JE, Gojo I, Burton M et al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010; 24(4): 699 – 705. doi: 10.1038/ leu.2009.292.
99. Wu YC, Yen WY, Lee TC et al. Heat shock protein inhibitors, 17-DMAG and KNK437, enhance arsenic trioxide-induced mitotic apoptosis. Toxicol Appl Pharmacol 2009; 236(2): 231 – 238. doi: 10.1016/ j.taap.2009.02.003.
100. Didelot C, Lanneau D, Brunet M et al. Anti-cancer therapeutic approaches based on intracellular and extracellular heat shock proteins. Curr Med Chem 2007; 14(27): 2839 – 2847.
101. Peng C, Brain J, Hu Y et al. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood 2007; 110(2): 678 – 685.
102. Turjap M, Juřica J, Demlová R. Možný klinický přínos terapeutického monitorování hladin imatinibu v onkologii. Klin Onkol 2015; 28(2): 105 – 111. doi: 10.14735/ amko2015105.
103. Barnes DJ, De S, van Hensbergen P et al. Different target range and cytotoxic specificity of adaphostin and 17-allylamino-17-demethoxygeldanamycin in imatinib-resistant and sensitive cell lines. Leukemia 2007; 21(3): 421 – 426.
104. Marcu MG, Chadli A, Bouhouche I et al. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem 2000; 275(47): 37181 – 37186.
105. Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst 2000; 92(3): 242 – 248.
106. Shelton SN, Shawgo ME, Matthews SB et al. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol Pharmacol 2009; 76(6): 1314 – 1322. doi: 10.1124/ mol.109.058545.
107. Delmotte P, Delmotte-Plaque J. A new antifungal substance of fungal origin. Nature 1953; 171(4347): 344.
108. Soga S, Shiotsu Y, Akinaga S et al. Development of radicicol analogues. Curr Cancer Drug Targets 2003; 3(5): 359 – 369.
109. Shiotsu Y, Neckers LM, Wortman I et al. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential G(1) phase accumulation against chronic myelogenous leukemia cells through destabilization of Bcr-Abl with Hsp90 complex. Blood 2000; 96 : 2284 – 2291.
110. Chiosis G, Timaul MN, Lucas B et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol 2001; 8(3): 289 – 299.
111. Boll B, Eltaib F, Reiners KS et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin Cancer Res 2009; 15(16): 5108 – 5116. doi: 10.1158/ 1078-0432.CCR-09-0213.
112. Elfiky A, Saif MW, Beeram M et al. BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: phase I experience. J Clin Oncol 2008; 26: abstr. 2503.
113. Plescia J, Salz W, Xia F et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005; 7(5): 457 – 468.
114. Gyurkocza B, Plescia J, Raskett CM et al. Antileukemic activity of shepherdin and molecular diversity of HSP90 inhibitors. J Natl Cancer Inst 2006; 98(15): 1068 – 1077.
115. Kaufmann SH, Karp JE, Litzow MR et al. Phase I and pharmacological study of cytarabine and tanespimycin in relapsed and refractory acute leukemia. Haematologica 2011; 96(11): 1619 – 1626. doi: 10.3324/ haematol.2011.049551.
116. Siegel D, Jagannath S, Vesole HD et al. A phase 1 study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 2011; 52(12): 2308 – 2315. doi: 10.3109/ 10428194.2011.600481.
117. Richardson PG, Mitsiades CS, Laubach JP et al. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol 2011; 152(4): 367 – 379. doi: 10.1111/ j.1365-2141.2010.08360.x.
118. George P, Bali P, Annavarapu S et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005; 105(4): 1768 – 1776.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue 1
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
- Current Insights into the Antispasmodic and Analgesic Effects of Metamizole on the Gastrointestinal Tract
- The Importance of Hydration in Wound Healing
Most read in this issue
- Survival Analysis Three-year Follow-up of Pacients with Head and Neck Cancer
- Genomic Tests as Predictors of Breast Cancer Patients’ Prognosis
- The Role of Heat Shock Proteins in Leukemia
- Potential of Long Non- coding RNA Molecules in Diagnosis of Tumors