
Molekulární genetika kolorektálního karcinomu
Molecular Pathogenesis of Colorectal Cancer
Background:
Colorectal cancer (CRC) remains a major health burden with an incidence of 1.3 million new cases worldwide and a mortality of almost 8.5%. It is the 2nd most common cancer in women (1st breast carcinoma) and 3rd most common in men (1st lung carcinoma, 2nd prostate carcinoma). CRC alongside breast, lung, prostate and stomach cancer is in the top five most common cancers in men and women worldwide. There are still more than 50% of CRC patients diagnosed with advanced disease (stage III and IV) in the Czech Republic. Genetically, CRC is a very heterogeneous disease with many factors playing key roles in pathogenesis. There are two types of CRC, hereditary with an incidence of between 5% and 10% with APC (FAP, aFAP) or MMR (HNPCC) genes affected, and sporadic colorectal cancer with an incidence of 90–95% with a lot of mutations in variable genes that accumulate during pathogenesis (APC, KRAS, MMR, microRNA, CIMP etc.). Knowledge of the molecular pathogenesis of CRC (hereditary, sporadic) is crucial for treatment, assessment of risk, prognosis, and patient follow-up.
Conclusion:
This article summarizes the molecular pathogenesis of sporadic and hereditary CRC.
Keywords:
colorectal cancer – pathogenesis – hereditary – sporadic – risk factors
This work was supported by grant of Grant Agency of the Czech Republic No. 15-08239S.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
3. 5. 2016
Accepted:
2. 6. 2016
:
J. Král 1,2; J. Slyšková 2; P. Vodička 2; J. Špičák 1
:
Klinika hepatogastroenterologie, Transplantcentrum, IKEM, Praha
1; Oddělení molekulární biologie nádorů, Ústav experimentální medicíny, AV ČR, v. v. i., Praha
2
:
Klin Onkol 2016; 29(6): 419-427
:
Review
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2016419
Východiska:
Kolorektální karcinom (colorectal cancer – CRC) je nádorové onemocnění tlustého střeva s každoročně diagnostikovanými téměř 1,3 miliony nových pacientů celosvětově. Mortalita na toto závažné nádorové onemocnění dosahuje již 8,5 %. Jedná se o 2. nečastější nádorové onemocnění u žen (1. nádor prsu) a 3. u mužů (1. nádor plic, 2. nádor prostaty). Tímto CRC patří vedle nádorů prsu, prostaty a plic k nejčastějším nádorovým onemocnění u mužů a žen na světě. V České republice je stále více než 50 % pacientů s CRC diagnostikováno v pokročilém stadiu (stadium III a IV). Z molekulárně genetického pohledu je CRC velice heterogenní onemocnění, na kterém se podílí mnoho faktorů. CRC má formu jak hereditární, kde dominují mutace v genech APC (FAP, aFAP), MMR (HNPCC) atd., jejíž výskyt se pohybuje okolo 5–10 %, tak formu sporadickou s výskytem 90–95 %, kde dochází k postupné akumulaci mutací v široké škále genů. Znalost genetické podstaty (APC, KRAS, MMR, mikroRNA, CIMP atd.) jak hereditární, tak sporadické formy CRC je nezbytná nejen pro zahájení léčby, ale také pro odhadnutí celkového rizika, prognózy a následného follow-up pacienta.
Závěr:
Tento článek shrnuje dosavadní molekulárně genetické aspekty patogeneze CRC.
Klíčová slova:
kolorektální karcinom – patogeneze – hereditární – sporadický – rizikové faktory
Epidemiologie kolorektálního karcinomu
Kolorektální karcinom (colorectal cancer – CRC) je mimořádně závažné nádorové onemocnění. V roce 2012 bylo celosvětově diagnostikováno přes 1,2 milionů pacientů s nádorem tlustého střeva, což činí 8 % ze všech nádorových onemocnění [1]. V rámci Evropy zaujímá ČR ve výskytu CRC 5. místo (1. Slovensko, 2. Maďarsko, 3. Dánsko, 4. Holandsko) a stejnou příčku drží i ve světě. Každoročně je v ČR diagnostikováno přes 8 000 nových pacientů a okolo 4 000 pacientů v jeho důsledku umírá. Jedná se o 2. nejčastější nádorové onemocnění u žen a 3. u mužů. Roční nárůst pacientů s daným onemocněním činil v minulých dekádách 2–3 %. Více než 50 % pacientů je diagnostikováno v pokročilém stadiu (stadium III a IV), které má horší prognózu nežli stadia časná. V posledních letech lze pozorovat trend snížené incidence, mortality a častější detekce časnějších stadií, přičemž jednou z možných příčin může být i screening. Veškeré klinické aspekty tohoto onemocnění se odvíjejí od molekulární genetiky a přehled této problematiky je cílem následujících řádků.
Formy CRC
CRC lze rozdělit na sporadický (90–95 % CRC) a hereditární (5–10 % CRC).
Sporadický CRC vzniká v důsledku postupné akumulace mutací. Tento proces trvající 10–15 let je znám jako tzv. adenom-karcinom sekvence a byl prvně popsán Fearonem a Volgelsteinem v roce 1990 [2,3]. Jedná se o nejčastější cestu vzniku CRC. Další sekvence, které mohou vést ke vzniku sporadického nádoru, jsou tzv. de novo, kde dochází ke vzniku nádoru na podkladě nově vzniklé mutace zpravidla genu TP53 bez předchozí mutace v APC genu. Kromě této cesty vzniká také CRC v zánětlivém terénu, např. u pacientů s idiopatickým střevním zánětem (inflammatory bowel disease – IBD), kde se zvyšuje riziko vzniku mutace vedoucí ke CRC nebo serátnímu adenomu (serrated adenoma). CRC u serátních adenomů (pilovité adenomy) vzniká nejčastěji časnou mutací v KRAS a BRAF genech [4,5].
Hereditární formy vznikají na podkladě zděděné mutace [6]. Zahrnují familiární adenomatózní polypózu (FAP), atenuovanou formu FAP (aFAP), hereditární nepolypózní formu CRC (hereditary nonpolyposis colorectal cancer – HNPCC), MYH asociovanou polypózu a polypózní syndromy [7].
FAP je autozomálně dominantní onemocnění, které je charakteristické vrozenou mutací v APC genu, což vede k narušení Wnt/β-kateninové signální dráhy (viz dále). Nicméně u 5–30 % pacientů s FAP není mutace v APC genu identifikována, ale byla u nich zjištěna mutace v MYH genu (viz dále) [8–10]. FAP zaujímá celkem 1 % ze všech CRC a je charakteristická výskytem velkého počtu polypů (stovky až tisíce) v druhé dekádě života s prakticky 100% rizikem malignizace. Z tohoto důvodu se u těchto pacientů doporučuje časná kolektomie. Součástí syndromu jsou rovněž extraintestinální projevy jako kožní léze, osteomy a extraintestinální nádorová onemocnění. Atenuovaná forma FAP je typická menším počtem polypů, pozdějším začátkem onemocnění a také menší frekvencí extraintestinálních příznaků.
