
Epitelo-mezenchymální tranzice v nádorové tkáni a její role při metastatickém šíření karcinomů
Epithelial-mesenchymal Transition in Tumor Tissue and Its Role for Metastatic Spread of Cancer
Background:
Metastasis, recurrence, and resistance to chemotherapy are leading causes of the majority of cancer-related mortality worldwide. The process of metastasis can be artificially divided into a series of sequential, highly organized, and organ-specific steps. The underlying mechanisms are still poorly understood, but are believed to be mediated by epithelial-mesenchymal transition (EMT). First described in embryogenesis, EMT is a cellular reprogramming process in which epithelial cells acquire a mesenchymal phenotype. During this transformation, epithelial cells lose their shape, epithelial markers, and ability to grow in colonies. They acquire a spindle-shaped morphology and exhibit more motile and invasive behavior. These phenotypic changes are associated with modifications in different interconnected protein and gene families, such as transcription factors, cadherins, catenins, matrix metalloproteases, and growth receptors. EMT has been observed in many cancers, such as breast, ovarian, colon, and esophageal cancers, and is associated with poor prognosis and metastasis. Also, resistance to cytotoxic treatments is associated with reactivation of embryonic programs. Understanding this process is necessary to provide a better understanding of cancer progression and could lead to the development of new therapeutic or prognostic strategies for the treatment of cancer.
Conclusion:
This article summarizes the known molecular pathways involved in EMT in cancer.
Key words:
epithelial-mesenchymal transition – carcinoma – metastasis
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
24. 6. 2016
Accepted:
14. 11. 2016
Authors:
V. M. Matějka 1,2; J. Fínek 1; M. Králíčková 2
Authors‘ workplace:
Onkologická a radioterapeutická klinika LF UK a FN Plzeň
1; Ústav histologie a embryologie, LF UK v Plzni
2
Published in:
Klin Onkol 2017; 30(1): 20-27
Category:
Review
doi:
https://doi.org/10.14735/amko201720
Overview
Východiska:
Metastazování, relaps a rezistence k chemoterapii jsou celosvětově hlavními důvody většiny úmrtí na malignitu. Proces metastazování je velmi precizně organizovaný a orgánově specifický. Mechanizmy metastazování nejsou stále přesně známy, ale některé z nich jsou zprostředkovány programem epitelo-mezenchymální tranzice (EMT). Poprvé byl tento proces popsán v průběhu embryogeneze. Tento buněčný program vede ke změně epiteliálního fenotypu buňky v mezenchymální typ. V průběhu této tranzice dochází ke změně tvaru epiteliální buňky, která ztrácí svoji polaritu, závislost na buněčných spojích a celou řadu epiteliálních markerů. Naopak získává vřetenovitý tvar a stává se mechanicky odolnější a je schopnější vycestovat z buněčné kolonie. Tato změna fenotypu je provázena i změnou exprese proteinů a genů, jako jsou transkripční faktory, cadheriny, cateniny, matrix metaloproteinázy nebo receptory pro růstové faktory. Známky aktivace programu EMT byly již popsány v různých typech nádorů, např. u karcinomu prsu, vaječníků, karcinomu tlustého střeva a jícnu, a jejich přítomnost byla asociována s horší prognózou onemocnění a častějším metastatickým šířením. Také rezistence k cytostatické léčbě je spojována s reaktivací těchto embryonálních procesů. Porozumění tomuto programu je nezbytné k lepšímu chápání procesu progrese maligního onemocnění, stejně jako k hledání nových léčebných cílů a prognostických faktorů.
Závěr:
Tento článek shrnuje dosud známé molekulární kaskády zapojené do procesu EMT u nádorových onemocnění.
Klíčová slova:
epitelo-mezenchymální tranzice – karcinom – metastatický
Úvod
Nádorová onemocnění jsou dnes druhou nejčastější příčinou smrti. Jen v ČR jimi onemocní každý třetí člověk a každý čtvrtý na ně zemře. Primární tumor je nicméně příčinou smrti jen výjimečně. V naprosté většině případů je úmrtí způsobeno selháním orgánů při jejich sekundárním postižení nemocí. Porozumění metastatické kaskádě a mechanizmům, které se při ní uplatňují, se tak stává nezbytností ve snaze o léčbu tohoto společensky závažného onemocnění [1,2].
Jedním z mechanizmů uplatňujících se při diseminaci nádorového onemocnění je v posledních letech studovaná epitelo-mezenchymální tranzice (EMT) [3]. Tento proces je stále více považován za jeden z hlavních a nezbytných mechanizmů uplatňujících se při diseminaci epiteliálních nádorů (karcinomů) do dalších, převážně parenchymatických orgánů těla. Při EMT dochází ke změně fenotypu buněk z epiteliálního na mezenchymový. Nádorové buňky tak získávají větší mechanickou odolnost, nezávislost na bazální membráně, motilitu a v neposlední řadě i odolnost k protinádorové léčbě [2–4].
Takto pozměněné buňky aktivně vycestovávají do krevního oběhu, kterým jsou pak bez nebezpečí mechanického postižení unášeny do místa nidace. Následně se mechanizmem opačným, zvaným mezenchymo-epiteliální tranzice (MET), vracejí ke svému původnímu epiteliálnímu fenotypu a zakládají dceřiná ložiska, mikrometastázy, z nichž se při příznivých podmínkách může vyvinout plnohodnotné metastatické ložisko [2,4].
Avšak ani EMT není vše vysvětlujícím mechanizmem. Jsou známy i jiné způsoby nádorové diseminace, např. kolektivní migrace, kdy se do krevního oběhu uvolňují celé shluky nádorových buněk s vlastní bazální membránou. Stejně tak tímto způsobem nedokážeme vysvětlit generalizaci anaplastických karcinomů nebo mezenchymálních nádorů. Množství důkazů nicméně poukazuje na významnou úlohu tohoto fenoménu při generalizaci dobře a středně diferencovaných karcinomů.
Nádorová tkáň – typy buněk z hlediska fenotypu
Nádorová masa karcinomu není tvořena homogenní hmotou. Jedná se o velmi komplexní prostředí, v němž jsou zastoupeny dvě základní heterogenní složky. První složkou je nádorový parenchym. Ten je tvořen nádorovými buňkami, které se formují do více či méně organizovaných struktur napodobujících původní tkáň. Druhou složkou je pak nádorové stroma obklopující nádorové buňky. Stroma je tvořeno vazivovou tkání, kterou procházejí lymfatické a krevní cévy zásobující či drénující tumor. Dále jsou v něm lymfocyty zodpovědné za imunologickou odpověď těla na nádor [2,4].