U hereditární nepolypózní formy CRC (HNPCC, Lynchův syndrom) nacházíme vrozenou mutaci v tzv. genech opravy chybně spárovaných bazí (DNA mismatch repair genes – MMR) [11]. Mutací v MMR dochází k hromadění replikačních chyb, které nejsou opraveny, a tím dochází k větší pravděpodobnosti vzniku CRC. HNPCC je nejčastější formu vrozeného CRC (2–3 %). Pacienti s HNPCC jsou zpravidla nižšího věku s vyšším rizikem vzniku synchronních a extraintestinálních nádorů (ovaria, endometrium, žaludek, pankreas, atd.) [12].
MYH-asociovaná polypóza je autozomálně recesivní onemocnění zapříčiněné mutací v MYH genech vzniklé poškozením bázové excizní reparace (BER). Tato forma je charakteristická vysokým počtem mutací v APC genu, vysokou frekvencí tzv. mikrosatelitů (sekvence opakujících se nukleotidů) vznikající poruchou systému opravy chybného párování bazí (DNA mismatch repair systém) a nízkou frekvencí ztráty jedné alely genu – tzv. ztráta heterozygozity (loss of heterozygosity – LOH). U pacientů nacházíme zpravidla desítky polypů se zhruba 65% pravděpodobností vzniku karcinomu [13].
Hamartogenní polypózní syndromy zahrnují Peutz-Jaeghersův syndrom (PJS), juvenilní polypózní syndrom (JPS) a Cowdenův syndrom. PJS je syndrom autozomálně dominantní s výskytem polypů v gastrointestinálním traktu (GIT) a mukokutánní pigmentací. JPS je rovněž autozomálně dominantní syndrom s výskytem četných juvenilních polypů, které jsou spojené s vyšším rizikem nádorů pankreatu. Hlavním znakem Cowdenova syndromu, který s sebou nese i vyšší riziko vzniku nádoru prsu, štítné žlázy a endometria, je výskyt hamartogenních polypů v GIT.
Patogeneze CRC
CRC vzniká postupnou akumulací genetických mutací a epigenetických změn, které vedou k přeměně normální sliznice tlustého střeva v nádorovou tkáň. Nejčastější molekulární a genetické změny vedoucí ke vzniku nádoru jsou strukturální a numerické změny chromozomů označované jako chromozomální instabilita (CIN), dále pak zvyšování počtu kopií opakujících se sekvencí DNA (tzv. mikrosatelity) způsobené defektní opravou špatně párovaných bází, označované jako mikrosatelitní instabilita (MSI), a nakonec aberantní hypermetylace promotorových oblastí genů, CpG island methylator phenotype (CIMP) [13–15]. Přítomnost CIN vylučuje přítomnost MSI a vice versa, nicméně některé studie poukazují, že oba fenomény se mohou prolínat [16].
Chromozomální instabilita (CIN)
CIN se spojuje se vznikem CRC v cca 65–70 % případů. Charakterizuje ji nadbytek kopií či ztráta celých chromozomů či jejich oblastí, které se podílejí na patogenezi CRC. Důsledkem CIN je aneuploidie (chybný počet chromozomů), chromozomální amplifikace (zmnožení DNA) či LOH (ztráta jedné alely daného lokusu). Příznačně jsou takto postiženy oblasti onkogenů a tumor supresorových genů APC, KRAS či TP53 [13,17].
Mikrosatelitní instabilita (MSI)
Mikrosatelity jsou krátké sekvence opakujících se nukleotidů, které se nacházejí v celém genomu a jsou náchylné k chybám, ke kterým dochází během replikace. Chyby, které vzniknou v mikrosatelitech, jsou rozpoznány systémem DNA mismatch repair (MMR, viz výše), který opravuje chyby v párování bazí. MSI je odrazem poškození MMR systému. Na funkci MMR systému se podílejí geny MSH2, MLH1, MSH6, PMS2, MLH3, MSH3, PMS1 a Exol. MSI jako důsledek poruchy MMR systému je spojena jak s hereditární formou (HNPCC), tak i se sporadickou formou CRC (15 % nádorů). V patogenezi rozlišujeme CRC s MSI-high a MSI-low, viz níže [13,17].
Metylace CpG ostrůvků – CIMP
CpG metylace je kromě modifikace histonů další epigenetická změna, která nevede ke změně DNA sekvence, ale ovlivňuje promotorovou oblast daného genu, a tím ovlivní jeho expresi zpravidla snížením. Nejčastěji k metylaci DNA dochází v oblasti 5’-CG-3’ dinukleotidu, resp. vzniká pevná kovalentní vazba -CH3 skupiny na pozici 5’ cytozinu v opakujícím se CG dinukleotidu. Geny, které jsou ovlivněny hypermetylací promotorové oblasti, jsou především APC, MCC, MLH1, MGMT a další [18]. Tumory s CIMP bývají spojeny s přítomností BRAF mutace a MSI. CIMP je příčinou cca 15–20 % sporadického CRC. Vyskytuje se častěji u žen, starších pacientů, kuřáků. Špatně diferencované a mucinózní tumory vycházejí nejčastěji ze serátních adenomů pravého tračníku [19,20].
Jedním z dalších důležitých reparačních genů, který je v patogenezi CRC metylován, je MGMT (O6-metylguanin DNA metyltransferáza). Ztráta funkce MGMT je téměř výhradně spojena s CpG metylací. Funkcí tohoto DNA reparačního enzymu je ochraňovat buňky před působením exogenních karcinogenů mechanizmem odstraňování alkyl skupiny z pozice O6 guaninu. Metylaci promotorové oblasti MGMT můžeme nalézt až ve 40 % CRC, nejčastěji v serátních adenomech. Předpokládá se, že metylace MGMT je v onkogenezi jednou z prvních událostí. Pacienti s defektním MGMT mají větší procento mutací v KRAS genu a lépe reagují na chemoterapii. Pacienti bez MGMT metylace přežívají déle nežli pacienti s metylací [21,22].
Genetické změny vedoucí ke CRC
Mutace v tumor supresorových genech
APC gen
APC gen je tumor supresorový gen lokalizovaný na dlouhém raménku 5. chromozomu (5q21). Byl objeven v roce 1987 a poprvé naklonován v roce 1991 [23]. Podílí se nejenom na vzniku sporadického CRC, ale rovněž hraje významnou roli v patogenezi hereditárních forem CRC, konkrétně při vzniku FAP a také aFAP [24].
APC gen s 15 exony hraje důležitou roli v proliferaci, diferenciaci, migraci, apoptóze a také v řízení buněčného cyklu vč. stabilizace mikrotubulů během mitózy. Mutace v APC genu se vyskytuje u časných stadií nádoru a zpravidla se jedná o bodové mutace či LOH. K manifestaci mutace APC je důležité, aby dle Knudsonovy hypotézy dvou nezávislých „hitů“ byly mutovány obě dvě alely tohoto genu, což vede ke ztrátě tumor supresivního účinku genu [25]. Výsledkem je poté zkrácený APC protein, který částečně ztrácí svou funkci.
APC gen řídí Wnt/β-kateninovou signální dráhu (obr. 1). Za fyziologických podmínek APC gen tvoří protein, který společně s Axin/Axin2 a GSK-3β vytváří tzv. destruktivní komplex, který iniciuje ubikvitaci (označení) β-kateninu s jeho následnou degradací v proteazomu (proteinový komplex degradující nepotřebné či poškozené proteiny). Mutace v APC genu vede ke snížení degradace β-kateninu (protein koordinující buněčnou adhezi a genovou transkripci) a následně k jeho hromadění. Nahromaděný β-katenin vstupuje do jádra, váže se na jaderné receptory a indukuje transkripci mnoha genů vč. cyklinu D1, c-myc a CRD-BP. Důsledkem je nejenom nekontrolovatelná proliferace a růst buňky, ale rovněž i indukce apoptózy. Poslední studie dokazují, že ztráta genu vedoucí k proliferaci a vyššímu přežívání buňky je závislá na jejich původní diferenciaci [24,26–29].
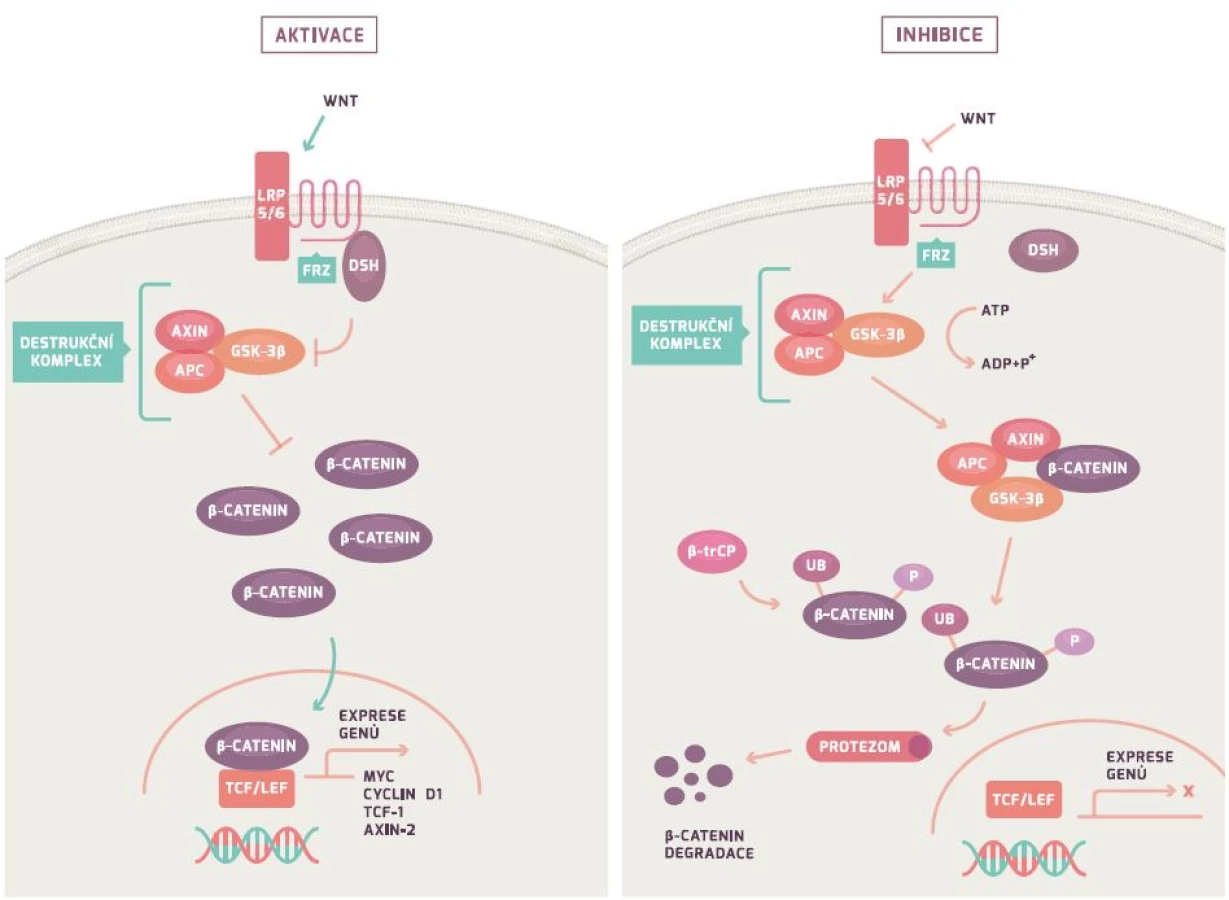
TP53
TP53 zvaný taktéž „strážný anděl“ je tumor supresivní gen, lokalizovaný na krátkém raménku 17. chromozomu (17p13.1). Základní funkcí TP53 je regulace buněčného cyklu (zastavení cyklu a možnost zahájení opravy DNA), apoptózy a buněčného metabolizmu [14,30,31]. V případě poškození DNA TP53 koordinuje opravu DNA, a pokud ji nelze opravit, tak navodí apoptózu buňky. TP53 je nejčastěji postiženým genem v mutagenezi mnoha nádorových onemocnění. Mutace bývá nalézána až u 50 % všech nádorů [32]. Je známo celkem 2 314 mutací TP53, které jsou buď aktivační (cca 71 %, protein se váže na Mdm2 a dojde k jeho degradaci), nebo inaktivační (29 %, nemožnost regulace genové exprese) [33]. Stejně jako u APC genu, tak i u TP53 je k funkčnímu projevu nutná mutace obou alel.
Zpravidla dochází ke ztrátě alely na 17. chromozomu a zároveň k mutaci druhé alely na 2. chromozomu. Až 80 % mutací představuje missense (zařazení odlišné aminokyseliny vedoucí ke změně až nefunkčnosti výsledného proteinu) vedoucí k expresi stabilního, stejně dlouhého proteinu, který ale ztrácí možnost vazby na DNA a její následnou regulaci [34]. Mutace genu bývá přítomna v pozdější fázi vývoje CRC a zpravidla vede k přechodu adenomu do adenokarcinomu. Pokročilé formy CRC (T1–4 N1–2 M0–1) mají ve srovnání se stadii T1–4 N0 M0 vyšší frekvenci mutací. Mutace v TP53 bývá přítomna až v 60 % tumorů a pro významnou heterogenitu mutantů TP53 lze jen těžko odhadnout jejich prediktivní a prognostický význam. Pacienti s mutací TP53 (aktivační i inaktivační) více profitují z chemoterapie.
Mutace v onkogenech
KRAS
KRAS gen patří do rodiny proto-onkogenů (HRAS, NRAS) a je mutován (nejčastěji exon 2 a exon 3) u 30–60 % pacientů se sporadickým CRC [13]. S mutací KRAS u familiární formy se setkáváme jen zřídka. Z hlediska posloupnosti jeho mutaci předchází mutace APC genu. Různé mutace KRAS genu (exon 2, exon 3) mohou mít odlišné důsledky, např. odlišný účinek biologické terapie.
KRAS protein je 21KDa velký membránový protein, který hraje roli v buněčné signalizaci ovlivňující buněčný růst, přežití, diferenciaci, proliferaci a řadu dalších buněčných dějů. Konkrétně KRAS a BRAF (viz níže) jsou součástí RAS/RAF/MAPK signální dráhy (obr. 2) [35]. Mutovaný protein si zachovává aktivní formu z důvodu nefunkční GTPázové aktivity, která mění GTP na GDP, a tím má stimulační efekt na proliferaci a růst buňky. KRAS je aktivován přes EGFR receptor, který je během mutageneze CRC častokrát více exprimován a po jeho stimulaci dochází k aktivaci KRAS navázáním TGF. Při aktivaci receptoru EGFR dochází také ke stimulaci intracelulární kinázové domény. Ta poté aktivuje SOS, dále GRB a ten následně KRAS, který poté stimuluje BRAF. Dále signální kaskáda pokračuje přes MEK, ERK, až dojde k ovlivnění genové exprese a proliferace buňky [14]. Během aktivace EGFR receptoru dochází ke stimulaci PI3K (fostatidil inositol 4–5 bisfofát), který inhibuje apoptózu buňky. Tato dráha je poté regulována pomocí PTEN proteinu, který blokuje funkci PI3K [36,37].