Tyto obě složky spolu stále na molekulární úrovni komunikují. Jedním z hlavních léčebných mechanizmů v onkologii je narušení této komunikace nebo využití jejích mechanizmů k cílenému poškození nádorových buněk. Iniciace EMT v nádorové tkáni je právě jedním z důsledků vzájemné komunikace mezi nádorovými buňkami a obklopujícím stromatem [2,5].
EMT
EMT je proces, při kterém dochází ke změně epiteliálního fenotypu ve fenotyp mezenchymální. V průběhu tohoto procesu dochází k celé řadě změn – ztráta polarity buněk, změna tvaru buňky ve vřetenovitý, ztráta mezibuněčných spojů a závislosti na nich, zvýšená produkce složek extracelulární matrix (ECM), ztráta závislosti na bazální membráně, zvýšená schopnost motility a v neposlední řadě zvýšená odolnost vůči apoptóze. Dokončení procesu EMT je pak signalizováno degradací základní bazální membrány a vytvořením buňky, která je schopna vycestovat z buněčné vrstvy, ve které vznikla [2,5].
Intracelulárně se aktivují transkripční faktory nezbytné ke změně specifických buněčných proteinů, což vede k reorganizaci a expresi cytoskeletu bílkovin, produkci ECM degradujících enzymů a změny v expresi specifických mikroRNA (miRNA). Tyto faktory se také samozřejmě používají jako specifické biomarkery, které můžeme použít k demonstraci tohoto procesu [2,5,6].
Dosud byly popsány tři základní typy EMT.
EMT I
Vůbec poprvé byla EMT pozorována E. Hayem, který popsal změny fenotypu buněk v embryu kuřete [7]. Jev, při kterém zárodečný epitel měnil své vlastnosti a tvar v typický pro mezenchymální buňky, byl tehdy popsán jako transformace. Když bylo později zjištěno, že buňky, které tento proces podstoupily, jsou schopné vrátit se ke svému původnímu fenotypu, byl tento dynamický jev přejmenován na tranzici.
Jako EMT I. typu nebo také vývojová EMT jsou souhrnně označovány všechny tranzice, které se odehrávají v průběhu vývoje zárodku. V embryu pak na tomto „switchi“ závisí množství dílčích kroků důležitých pro vývoj jedince. Například gastrulace, zakládání neurální lišty, vývoj patra, formování srdečních chlopní, vývoj nefronů a končetinového svalstva [2,8].
Vývojová EMT je velmi precizně a specificky řízená signálními molekulami produkovanými Spemann-Mangoldovým organizérem, což je oblast embrya odpovědná za zachování osové souměrnosti organizmu [9]. První případ EMT je pozorovatelný již v průběhu gastrulace, při které dochází ke ztrátě bazální membrány pod epiblastem. Buňky následně ztrácejí E-cadherin z povrchu buněčné membrány, a tím dochází k destabilizaci buněčných spojů. Buňky entodermu migrují a zakládají mezoderm a následně po reverzi pomocí MET i entoderm. Tento proces je řízen primárně fibroblastickým růstovým faktorem přes transkripční faktor Snail [10].
Dalším případem EMT I. typu je proces zakládání neurální trubice. Neuroektoderm generuje populaci mezenchymálních buněk obklopující neurální trubici, z nichž následně vznikají další struktury, jako neurony periferního nervového systému, pigmentové buňky a buňky dřeně nadledvin [11]. Molekulárně zde opět došlo k prokazatelné expresi Snail faktoru, který inhibuje E-cadherin. Zpočátku byl v tomto případě proces definován jako kolektivní migrace buněk, dokud ovšem nedošlo k potvrzení mezenchymálního fenotypu výsledných buněk [11,12].
V průběhu vývoje srdečních chlopní byla popsána přeměna endokardiálních buněk vlivem působků produkovaných myokardem. U endokardiálních polštářů dochází k aktivní migraci buněk přes bazální membránu a jejich přeměnu na fibroblasty [13]. Tentokrát buňky, které projdou EMT, nadále zůstávají mezenchymální a formují kardiální skelet. V průběhu formování kardiálních chlopní byly jako regulátory EMT identifikovány TGF-β, BMP, β-catenin, MSX1/2 a Slug [13–17].
EMT II
Jako EMT II. typu nazýváme tento program, pokud je spojený s hojením ran, regenerací tkání, ale také fibrózou tkání a orgánů. V případě zánětlivé reakce nebo poškození orgánů dochází ke spuštění programu EMT, který napomáhá reparaci tkáně. Důsledkem je produkce fibroblastů a jiných příbuzných buněk, které se účastní rekonstrukce tkání a zánětlivé odpovědi. Na rozdíl od I. typu je tento typ asociován se zánětem a je ukončen, jakmile je zánětlivá reakce zmírněna. To je patrné na průběhu hojení ran a při regeneraci tkání [18–20].
Pokud není zánětlivý proces ukončen, hromadí se v tkáni zánětlivé buňky a fibroblasty, a stejně tak proces EMT nadále běží. Zdravá tkáň je destruována a nahrazována tkání fibrózní. Spojení tohoto typu EMT s fibrózou již bylo prokázáno v ledvinách, játrech, plicích a zažívací trubici [20]. Epiteliální buňky ledvin, jater, plic a střeva, které prošly EMT v průběhu chronického zánětu, byly identifikovány pomocí markerů jako vimentin, desmin, DDR2 (discoidin domain receptor tyrosine kinase 2), FSP1 (fibroblast specifický protein 1), třída S100 proteinů cytoskeletu a α-SMA (α-smooth muscle actin) [21–23].
Kromě markerů pro mezenchymální typ totiž tyto buňky nadále vykazují specifické morfologické a molekulární znaky pro epiteliální fenotyp, jako epiteliální cytokeratiny a E-cadherin. Kombinace markerů jak epiteliálních, tak mezenchymálních potvrzuje jejich fenotypový přechod [24].
Proces EMT je v tomto případě vícestupňový. Buňky pod tlakem zánětlivé reakce podstoupí několik prvních kroků k změně svého fenotypu, tzv. dílčí EMT. Poté opustí epiteliální vrstvu, projdou bazální membránou a začnou se hromadit v intersticiu tkáně, kde nakonec dochází ke ztrátě i zbylých epiteliálních znaků a převládne u nich plně fibroblastický fenotyp [24,25].
EMT II. typu byla prokázána např. ve studiích fibrózy ledvin u člověka [26]. Rastaldi et al v souboru 133 pacientů s fibrózou ledvin prokázal tento proces v podstatném počtu vzorků. Průkazu bylo dosaženo dvojitým označováním tubulárních epitelových buněk na cytokeratin, vimentin, α-SMA nebo zona occludens 1 (ZO-1). Další studie prokázala EMT u pacientů s Crohnovou chorobou, kde se podílí na fibrotizaci tlustého střeva [27]. Studium EMT II. typu se tak stává nezbytným pro budoucí terapeutický zásah do mechanizmu fibrotizace u chronických zánětlivých procesů.
EMT III
Pro karcinomy je charakteristickým rysem nadměrná buněčná proliferace a přítomnost angiogeneze [28]. Jelikož i epiteliální nádorové buňky jsou závislé na přítomnosti bazální membrány, je schopnost jejího překonání nezbytným krokem metastatické kaskády. I tento proces přestupu přes bazální membránu je vícefázový. Prvním krokem je získání nezávislosti na vazbě k bazální membráně a následně na mezibuněčných spojích s okolními buňkami. Získání těchto schopností je podmíněno právě aktivací programu EMT.
Buňky, které získaly schopnost přecházet přes bazální membránu a exprimují mezenchymální markery, jako α-SMA, FSP1, vimentin a desmin, byly prokázány hlavně na okrajích nádoru. Zde tvoří invazivní předvoj a jsou asociovány s tzv. tumor buddingem [2,3,29].
O těchto buňkách se uvažuje jako o těch hlavních, které vstupují do metastatické kaskády, protože získání daných vlastností jim zajišťuje dostatečnou motilitu pro přestup do krevního oběhu (intravazaci). Dále získávají touto změnou dostatečnou mechanickou odolnost, aby vydržely cestu krevním oběhem, kde panují tlaky, pro které nejsou typicky epiteliální buňky stavěny. V neposlední řadě pak je pro šíření nemoci nezbytná schopnost přestupu přes cévní stěnu do parenchymatických orgánů (extravazace), kde vytvářejí mikrometastázy, z kterých se mohou dále vyvinout metastázy [30–32].
Při histologickém vyšetření metastáz nacházíme buňky karcinomu typicky epiteliálního fenotypu, v převážné většině případů velmi podobné zdrojové nádorové tkáni. Tento fenomén vysvětluje mechanizmus MET. MET je velmi pravděpodobně způsobena lokálním mikroprostředím, kde po extravazaci nádorových buněk mezenchymálního fenotypu nejsou již tyto buňky pod vlivem signálů, které byly zodpovědné za indukci EMT v primárním ložisku [33–35]. To naznačuje, že indukce EMT je pravděpodobně centrálně důležitý mechanizmus pro progresi karcinomu do metastatického stadia. Nicméně mnoho kroků tohoto mechanického modelu stále vyžaduje přímé experimentální ověření.
Signální dráhy zapojené do EMT v nádorovém onemocnění
I když již bylo množství indukčních a signalizačních molekul podílejících se na spuštění programu EMT v nádorové tkáni popsáno, celý mechanizmus aktivace a průběhu tohoto procesu zůstává nejasný. Nejvíce zvažovaná teorie předpokládá vliv genetických a epigenetických změn v nádorových buňkách na citlivost pro signály indukující EMT. Ty pak přicházejí ve zvýšené míře hlavně z nádorového stromatu.
V řadě karcinomů byly popsány vlivy růstových a transformačních faktorů přicházejících právě z nádorového stromatu, jako HGF, EGF, PDGF a TGF-β. Tyto faktory indukují transkripční faktory jako Snail, Slug, ZEB1 (zinc finger E-box binding homeobox 1), Twist, Goosecoid a FOXC2 [30,36–40]. Každý z těchto transkripčních faktorů může způsobit aktivaci EMT programu. Málokdy jde ale o vliv jednoho z těchto faktorů samotného, mnohem častěji je aktivace multifaktoriální.
Realizace programu je pak závislá na celé řadě intracelulárních signálů. Ty jsou realizovány transdukčními proteiny jako ERK, MAPK, PI3K, Akt, Smads, RhoB, β-catenin, Ras a c-Fos a proteiny buněčné membrány – integriny (hlavně β4 α5β1 integrin, a αVβ6 integrin) [41]. Aktivace a průběh EMT jsou také usnadněny primárním narušením mezibuněčných spojů a spojů mezi buňkou a ECM. Toho se pak účastní hlavně celá další řada integrinů [41–46].
TGF-β
Nejvýznamnějším faktorem v procesu aktivace EMT je TGF-β. Jeho role v nádorových onemocněních není ale jednoduchá. V první fázi funguje jako inhibiční faktor epiteliální buněčné proliferace a induktor apoptózy. V pokročilém epiteliálním nádoru pak má naopak roli pozitivního regulátoru progrese nádoru a metastáz [47–49]. Toto bylo potvrzeno již studiemi in vitro, kde bylo prokázáno i spuštění programu EMT [50].
V aktivaci EMT přes TGF-β byly identifikovány dvě základní signální dráhy. První z nich zahrnuje Smad proteiny, které zprostředkovávají TGF-β kroky k vyvolání EMT přes ALK-5 receptor [51–55]. Tato cesta vede k usnadnění motility buňky a navíc inhibiční vliv Smad moduluje účinky příslušných transkripčních faktorů a cytoplazmatických kináz, a tím indukuje autokrinní produkci TGF-β, což vede k zesílení EMT programu [56,57].
Druhá cesta indukce EMT probíhá přes p38 MAPK a RhoA, které vedou opět k autokrinní produkci TGF-β. Na této signální dráze se také podílejí integriny β1a αVβ6 a Fibulin-5 [56–58]. Některá pozorování navíc potvrdila, že dráhy ERK/MAPK a PI3K/Akt zprostředkovávají EMT vyvolané mutantním statutem RAS. Takto spuštěný program EMT může být blokován např. MEK1 inhibitory [59,60]. Také onkogen Raf zprostředkovává TGF-β indukovanou EMT, a podporuje tím invazivnost nádorových buněk. V myších modelech karcinomu kůže a lidské rakoviny tlustého střeva absence TGF-β exprese receptoru skutečně významně zlepšovala prognózu [61,62].
Obě tyto dráhy končí aktivací transkripčních faktorů Snail, Slug, Twist, ZEB1 a TCF3 [63], která vede k inhibici exprese E-cadherinu a zvýšené expresi mezenchymálních markerů jako N-cadherin a vimentin, a navíc ke zvýšené produkci matrix-metaloproteináz.