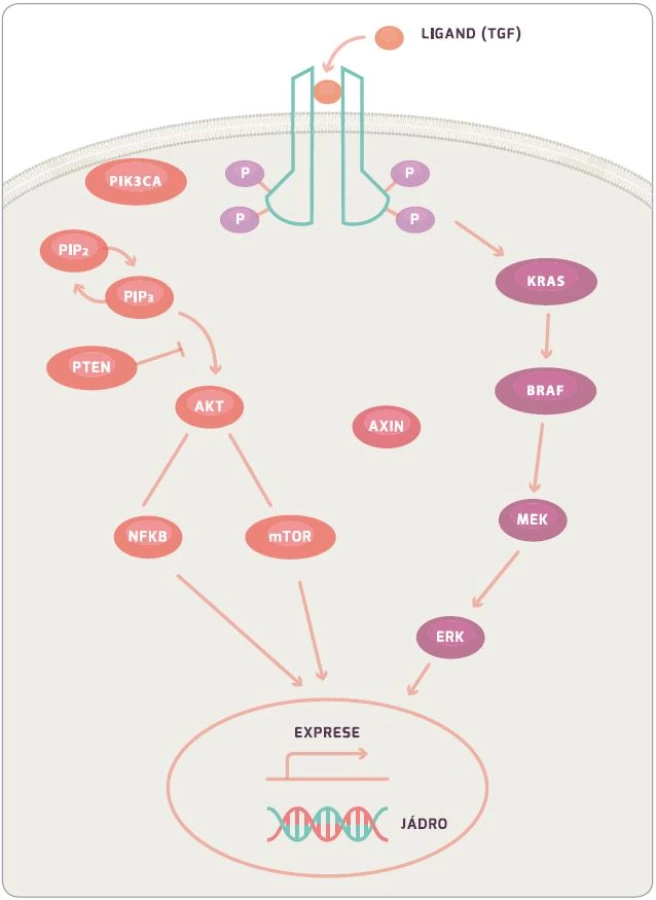
Informace, zda pacient má, či nemá mutaci v KRAS, je velice důležitá z důvodu následné léčby monoklonálními protilátkami proti EGFR. I přes inhibici EGFR receptoru monoklonálními protilátkami mutují KRAS nebo BRAF. Geny mají aktivační konformaci a stále mohou stimulovat buněčnou proliferaci. Důsledkem je snížená odpověď na biologickou léčbu [15,38].
BRAF gen, stejně jako KRAS, je proto-onkogen, který je součástí RAF rodiny serin/treonin kináz a reguluje růst buňky skrze RAF-MAP signální dráhu. V této signální dráze zajišťuje tzv. down-regulaci KRAS a up-regulaci MEK. U sporadické formy nádorů se tato mutace vyskytuje cca v 10 %. Jedná se převážně o hotspot mutaci ve V600E. BRAF mutace byly identifikovány u 4 % pacientů s MSI-low a u 40 % s MSI-high. Stejně jako u KRAS pacienti s BRAF mutací mají horší odpověď na léčbu monoklonálními protilátkami proti EGFR. BRAF způsobuje sníženou expresi KRAS genu. Tento fakt je klinicky významný pouze tehdy, pokud KRAS mutace není přítomna [14,39,40].
I přes nepřítomnost mutace v KRAS či BRAF není léčba monoklonálními protilátkami 100% úspěšná. Byla identifikována další signální dráha, která hraje roli v patogenezi CRC, a to PI3K dráha. PIK3CA gen je mutován u více než 25 % pacientů s CRC. Při této mutaci dochází k aktivaci signální dráhy, která v konečném důsledku vede k inhibici apoptózy. Na druhou stranu protein PTEN má downregulující funkci na PI3K, a tudíž vede ke snížení aktivity signální dráhy. Nicméně setkáváme se i s mutací v PTEN genu, která má za následek neschopnost regulace PI3K signální dráhy vedoucí k horší odpovědi na biologickou léčbu a horšímu celkovému přežití (overall survival – OS) pacienta [15,41,42].
Mutaci můžeme nalézt rovněž v receptoru pro růstový faktor (TGFR). Známy jsou celkem tři formy tohoto receptoru, ale nejčastěji se setkáváme s mutací u TGF-βRII. Tato mutace je přítomna až u 90 % pacientů s MSI. Mutace TGF-βRII vede k aktivaci PI3K, která vyústí v inhibici apoptózy, a rovněž povede k epiteliální/mezenchymální transformaci (proces, při kterém epitelové buňky ztrácejí buněčnou adhezi, a tím získávají možnost migrovat a stávají se z nich mezenchymální zárodečné buňky s možností diferenciace u mnoha buněčných typů; proces lze identifikovat při hojení ran, fibróze a v procesu metastazování). Transformace má za následek progresi, invazi a metastazování CRC.
Ztráta heterozygozity (LOH)
LOH byla popsána Knudsonem [43] a je dalším mechanizmem, který může vést ke vzniku CRC. Nejčastěji dochází k LOH na 18q chromozomu. Další chromozomy postiženy mechanizmem LOH jsou 1p, 5q, 17p. Na 5q se jedná o ztrátu alely pro APC gen, zatímco na 17p chromozomu se jedná o ztrátu alely pro TP53 [14].
DCC
Jeden z genů, který může být postižen na 18. chromozomu, je DCC gen, který je zodpovědný zejména za expresi DCC transmembránového proteinu, ale rovněž i za jiné produkty díky alternativnímu sestřihu (splicingu) [44]. Téměř 70 % CRC má přítomnou LOH genu DCC (exon 7) [44]. Kromě LOH DCC se v patogenezi uplatňují i jiné somatické mutace, např. bodová mutace či delece DCC [45]. DCC je tumor supresorový gen, ačkoliv o jeho jasném zařazení se vedou stále velké diskuze. Někdy je označován jako poslední obránce (Late Gatekeeper), který limituje progresi tumoru navozením apoptózy [46]. Jeho funkcí je blokovat růst buňky v případě absence svého ligandu (netrin-1). Netrin patří do rodiny difuzibilních proteinů podobných lamininu, který hraje důležitou roli v určování směru růstu a migrace axonů. Netrin-1 byl popsán jako faktor buněčného přežití [47]. Váže se na transmembránové receptory DCC a UNC5H (UNC5A, UNC5B, UNC5C) [47]. Oba receptory jsou schopné indukovat buněčnou smrt v nepřítomnosti svého ligandu, a proto receptory hrají roli v patogenezi CRC.
Koncentrace netrinu-1 je nejvyšší v oblasti báze krypt tlustého střeva a klesá apikálním směrem. V případě mutace DCC nebude přítomen transmembránový receptor pro netrin-1, a tudíž nebude navozena apoptóza a buňka bude nadále proliferovat. V nepřítomnosti netrinu-1 či při jeho nižší koncentraci (fyziologický děj) dochází k odštěpení intracelulární domény DCC proteinu neznámou kaspázou za vzniku ADD domény (addiction/dependent domain), která je schopná vazby na kaspázu 9, a tím aktivaci buněčné smrti [44,48]. Mutace v tomto genu nalézáme zpravidla až v pokročilých formách CRC, nikoliv v úvodní fázi patogeneze. Otázkou je, jak je tomu u mutace transmembránového proteinu UNC5H. LOH na 18. chromozomu můžeme najít v patogenezi i jiných nádorových onemocnění, jako jsou tumory žaludku, prostaty, endometria, ovarií atd. Ze studií vyplývá, že nemocní s LOH DCC mají horší prognózu nežli pacienti bez ní, a to z důvodu vyšší agresivity tumorózního procesu.
Jak již bylo řečeno výše, netrin-1 se váže kromě DCC i na UNC5H receptory (A, B, C), které při nepřítomnosti netrinu-1 indukují apoptózu. Mechanizmus indukce apoptózy je odlišný od DCC receptoru. Při absenci netrinu-1 dochází ke štěpení intracelulární části UNC5H receptoru pomocí neznámé proteázy či kaspázy 3, která odhalí doménu, která následně zahájí apoptózu. Mutace této intracelulární části UNC5H receptoru povede k inhibici apoptózy. Exprese UNC5H receptoru je fyziologicky řízena pomocí genu TP53. V případě poškození DNA dochází ke zvýšené expresi UNC5H receptoru a následnému odštěpení intracelulární domény a navození apoptózy. TP53 gen řídí expresi netrinu-1, který společně s UNC5H zpětně downreguluje expresi TP53 genu, a díky vazbě netrin-1 a UNC5H inhibuje TP53 indukovanou apoptózu buňky [45].
SMAD
Dalšími geny, které jsou lokalizovány na 18. chromozomu a mohou se podílet na patogenezi CRC, jsou SMAD2 a SMAD4. Hrají zásadní roli v TGF-β signální dráze. TGF-β rodina zahrnuje TGF-β, aktiviny a kostní morfogenetický protein. Tyto proteiny se vážou na TGF-β receptory (TβRI, TβRII) mající široké spektrum účinku, jako je řízení buněčné proliferace, diferenciace, apoptózy, migrace a tvorby extracelulární mezibuněčné hmoty [49]. Po vazbě TGF-β na receptor dochází k aktivaci SMAD proteinu (Smad anchor for receptor activation), který zaktivuje intracelulární proteiny SMAD2 a SMAD3. Komplex SMAD2 a SMAD3 se naváže na SMAD4 a tento komplex SMAD2–4 putuje do jádra, kde se váže přímo na DNA sekvenci či na DNA transkripční faktory. Výsledkem vazby je inhibice růstu buňky či navození apoptózy. Buněčný cyklus se po aktivaci TGF-β zastaví v G1 fázi. SMAD gen je tudíž genem tumor supresorovým.
Výsledkem exprese SMAD jsou SMAD proteiny různých funkcí. R-SMAD (receptorové SMAD proteiny: 1, 2, 3, 5, 8), Co-SMAD (mediátorové SMAD proteiny: 4α, 4β), I-SMAD (inhibitorové SMAD proteiny: 6, 7) regulují genovou expresi. TGF-β/SMAD signální dráha musí být pečlivě regulována, což mají za úkol SMAD6 a 7. SMAD7 protein se váže na SMURF1,2 (E3 ubikvitin protein ligáza) a aktivuje degradaci SMAD4, čímž reguluje TGF-β/SMAD signální dráhu. Růstové faktory jako EGF a HGF dokážou inhibovat TGF-β/SMAD signální dráhu skrze aktivaci Ras, která bude indukovat degradaci SMAD4 v proteazomu. TGF-β v pozdější fázi nádorového procesu nemusí hrát pouze roli tumorsupresivní dráhy, ale může být promotorem nádorového bujení, a to skrze aktivaci RhoA kinázy. Kináza následně způsobí uvolnění buněčných spojů, snížení exprese E-cadherinu a zvýšení motility buňky. Následkem je epiteliální/mezenchymální transformace (EMT) a možnost šíření nádorových buněk a zakládání metastáz. Tento fakt dokládá možnost tvorby metastáz nezávisle na funkci TGF-β/ /SMAD signální dráhy.
TGF-β signální dráha dokáže aktivovat i jiné signální dráhy mimo RhoA či Ras. Příkladem mohou být ERK, PI3K, JNK. Tyto dráhy na rozdíl od SMAD signální dráhy mají proto-onkogenní účinek. U CRC je frekvence mutace v SMAD2 výrazně menší nežli mutace v SMAD4 [45]. Ta v SMAD4 bývá přítomna u jedné třetiny CRC pacientů s LOH [44]. Důsledkem této mutace je nefunkčnost TGF-β/SMAD signální dráhy, která povede k proliferaci buněk a růstu tumoru. Nemocní s CRC a vysokou expresí SMAD4 mají signifikantně delší OS než pacienti s nízkou expresí. Tento fakt však nemusí platit u jiných malignit, jako je nádorové onemocnění prsu, kde vysoká exprese SMAD naopak vede k tvorbě kostních metastáz [50].
MikroRNA a CRC
MikroRNA (miRNA) může v patogenezi CRC hrát roli jak tumor supresivní, tak i proto-onkogenní. Role miRNA je dána typem tkáně a charakterem genu, který reguluje. MiRNA je krátká nekódující RNA o délce 22–23 nukleotidů, která je kódována v DNA [51,52]. MiRNA je kódována strukturálními geny (70 %), ale také intergenovou nekódující DNA (30 %), která není vázána na expresi genů jako takových. Předpokládá se, že miRNA je zodpovědná za regulaci cca 30 % genů (některé zdroje uvádějí 30–60 %). Je známo, že polovina genů kódujících miRNA je v oblasti genetické informace, která je lokalizována v místech častých amplifikací, LOH nebo mutací. Po přepisu DNA kódující miRNA vznikne pri-miRNA, která je v jádře upravena na pre-miRNA, která je dále exportována z jádra do cytozolu. V cytozolu podléhá další úpravě za vzniku jednovláknové miRNA, která plní svou funkci [53,54]. Funkcí miRNA je regulace genové exprese, která probíhá několika možnými způsoby. Jedním ze způsobů je interakce s mRNA, kde dojde k navození degradace mRNA nebo inhibice translace (častější u živočišných buněk).