Význam E-cadherinu
Význam ztráty exprese E-cadherinu v nádorových buňkách pro aktivaci EMT programu byla potvrzena již v mnoha studiích (obr. 1) [64,65]. Pokud zůstává komplex E-cadherin v buněčné membráně, je potlačena buněčná proliferace a buňky si zachovávají epiteliální fenotyp. Naopak, pokud je z buněčné membrány E-cadherin uvolněn, je uvolněn do cytoplazmy také β-catenin, jenž se následně cytoplazmou přesouvá do jádra. Tam pak působí jako transkripční faktor. S tímto přesunem koreluje i změna fenotypu z epiteliálního na mezenchymální [66]. Akumulace β-cateninu v jádru spojená se ztrátou E-cadherinu koreluje se vznikem invazivního fenotypu a vstupem buňky do programu EMT. Buňky, jež ztrácejí E-cadherin ze svého buněčného povrchu, se tak stávají citlivějšími na impulzy růstových faktorů vedoucí ke spuštění EMT [67].
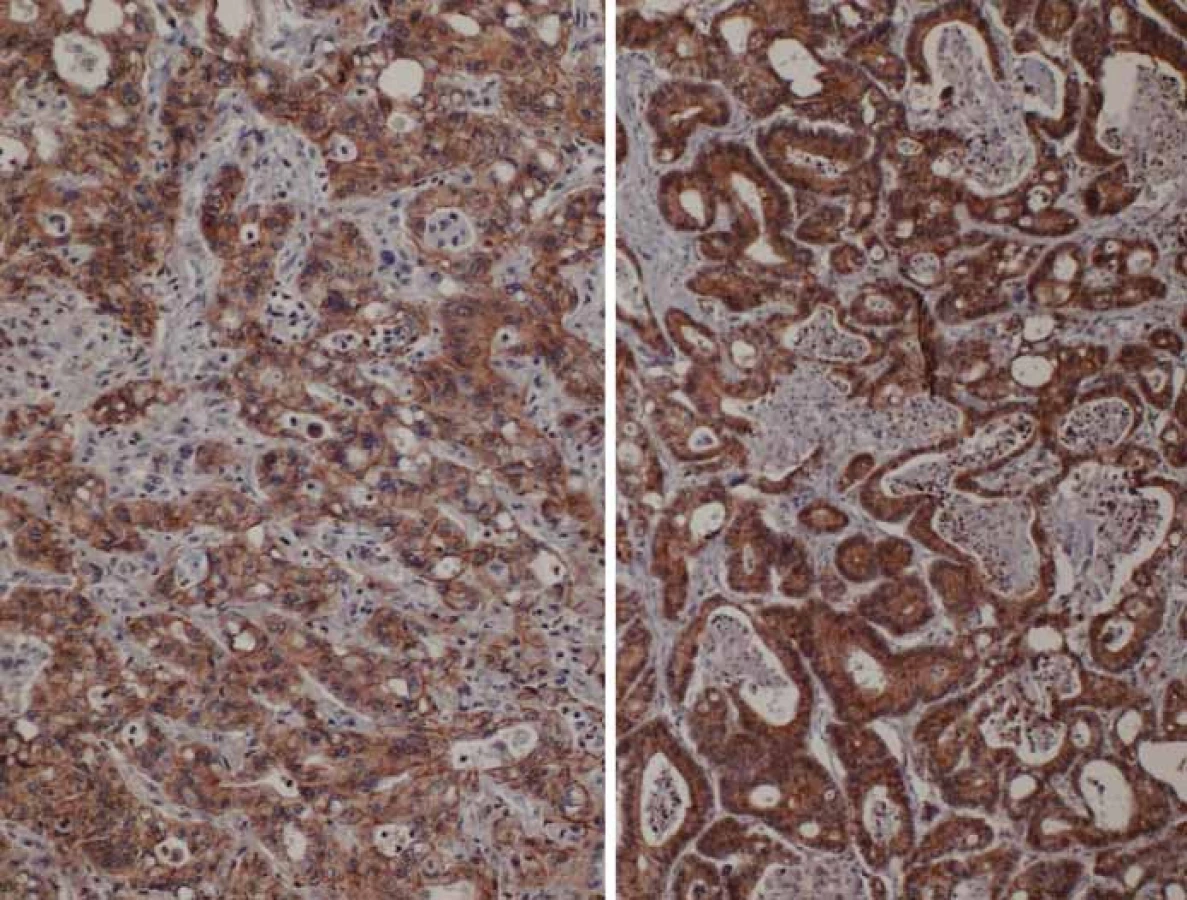
Některé studie prokázaly, že epigenetická regulace aktivity E-cadherinu a β-cateninu je důležitá při stanovení metastatického potenciálu rakovinných buněk [68–71]. Hladiny exprese E-cadherinu jsou odlišné v různých lidských nádorech. Důležitou roli hrají nejen hladiny exprese, ale také mutace genu pro E-cadherin, které byly identifikovány v různých nádorových buňkách, častěji pak u nádorů agresivních a metastatických [68–70].
Význam ztráty E-cadherinu pro proces EMT je dobře ilustrován několika EMT vyvolávajícími transkripčními faktory, které usnadňují akvizici mezenchymálního fenotypu, jako je Snail a Slug, ZEB1, SIP1 a E12 (také známý jako E47-E2A).
Tyto transkripční faktory jsou indukované TGF-β a jednou exprimované potlačují expresi E-cadherinu. Ztráta E-cadherinu podporuje Wnt signalizaci a je spojena s vysokou úrovní Snail v jádru [72]. SIP1 potlačuje expresi E-cadherinu a váže se spolu se Snail na promotor E-cadherinu. Exprese Snail a E-cadherinu je nepřímo úměrná prognóze u pacientů, kteří trpí karcinomem prsu nebo spinocelulárním karcinomem dutiny ústní [72,73].
Notch
Signální dráha Notch je další drahou zapojenou do procesu aktivace EMT. Jedná se o hlavní dráhu řady buněčných procesů, vč. apoptózy, schopnosti migrace, invaze a angiogeneze [74,75]. Aktivace této dráhy začíná aktivací receptoru Notch ligandem (Jagged, Delta), který způsobí proteolytické rozštěpení receptoru, kdy intracelulární část receptoru působí jako transkripční aktivátor. V mnoha nádorových onemocněních byla popsána zvýšená exprese receptoru Notch [76]. Výsledkem je aktivace EMT pomocí transkripčního faktoru Snail [77]. Aktivace EMT touto cestou byla popsána hlavně v případech hypoxií indukovanou EMT [78].