Dalšími diskutovanými mechanizmy, kterými by miRNA mohla regulovat genovou expresi, je metylace promotorové oblasti genu či ovlivnění konformace histonů (metylace), a tím kondenzaci heterochromatinu [55,56]. Ke ztrátě funkce miRNA dochází několika způsoby – mutace genu pro miRNA, miRNA posttranskripční úprava a epigenetické změny. Vzhledem k tomu, že miRNA se podílí na řízení buněčného cyklu, metabolizmu a vývoji buňky, tak změna exprese (up - či down-regulace exprese miRNA) bude mít za následek proto-onkogenní (progrese a růst nádoru) či tumor supresivní (inhibice růstu a proliferace) důsledky pro buňky [51,52,55,56], jelikož miRNA, která je nadmíru exprimována v nádorových buňkách, pravděpodobně funguje jako inhibitor tumor supresivních genů. Na druhou stranu miRNA, která má sníženou expresi v nádorových buňkách, může zapříčinit snížení exprese onkogenů v normální tkáni. V patogenezi CRC byla zjištěna změna exprese miRNA, např. miR-145, miR-143, let7 či zvýšená exprese miR-21, miR-135 atd. MiR-145 a miR-143 byly jedny z prvních miRNA identifikovaných u nádorového onemocnění tlustého střeva. MiR-143 se podílí na inhibici onkogenu KRAS a pravděpodobně miR-145 funguje tumor supresivně přímou inhibicí p70S6K1 (serin/threonin kináza, která snižuje aktivitu PI3 signální dráhy). MiR-21 je spojena s invazí a metastazováním CRC díky inhibici tumor supresivního genu PDCD4 [57]. Vzhledem k velké specificitě exprese jednotlivých miRNA a typu tumoru se zde skýtá možnost využití miRNA jako diagnostického či prognostického markeru [58]. Další výhodou miRNA je její stabilita v biologické tkáni, ať už se jedná o plazmu, nebo stolici [59,60].
Geny opravy chybného párováni (MMR)
Jak již bylo řečeno v úvodu, CRC vzniká na podkladě CIN, CpG metylace či mutací v tzv. DNA mismatch repair genech (MMR), které vedou k MSI. MMR je systém, který opravuje chybné párování bazí při replikaci genetické informace, zjednodušeně opravuje chyby, které nejsou rozpoznány proof-reading aktivitou DNA polymerázy během replikace. Nejedná se pouze o tuto funkci, ale MMR systém má i další funkce jako regulace buněčného cyklu a TP53 dependentní apoptotickou odpověď na odlišná poškození DNA [61].
V patogenezi CRC dochází vlivem mutace v MMR genech nejčastěji ke vzniku tzv. MSI. K tomu, aby byl MMR gen vyřazen z funkce, je zapotřebí mutace v obou dvou alelách genu. Proto se MMR geny chovají jako tumor supresorové geny s výjimkou EXO1, u kterého je dostatečná mutace jedné alely. Mikrosatelity jsou krátké opakující se nukleotidové sekvence umístěné po celém genomu. Jsou náchylné na chyby způsobené během replikace. V genomu se opakují každých 30–60 kilobází [14]. Právě MMR systém má za úkol takové chyby opravovat a zabránit tak vzniku MSI [13]. Mutace v MMR systému mohou být jak vrozené (germinální), tak i získané. Právě vrozené mutace v MMR systému jsou klíčem ke vzniku HNPCC, kde nacházíme mutaci v MMR genech u 50–70 % případů [14,62]. Získané mutace (delece, inzerce atd.) či epigenetické změny (metylace) v MMR jsou zodpovědné za vznik cca 15–20 % sporadického CRC [13,14,63]. Jedná se o mutace či metylace v těchto genech: MSH2 (2p16), MLH1 (3p21), MSH6 (2p16), PMS2 (7p22), MLH3 (14q24.3), MSH3 (5q11-q12), PMS1 (2q31-33) a Exo1 (1q--42-43) [13,14,61,63,64].
Tumory s MSI můžeme dělit na MSI-high tumory a MSI-low tumory. MSI-high tumory jsou specifikovány instabilitou ve více než dvou mikrosatelitech, zatímco MSI-low tumory mají mutaci pouze v jednom sledovaném mikrosatelitu. Pacienti s CRC na podkladě MSI bývají mladšího věku a překvapivě mají lepší OS nežli ostatní formy CRC. Pacienti s MSI-high mutacemi jsou převážně ženy s pravostrannou lokalizací tumoru. Z patologického hlediska se většinou jedná o špatně diferencovaný, mucinózní karcinom se zvýšenou infiltrací lymfocyty. MSI-high tumory mívají méně KRAS a TP53 mutací, zato BRAF mutace jsou častější. Pacienti rovněž s MSI-high mají horší terapeutickou odpověď na léčbu 5-fluorouracilem [15]. Navzdory těmto faktům pacienti s MSI-high mají lepší OS nežli ostatní pacienti s CRC [13,14,65]. U sporadické formy CRC dochází nejčastěji k inaktivaci promotorové oblasti genu MLH1 metylací s následkem zabránění exprese genu [13].
Common disease – Common variant (CD-CV)
V lidské populaci existují varianty genů, jež mohou souviset se vznikem závažných onemocnění. Tyto varianty bývají v populaci přítomny s frekvencí menší než 1 %. Nicméně existují názory, že i běžné varianty lidských genů (jednonukleotidové polymorfizmy – SNPs) se mohou významně podílet na vzniku závažných onemocnění. Jedná se o takzvanou CD-CV hypotézu (common disease – common variant hypothesis). Nejinak tomu je i u CRC. V roce 2008 Albert Tenesa a další uskutečnili studii, kde se snažili identifikovat lokusy spojené s rizikem vzniku CRC. Provedli genotypizaci celkem 555 510 SNPs u více než 1 000 pacientů s CRC. Po podrobné analýze těchto SNPs identifikovali tři SNPs, které byly asociovány s vyšším rizikem vzniku nádoru v různých populacích v lokusech 11q23 (rs3802842), 8q24 (rs7014346) a 18q21 (rs4939827) [66]. Ze studie COGENT z roku 2011, na které se podílelo více než 20 výzkumných skupin z celé Evropy, Ameriky, Číny, Austrálie a Japonska identifikovali celkem 14 SNPs asociovaných s vyšším rizikem vzniku CRC [67].
Rizikové faktory a prevence CRC
Na patogenezi CRC se významnou měrou podílejí i vlivy životního prostředí, mezi něž patří především kouření cigaret, vysoká konzumace červeného masa, živočišných tuků, snížená konzumace vlákniny, alkohol, diabetes mellitus a některé další.
V roce 2009 byla provedena metaanalýza studií zabývající se vlivem kouření na vznik CRC v rozmezí let 1950–2008 zahrnující 28 prospektivních kohortových studií s celkem 1 463 796 jedinci z Ameriky, Evropy a Asie, s mediánem pozorování 13 let. Kuřáci vykázali vyšší riziko (relativní riziko – RR 1,20; 95% CI 1,10–1,30) vzniku nádoru než nekuřáci. Riziko u mužů kuřáků bylo vyšší nežli u žen kuřaček (RR 1,38 vs. 1,06). Prokázalo se, že zvýšené riziko výskytu vzniku nádoru tlustého střeva závisí na množství vykouřených cigaret za den a počtu let aktivního kouření [68].
Metaanalýza prospektivních studií z roku 2011 zkoumala riziko zvýšené konzumace červeného masa a její vliv na vznik karcinomu tlustého střeva během let 1966–2011. Prokázalo se, že konzumace 140 g červeného masa denně je spojena s vyšším rizikem vzniku CRC (RR 1,22; 95% CI 1,11–1,34) [69].
Nižší příjem vlákniny v dietě je ve spojení se zvyšujícím se věkem spojen s vyšším rizikem vzniku CRC, jak bylo prokázáno z metaanalýzy 13 prospektivních kohortových studií. Nicméně vyšší příjem vlákniny není spojen s nižším rizikem vzniku CRC [70].
Již v minulosti byla pokládána otázka vlivu aspirinu (acetylsalicylová kyselina) na riziko vzniku CRC. Z randomizovaných studií vyplývá, že užívání aspirinu déle než 10 let snižuje riziko vzniku nádoru. Tento efekt byl však asociován s užíváním min. 300 mg aspirinu denně. Užívání aspirinu déle než 20 let nevedlo k dalšímu snížení tohoto rizika. Také nebyl pozorován rozdíl mezi aspirinem či jinými nesteroidními antirevmatiky. Menší dávky aspirinu tento efekt však jasně nepotvrdily a data jsou inkonzistentní [71].
Chronická konzumace alkoholu je jeden z hlavních rizikových faktorů vzniku nádorů trávicí trubice (orofarynx, jícen) a rovněž nádoru tlustého střeva. Již mírný pravidelný přísun alkoholu denně může vést k většímu riziku vzniku nádoru [72]. V roce 2011 byla provedena metaanalýza 34 studií, která předložila jasné důkazy o souvislosti mezi konzumací jednoho alkoholického nápoje denně a vyšším rizikem vzniku CRC [73].
Riziko vzniku nádoru tlustého střeva je rovněž vyšší u pacientů s diabetem mellitem 2. typu. Toto riziko je zvýšené jak u mužů, tak i u žen, stejně pro kolonickou, tak i rektální formu. U takových pacientů by měl být proveden screening před zahájením inzulinové terapie a intervaly mezi kolonoskopickými kontrolami by neměly přesáhnout dobu pěti let [74,75].
Závěr
Z genetického a molekulárního hlediska představují nádory tlustého střeva a konečníku značně heterogenní skupinu onemocnění. To se v praxi projevuje různou mírou rizika, reakcí na léčbu a vznikem rezistence na jednotlivé léky, tedy celkovou prognózou. Významnou roli v patogenezi CRC rovněž hrají rozmanité epigenetické faktory a vlivy zevního prostředí, jejichž vliv je opět individuální. Praktickým dopadem preklinického výzkumu bude dokonalé poznání patogeneze s cílem určit biomarkery, které přispějí k časné detekci a pomohou nastavit individuálně optimální léčebný režim.
Práce byla podpořena grantem GA ČR č. 15-082-39S.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Jan Král
Klinika hepatogastroenterologie
Transplantcentrum, IKEM
Vídeňská 1958/9
140 21 Praha 4
e-mail: jan.kral@centrum.cz
Obdrženo: 3. 5. 2016
Přijato: 2. 6. 2016
Sources
1. Ferlay J, Soerjomataram I, Ervik M et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11; 2013 [online]. Available from: http://globocan.iarc.fr/Pages/references.aspx.
2. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61 (5): 759–767.
3. Vogelstein B, Papadopoulos N, Velculescu VE et al. Cancer genome landscapes. Science 2013; 339 (6127): 1546–1558. doi: 10.1126/science.1235122.
4. Gala MK, Mizukami Y, Le LP et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014; 146 (2): 520–529.
5. Bettington M, Walker N, Clouston A et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology 2013; 62 (3): 367–386. doi: 10.1111/his.12055.
6. Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol 2014; 27 (1): 9–14.
7. Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 2007; 21 (20): 2525–2538.
8. Varesco L. Familial adenomatous polyposis: genetics and epidemiology. Tech Coloproctol 2004; 8 (Suppl 2): 305–308.
9. Pezzi A, Roncucci L, Benatti P et al. Relative role of APC and MUTYH mutations in the pathogenesis of familial adenomatous polyposis. Scand J Gastroenterol 2009; 44 (9): 1092–1100. doi: 10.1080/00365520903100481.
10. Kartheuser A, West S, Walon C et al. The genetic background of familial adenomatous polyposis. Linkage analysis, the APC gene identification and mutation screening. Acta Gastroenterol Belg 1995; 58 (5–6): 433–451.
11. Chew MH, Tan WS, Liu Y et al. Genomics of hereditary colorectal cancer: lessons learnt from 25 years of the Singapore polyposis registry. Ann Acad Med Singapore 2015; 44 (8): 290–296.
12. Vaja R, McNicol L, Sisley I. Anaesthesia for patients with liver disease. Contin Educ Anaesth Crit Care Pain 2010; 10 (1): 15–19. doi: 10.1093/bjaceaccp/mkp040.
13. Al-Sohaily S, Biankin A, Leong R et al. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol 2012; 27 (9): 1423–1431. doi: 10.1111/j.1440-1746.2012.07 200.x.
14. Armaghany T, Wilson JD, Chu Q et al. Genetic alterations in colorectal cancer. Gastrointest Cancer Res 2012; 5 (1): 19–27.
15. Strimpakos AS, Syrigos KN, Saif MW. Pharmacogenetics and biomarkers in colorectal cancer. Pharmacogenomics J 2009; 9 (3): 147–160. doi: 10.1038/tpj.2009.8.
16. Trautmann K, Terdiman JP, French AJ et al. Chromosomal instability in microsatellite-unstable and stable colon cancer. Clin Cancer Res 2006; 12 (21): 6379–6385.
17. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 2009; 361 (25): 2449–2460. doi: 10.1056/NEJMra0804588.
18. Farkas SA, Vymetalkova V, Vodickova L et al. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/beta-catenin signaling pathway genes. Epigenomics 2014; 6 (2): 179–191. doi: 10.2217/epi.14.7.
19. Noffsinger AE. Serrated polyps and colorectal cancer: new pathway to malignancy. Annu Rev Pathol 2009; 4 (1): 343–364. doi: 10.1146/annurev.pathol.4.110807.092317.
20. East JE, Saunders BP, Jass JR. Sporadic and syndromic hyperplastic polyps and serrated adenomas of the colon: classification, molecular genetics, natural history, and clinical management. Gastroenterol Clin North Am 2008; 37 (1): 25–46. doi: 10.1016/j.gtc.2007.12.014.
21. Minoo P. Toward a molecular classification of colorectal cancer: the role of MGMT. Front Oncol 2013; 3 : 266. doi: 10.3389/fonc.2013.00266.
22. Shen L, Kondo Y, Rosner GL et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst 2005; 97 (18): 1330–1338.
23. Bodmer WF, Bailey CJ, Bodmer J et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987; 328 (6131): 614–616.
24. Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol 2009; 656 : 85–106.
25. Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature 2011; 476 (7359): 163–169. doi: 10.1038/nature10275.
26. Fearnhead NS, Wilding JL, Bodmer WF. Genetics of colorectal cancer: hereditary aspects and overview of colorectal tumorigenesis. Br Med Bull 2002; 64 : 27–43.
27. Benchabane H, Ahmed Y. The adenomatous polyposis coli tumor suppressor and Wnt signaling in the regulation of apoptosis. Adv Exp Med Biol 2009; 656 : 75–84.
28. Walther A, Johnstone E, Swanton C et al. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 2009; 9 (7): 489–499. doi: 10.1038/nrc2645.
29. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 2001; 1 (1): 55–67.
30. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408 (6810): 307–310.
31. Naccarati A, Polakova V, Pardini B et al. Mutations and polymorphisms in TP53 gene – an overview on the role in colorectal cancer. Mutagenesis 2012; 27 (2): 211–218.
32. Royds JA, Iacopetta B. p53 and disease: when the guardian angel fails. Cell Death Differ 2006; 13 (6): 1017–1026.
33. Soussi T, Kato S, Levy PP et al. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Hum Mutat 2005; 25 (1): 6–17.
34. Iacopetta B, Russo A, Bazan V et al. Functional categories of TP53 mutation in colorectal cancer: results of an International Collaborative Study. Ann Oncol 2006; 17 (5): 842–847.