WNT/β-catenin
Signální dráha Wnt hraje také jednu z hlavních rolí v celé řadě vývojových procesů. Mezi hlavní patří gastrulace, buněčná polarita, organogeneze, osové formování a další [79]. Centrálním regulátorem této dráhy je β-catenin, protein ukotvený do E-cadherinu v buněčné membráně. Aktivace Wnt jeho ligandy vede k inhibici aktivity komplexu zodpovědného za odbourávání β-cateninu, a tím k jeho navýšení v cytoplazmě, což vede k jeho translokaci do jádra, kde reguluje expresi cílových genů Wnt [80]. Kromě toho tato dráha může zkříženě aktivovat dráhu TGF-β, Notch a CTGF (connective tissue growth factor). Translokace β-cateninu (obr. 2, 3) do jádra, kde působí jako transkripční faktor na cílové geny, které jsou zodpovědné za zvýšení invazivity, schopnosti migrace a buněčné proliferace, je jedním z hlavních mechanizmů aktivace programu EMT [81].
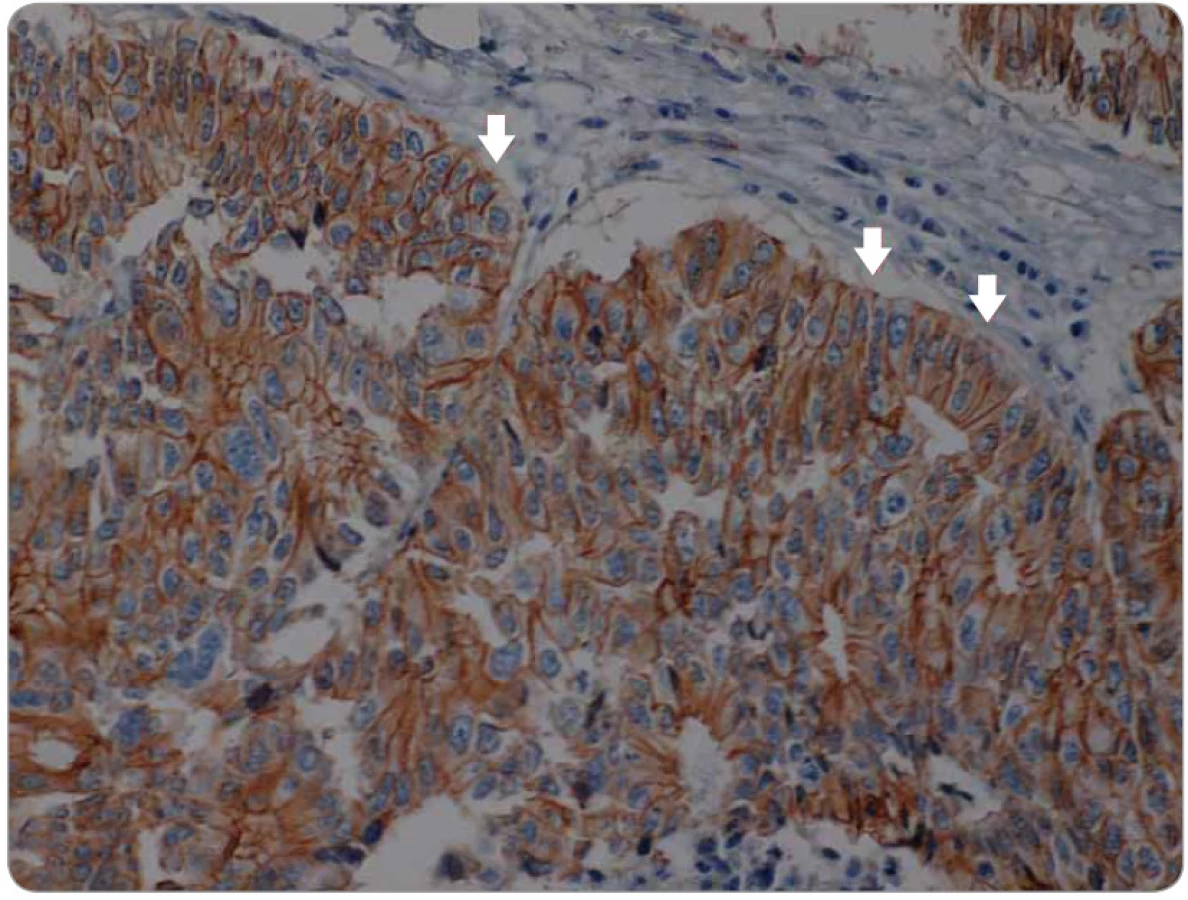
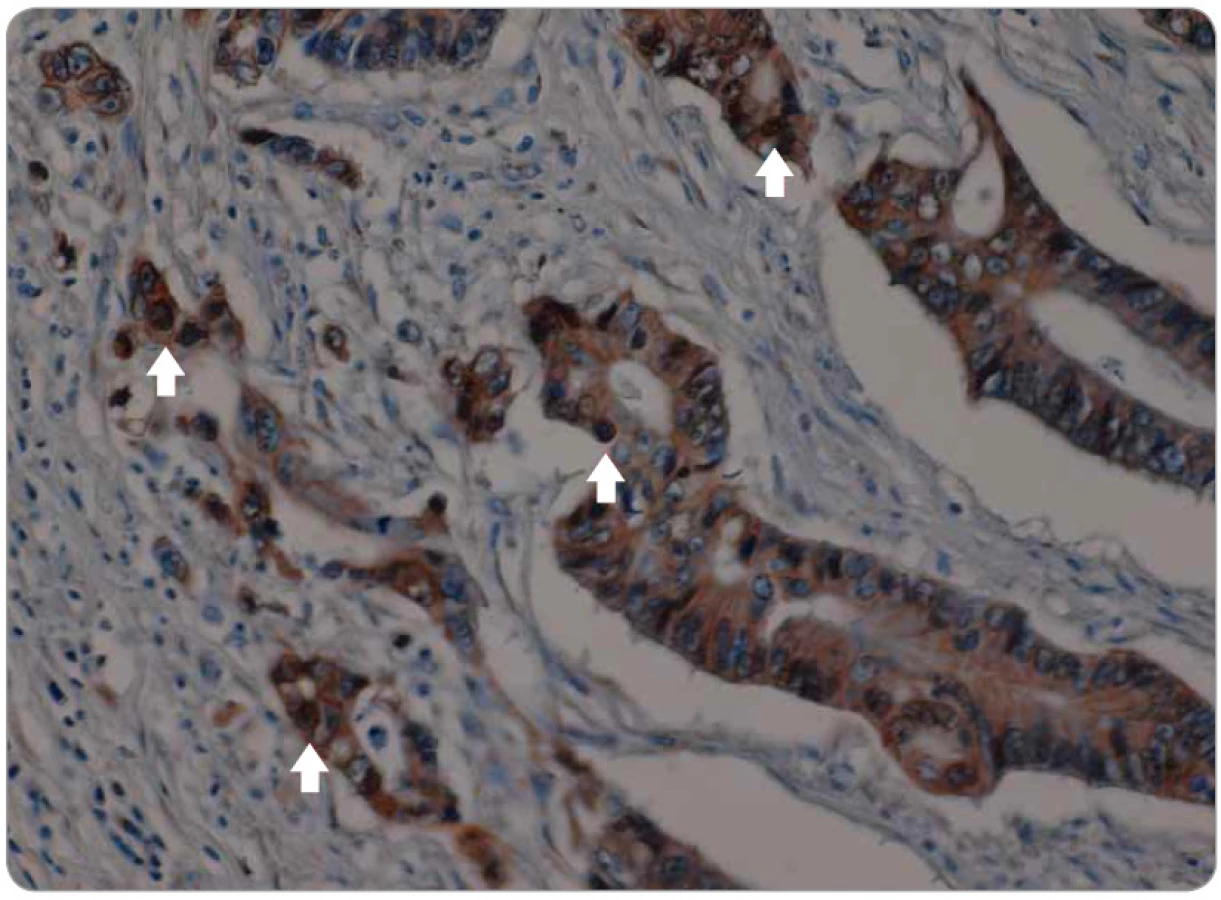
Hedgehog
Signální dráha Hedgehog (Hh) je nezbytná pro celou řadu procesů v embryogenezi, nejvíce se pak podílí na buněčném růstu, buněčné proliferaci, diferenciaci a organogenezi. Ve zdravé dospělé tkáni je pak zásadní pro udržování procesů v populaci kmenových buněk, buněčné opravě a regeneraci [82]. Tato dráha je aktivována pomocí peptidových ligandů nazývaných Hedgehog, z nichž jsou dosud popsány tři homology a dva transmembránové receptory (Ptch a SMO). Hypoxie aktivuje signální dráhu Hh zvýšením exprese SMO [83].