35. Lamy A, Blanchard F, Le Pessot F et al. Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol 2011; 24 (8): 1090–1100. doi: 10.1038/modpathol.2011.60.
36. Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol 2010; 347 : 21–41. doi: 10.1007/82_2010_68.
37. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 2006; 94 (4): 455–459.
38. Demes M, Scheil-Bertram S, Bartsch H et al. Signature of microsatellite instability, KRAS and BRAF gene mutations in German patients with locally advanced rectal adenocarcinoma before and after neoadjuvant 5-FU radiochemotherapy. J Gastrointest Oncol 2013; 4 (2): 182–192. doi: 10.3978/j.issn.2078-6891.2013.012.
39. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417 (6892): 949–954.
40. Rosty C, Young JP, Walsh MD et al. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol 2013; 26 (6): 825–834. doi: 10.1038/modpathol.2012.240.
41. Sartore-Bianchi A, Martini M, Molinari F et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009; 69 (5): 1851–1857. doi: 10.1158/0008-5472.CAN-08-2466.
42. Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene 2008; 27 (41): 5443–5453. doi: 10.1038/onc.2008.241.
43. Knudson AG Jr. Hereditary cancer, oncogenes, and antioncogenes. Cancer Res 1985; 45 (4): 1437–1443.
44. Mehlen P, Fearon ER. Role of the dependence receptor DCC in colorectal cancer pathogenesis. J Clin Oncol 2004; 22 (16): 3420–3428.
45. Arakawa H. Netrin-1 and its receptors in tumorigenesis. Nat Rev Cancer 2004; 4 (12): 978–987.
46. Castets M, Broutier L, Molin Y et al. DCC constrains tumour progression via its dependence receptor activity. Nature 2012; 482 (7386): 534–537. doi: 10.1038/nature10708.
47. Mazelin L, Bernet A, Bonod-Bidaud C et al. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 2004; 431 (7004): 80–84.
48. Duman-Scheel M. Deleted in colorectal cancer (DCC) pathfinding: axon guidance gene finally turned tumor suppressor. Curr Drug Targets 2012; 13 (11): 1445–1453.
49. Ten Dijke P, Goumans MJ, Itoh F et al. Regulation of cell proliferation by Smad proteins. J Cell Physiol 2002; 191 (1): 1–16.
50. Zhang B, Halder SK, Kashikar ND et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 2010; 138 (3): 969–980. doi: 10.1053/j.gastro.2009.11.004.
51. Chin LJ, Slack FJ. A truth serum for cancer – microRNAs have major potential as cancer biomarkers. Cell Res 2008; 18 (10): 983–984. doi: 10.1038/cr.2008.290.
52. Bonfrate L, Altomare DF, Di Lena M et al. MicroRNA in colorectal cancer: new perspectives for diagnosis, prognosis and treatment. J Gastrointestin Liver Dis 2013; 22 (3): 311–320.
53. Wouters MD, van Gent DC, Hoeijmakers JH et al. Micro - RNAs, the DNA damage response and cancer. Mutat Res 2011; 717 (1–2): 54–66. doi: 10.1016/j.mrfmmm.2011.03.012.
54. Aslam MI, Patel M, Singh B et al. MicroRNA manipulation in colorectal cancer cells: from laboratory to clinical application. J Transl Med 2012; 10 : 128. doi: 10.1186/1479-5876-10-128.
55. Garzon R, Calin GA, Croce CM. MicroRNAs in cancer. Annu Rev Med 2009; 60 (1): 167–179. doi: 10.1146/annurev.med.59.053006.104707.
56. Kusenda B, Mraz M, Maer J et al. MicroRNA biogenesis, functionality and cancer relevance. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2006; 150 (2): 205–215.
57. Raisch J, Darfeuille-Michaud A, Nguyen HT. Role of microRNAs in the immune system, inflammation and cancer. World J Gastroenterol 2013; 19 (20): 2985–2996. doi: 10.3748/wjg.v19.i20.2985.
58. Hrašovec S, Glavač D. MicroRNAs as novel biomarkers in colorectal cancer. Front Genet 2012; 3 : 180. doi: 10.3389/fgene.2012.00180.
59. Slaby O, Svoboda M, Michalek J et al. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer 2009; 8 (1): 102. doi: 10.1186/1476-4598-8-102.
60. Svoboda M, Slyskova J, Schneiderova M et al. HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis 2014; 35 (7): 1510–1515. doi: 10.1093/carcin/bgu055.
61. Jacob S, Praz F. DNA mismatch repair defects: role in colorectal carcinogenesis. Biochimie 2002; 84 (1): 27–47.
62. Stigliano V, Assisi D, Cosimelli M et al. Survival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patients. J Exp Clin Cancer Res 2008; 27 (1): 39. doi: 10.1186/1756-9966 - 27-39.
63. Sandouk F, Al Jerf F, Al-Halabi MH. Precancerous lesions in colorectal cancer. Gastroenterol Res Pract 2013; 2013 : 457901. doi: 10.1155/2013/457901.
64. Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Hum Mutat 1997; 10 (2): 89–99.
65. Devaud N, Gallinger S. Chemotherapy of MMR-deficient colorectal cancer. Fam Cancer 2013; 12 (2): 301–306. doi: 10.1007/s10689-013-9633-z.
66. Tomlinson IP, Webb E, Carvajal-Carmona L et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 2008; 40 (5): 623–630. doi: 10.1038/ng.111.
67. Houlston RS, members of COGENT. COGENT (COlorectal cancer GENeTics) revisited. Mutagenesis 2012; 27 (2): 143–151. doi: 10.1093/mutage/ger059.
68. Tsoi KK, Pau CY, Wu WK et al Cigarette smoking and the risk of colorectal cancer: a meta-analysis of prospective cohort studies. Clin Gastroenterol Hepatol 2009; 7 (6): 682–688. doi: 10.1016/j.cgh.2009.02.016.
69. Chan DS, Lau R, Aune D et al. Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies. PLoS One 2011; 6 (6): e20456. doi: 10.1371/journal.pone.0020456.
70. Park Y, Hunter DJ, Spiegelman D et al. Dietary fiber intake and risk of colorectal cancer: a pooled analysis of prospective cohort studies. JAMA 2005; 294 (22): 2849–2857.
71. Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet 2007; 369 (9573): 1603–1613.
72. Pöschl G, Seitz HK. Alcohol and cancer. Alcohol Alcohol 2004; 39 (3): 155–165.
73. Fedirko V, Tramacere I, Bagnardi V et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol 2011; 22 (9): 1958–1972. doi: 10.1093/annonc/mdq653.
74. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol 2005; 3 (6): 587–594.
75. Berster JM, Göke B. Type 2 diabetes mellitus as risk factor for colorectal cancer. Arch Physiol Biochem 2008; 114 (1): 84–98. doi: 10.1080/13813450802008455.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue 6
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Molecular Genetic Testing for Acute Myeloid Leukemia
- Molecular Pathogenesis of Colorectal Cancer
- A Patient with Primary Intraventricular Gliosarcoma and Long-term Survival – a Case Report
- Quality of Life, Anxiety and Depression in Patients with Differentiated Thyroid Cancer under Short Term Hypothyroidism Induced by Levothyroxine Withdrawal