MiRNA a EMT
MiRNA jsou malé nekódující RNA dlouhé 19–24 nukleotidů, které posttranskripčně kontrolují genovou expresi degradací mRNA nebo inhibicí translace. V posledních letech byl prokázaný velký význam množství různých miRNA v procesu nádorového onemocnění. Zkoumán byl také vliv těchto molekul na program EMT.
Nejvíce byl prozkoumán vliv miRNA rodiny miRNA200 [84]. Například jedním z cílů miRNA200c a miRNA200b jsou transkripční faktory ZEB1 a ZEB2, jejichž inaktivace touto cestou vede ke stabilizaci epiteliálního fenotypu a inhibici aktivace EMT. MiRNA205 přímo inhibuje molekuly kaskády TGF-β. V karcinomu prsu ztráta miRNA200 koreluje se zvýšenou expresí vimentinu a snížením hladiny E-cadherinu v nádorových buňkách [84–88]. Na druhou stranu vliv těchto miRNA a jejich ektopická produkce se pravděpodobně podílejí na MET, tedy návratu fenotypu nádorové buňky k fenotypu epiteliálnímu. To prokázala např. studie porovnávající miRNA200 a miRNA141 v tkáni kolorektálního karcinomu a tkáni korelujících jaterních metastáz. Oproti centru ložisek byla exprese těchto miRNA na invazivním čele potlačena. Tento vliv miRNA na program EMT z nich dělá velmi významný potenciální klinický cíl. Kromě výše popsaných miRNA se na inhibici EMT podílejí také další – miRNA34, 137, 138, 212, 30b a 101 [84,87–89].
Naopak u mnohých miRNA byl prokázán zase prometastatický vliv. Zvýšená aktivita programu EMT asociovaného s TGF-β byla nalezena při zvýšené expresi miRNA21 a 31 [90–91]. U miRNA21 byla prokázána i signifikantně zhoršená prognóza. Naproti tomu u miRNA31 bylo pozorováno častější metastatické šíření lymfatickým systémem. MiRNA9 inhibuje přímo expresi E-cadherinu. Další prometastatické miRNA jsou miRNA130b, 29a a 103/107, u nichž bylo také pozorováno zvýšení znaků EMT v tkáni [90–94].
EMT a léková rezistence
Rezistence k cytostatikům je jedním z nejzávažnějších problémů moderní onkologie. V poslední době se množí důkazy, že proces EMT se podílí i na vývoji chemorezistence nádorových buněk. Například buněčné řady oxaliplatin rezistentních nádorových buněk vykazují znaky mezenchymálního fenotypu (pokles exprese E-cadherinu a zvýšení exprese vimentinu) [95]. Podobně zvýšení Snail a TGF-β v nádorových buňkách vedlo k zvýšení rezistence na 5-fluorouracil. Domněnku pak potvrzuje zjištění, že miR200c, výrazný inhibitor EMT, zvyšuje chemosenzitivitu [96,97].
Na druhou stranu bylo potvrzeno v nádorech u nemocných podstupujících neoadjuvantní léčbu, že v jejich biop - tických vzorcích byly popsány změny charakteru EMT, tzn. vyvíjela se v nich rezistence k léčbě [98].
Role EMT v rezistenci k léčbě není ale omezená jen na cytostatika. Například v buněčných liniích pankreatických a kolorektálních nádorových buněk rezistentních k cílené léčbě byla potvrzena vyšší exprese proteinů asociovaných s EMT programem – vimentin, ZEB1 a Snail. V buňkách s epiteliálním fenotypem je až 7× vyšší citlivost k léčbě než v těch, které prošly programem EMT a vykazují mezenchymální znaky [99].
I přes to, že není jasný mechanizmus aktivace EMT v průběhu léčby, je zvažováno několik možných mechanizmů. Nejpravděpodobnější se zdá být teze nádorových kmenových buněk. EMT produkuje buňky, které mají některé z jejích parametrů [100]. Nádorové kmenové buňky (cancer stem cells – CSC) jsou vysoce chemorezistentní buňky schopné sebeobnovy, které zůstávají dlouhodobě v G0 fázi buněčného cyklu. Jejich rezistence je způsobena několika mechanizmy – minimální vstupování do buněčného cyklu, vysoce efektivní proteiny DNA oprav a vysoce aktivní MDMT systém [100,101].
Možnosti léčebného ovlivnění EMT
Poznání procesu EMT vede nejen k odhalování molekulárních mechanizmů v nádorové tkáni, ale také nastiňuje nové možné cíle protinádorové terapie. Možnost zásahu do tohoto specifického programu je slibná jednak z důvodu ovlivnění metastatického potenciálu nádoru, jednak možností ovlivnění chemosenzitivity onemocnění.
Již byly provedeny první pokusy o ovlivnění programu EMT. Jedním z prvních bylo podání molekul ovlivňujících expresi E-cadherinu. Mezi tyto molekuly patří např. metotrexát, u kterého byl tento efekt pozorován. Další molekuly se připravují. Zasažení do přímé aktivace EMT je velmi náročné, neboť se jedná o transkripční faktory. Slibnou cestou se jeví spíše ovlivnění hladin miRNA [102].
Další možnou metodou ovlivnění EMT se zdá být T lymfocyty mediovaná imunoterapie. Brachyury je člen T-box transkripčního faktoru (rodina), spouštěč EMT programu, na který byla připravena vakcína, která nyní vstoupila do I. fáze klinického testování [103,104].
Nejvíce zkoumanou a testovanou cestou je zacílení na receptory extracelulárního signálu vedoucího k aktivaci EMT, např. inhibitory ALK5, MEK a SRC. Další cestou je pak ovlivnění cyklooxygenázy-2 (např. celecoxib), které může ovlivnit hypoxií nebo EGF aktivovaný proces EMT [105–107].
Jak je patrné, možnosti terapeutického ovlivnění EMT jsou široké a agens zasahujících do tohoto programu bude přibývat. Problémem však zůstává také správné načasování této terapie, neboť v různých situacích může být dopad takové léčby rozdílný. Během počáteční fáze onemocnění může léčba způsobit diferenciaci mezenchymálních buněk v nádoru. V pozdější fázi pak stejná léčba může uspíšit proces MET, a tím růst již přítomných metastatických ložisek.
Závěr
Jak je z výše zmíněného patrné, proces EMT hraje u nádorového onemocnění ústřední roli v procesu metastazování a získávání odolnosti k systémové onkologické léčbě. Vazbu má také k dalším negativním prognostickým faktorům, jako tumor budding, volné nádorové cirkulující buňky a obnově CSC. Stále ještě víme velmi málo o tomto fyziologickém jevu, který, pokud se dá do služeb nádorového onemocnění, je víceméně zodpovědný za hlavní pochody, které vedou k ohrožení pacienta na životě – selhání léčby a metastatické šíření. Příprava léčebných agens zasahujících do tohoto programu tak velmi pravděpodobně rozšíří armamentarium klinických onkologů a povede k omezeným schopnostem nádoru diseminovat.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Obdrženo: 24. 6. 2016
Přijato: 14. 11. 2016
MUDr. Vít Martin Matějka
Onkologická a radioterapeutická klinika
LF UK a FN Plzeň
alej Svobody 80
304 60 Plzeň
e-mail: matejkavm@fnplzen.cz
Sources
1. Dusek L, Muzik J, Maluskova D et al. Epidemiology of cancers with implemented screening programmes in an international comparison. Klin Onkol 2014; 27 (Suppl 2): 2S40–2S48. doi: 10.14735/amko20142S40.
2. Yeatman TJ, Nicolson GL. Molecular basis of tumor progression: mechanisms of organ-specific tumor metastasis. Sem Sur Onco 1993; 9 (3): 256–263.
3. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119 (6): 1420–1428. doi: 10.1172/JCI39104.
4. Chui MH. Insights into cancer metastasis from a clinicopathologicperspective: epithelial-mesenchymal transition is not a necessary step. Int J Cancer 2013; 132 (7): 1487–1495. doi: 10.1002/ijc.27745.
5. Wan L, Pantel K, Kang Y. Tumormetastasis: moving new biological insights into the clinic. Nat Med 2013; 19 (11): 1450–1464. doi: 10.1038/nm.3391.
6. Thiery JP, Sleeman JP. Complex networks orchestrace epithelial-mesenchymal transitions. Nat Rev Mol Cell Biology 2006; 7 (2): 131–142.
7. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995; 154 (1): 8–20.
8. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 2005; 233 (3): 706–720.
9. Heisenberg CP, Solnica-Krezel L. Back and forth between cell fate specification and movement during vertebrate gastrulation. Curr Opin Genet Dev 2008; 18 (4): 311–316. doi: 10.1016/j.gde.2008.07.011.
10. Solnica-Krezel L. Conserved patterns of cell movements during vertebrate gastrulation. Curr Biol 2005; 15 (6): 213–228.
11. Tucker RP. Neural crest cells: a model for invasive behavior. Int J Biochem Cell Biol 2004; 36 (2): 173–177.
12. Duband JL, Thiery JP. Appearance and distribution of fibronectin during chick embryo gastrulation and neurulation. Dev Biol 1982; 94 (2): 337–350.
13. Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007; 185 (1–3): 146–156.
14. Inai K, Norris RA, Hoffman S et al. BMP-2 induces cell migration and periostin expression during atrioventricular valvulogenesis. Dev Biol 2008; 315 (2): 383–396. doi: 10.1016/j.ydbio.2007.12.028.
15. Liebner S, Cattelino A, Gallini R et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 2004; 166 (3): 359–367.
16. Chen YH, Ishii M, Sucov HM et al. Msx1 and Msx2 are required for endothelial-mesenchymal transformation of the atrioventricular cushions and patterning of the atrioventricular myocardium. BMC Dev Biol 2008; 8 : 75. doi: 10.1186/1471-213X-8-75.
17. Romano LA, Runyan RB. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Dev Biol 2000; 223 (1): 91–102.
18. Thiery JP, Acloque H, Huang RY et al. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139 (5): 871–890. doi: 10.1016/j.cell.2009.11.007.
19. Strutz F, Okada H, Lo CW et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 1995; 130 (2): 393–405.
20. Okada H, Danoff TM, Kalluri R et al. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997; 273 (4 Pt 2): F563–F574.
21. Zeisberg EM, Tarnavski O, Zeisberg M et al. Endothelial-tomesenchymal transition contributes to cardiac fibrosis. Nat Med 2007; 13 (8): 952–961.
22. Kim KK, Kugler MC, Wolters PJ et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 2006; 103 (35): 13180–13185.
23. Rastaldi MP, Ferrario F, Giardino L et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int 2002; 62 (1): 137–146.
24. Zeisberg M, Yang C, Martino M et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 2007; 282 (32): 23337–23347.
25. Zeisberg M, Hanai J, Sugimoto H et al. BMP-7 counteracts TGFbeta1-induced epithelial-to-mesenchymal transitiv and reverses chronic renal injury. Nat Med 2003; 9 (7): 964–968.
26. Rastaldi MP, Ferrario F, Giardino L et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int 2002; 62 (1): 137–146.
27. Bataille F, Rohrmeier C, Bates R et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm Bowel Dis 2008; 14 (11): 1514–1527. doi: 10.1002/ibd.20590.
28. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100 (1): 57–70.
29. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2 (6): 442–454.
30. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14 (6): 818–829. doi: 10.1016/j.devcel.2008.05.009.
31. Fidler IJ, Poste G. The „seed and soil“ hypothesis revisited. Lancet Oncol 2008; 9 (8): 808. doi: 10.1016/S1470-2045 (08) 70201-8.
32. Brabletz T, Jung A, Reu S et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001; 98 (18): 10356–10361.
33. Zeisberg M, Shah AA, Kalluri R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J Biol Chem 2005; 280 (9): 8094–8100.
34. Jechlinger M, Grunert S, Beug H. Mechanisms in epithelial plasticity and metastasis: insights from 3D cultures and expression profiling. J Mammary Gland Biol Neoplasia 2002; 7 (4): 415–432.
35. Bissell MJ, Radisky DC, Rizki A et al. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 2002; 70 (9–10): 537–546.
36. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2 (6): 442–454.
37. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113 (6): 685–700.
38. Niessen K, Fu Y, Chang L et al. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol 2008; 182 (2): 315–325. doi: 10.1083/jcb.200710067.
39. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transitiv through beta-catenin-T-cell factor-4-dependent expression of transforming growth factorbeta3. Mol Biol Cell 2008; 19 : 4875–4887.
40. Kokudo T, Suzuki Y, Yoshimatsu Y et al. Snail is required for TGF{beta}-induced endothelial-mesenchymal transitiv of embryonic stem cell-derived endothelial cells. J Cell Sci 2008; 121 (Pt 20): 3317–3324. doi: 10.1242/jcs.028282.
41. Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transitiv and contribution of tumor microenvironment. J Cell Biochem 2007; 101 (4): 816–829.
42. Gupta PB, Mani S, Yang J et al. The evolving portrait of cancer metastasis. Cold Spring Harb Symp Quant Biol 2005; 70 : 291–297.
43. Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res 2006; 66 (9): 4549–4552.
44. Mani SA, Yang J, Brooks M et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A 2007; 104 (24): 10069–10074.
45. Mani SA, Guo W, Liao MJ et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133 (4): 704–715. doi: 10.1016/j.cell.2008.03.027.
46. Hartwell KA, Muir B, Reinhardt F et al. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A 2006; 103 (50): 18969–18974.
47. Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006; 6 (7): 506–520.
48. Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 1998; 8 (23): 1243–1252.
49. Hata A, Shi Y, Massague J. TGF-beta signaling and cancer: structural and functional consequences of mutations in Smads. Mol Med Today 1998; 4 (6): 257–262.
50. Song J. EMT or apoptosis: a decision for TGF-beta. Cell Res 2007; 17 (4): 289–290.
51. Miyazono K, ten Dijke P, Heldin CH. TGF-beta signaling by Smad proteins. Adv Immunol 2000; 75 : 115–157.
52. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 2001; 29 (2): 117–129.
53. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nukleus through SMAD proteins. Nature 1997; 390 (6659): 465–471.
54. Roberts AB, Tian F, Byfield SD et al. Smad3 is key to TGFbeta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev 2006; 17 (1–2): 19–27.
55. Piek E, Moustakas A, Kurisaki A et al. TGF - (beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112 (Pt 24): 4557–4568.
56. Bhowmick NA, Ghiassi M, Bakin A et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 2001; 12 (1): 27–36.
57. Saika S, Ikeda K, Yamanaka O et al. Transient adenoviral gene transfer of Smad7 prevents injury-induced epithelial-mesenchymal transition of lens epithelium in mice. Lab Invest 2004; 84 (10): 1259–1270.
58. Bhowmick NA, Zent R, Ghiassi M et al. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem 2001; 276 (50): 46707–46713.
59. Lee YH, Albig AR, Maryann R et al. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 2008; 29 (12): 2243–2251. doi: 10.1093/carcin/bgn 199.
60. Janda E, Lehmann K, Killisch I et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002; 156 (2): 299–313.
61. Cui W, Fowlis DJ, Bryson S et al. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996; 86 (4): 531–542.
62. Watanabe T, Wu TT, Catalano PJ et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med 2001; 344 (16): 1196–1206.
63. Katsuno Y, Lamouille S, Derynck R et al. TGF-beta signaling end epithelial-mezenchymal transition in cancer progression. Curr Opin Oncol 2013; 25 (1): 76–84. doi: 10.1097/CCO.0b013e32835b6371.
64. Tepass U, Truong K, Godt D et al. Cadherins in embryonic and neural morphogenesis. Nat Rev Mol Cell Biol 2000; 1 (2): 91–100.
65. Edelman GM, Gallin WJ, Delouvee A et al. Early epochal maps of two different cell adhesion molecules. Proc Natl Acad Sci U S A 1983; 80 (14): 4384–4388.
66. Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesionindependent manner. J Cell Biol 2001; 153 (5): 1049–1060.
67. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int 2002; 26 (5): 463–476.
68. Muta H, Noguchi M, Kanai Y et al. E-cadherin gene mutations in signet ring cell carcinoma of the stomach. Jpn J Cancer Res 1995; 87 (8): 843–848.
69. Saito A, Kanai Y, Maesawa C et al. Disruption of E-cadherinmediated cell adhesion systems in gastric cancers in young patients. Jpn J Cancer Res 1999; 90 (9): 993–999.
70. Hirohashi S. Inactivation of the E-cadherinmediated cell adhesion system in human cancers. Am J Pathol 1998; 153 (2): 333–339.
71. Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta 1994; 1198 (1): 11–26.
72. Blanco MJ, Moreno-Bueno G, Sarrio D et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 2002; 21 (20): 3241–3246.
73. Yokoyama K, Kamata N, Hayashi E et al. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol 2001; 37 (1): 65–71.
74. Miele L, Golde T, Osborne B et al. NOTCH signaling ain cancer. Curr Mol Med 2006; 6 (8): 905–918.
75. Miele L, Miao H, Nickoloff BJ et al. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets 2006; 6 (4): 313–323.
76. Penton AL, Leonard LD, Spinner NB et al. Notch signaling in human development and disease. Semin Cell Dev Biol 2012; 23 (4): 450–457. doi: 10.1016/j.semcdb.2012.01.010.
77. Wang T, Xuan X, Pian L et al. Notch-1-mediated esophageal carcinoma EC-9706 cell invasion and metastasis by inducing epithelial-mesenchymal transition through Snail. Tumour Biol 2014; 35 (2): 1193–1201.
78. Ishida T, Hijioka H, Kume K et al. Notch signaling induces EMT in OSCC cell lines in a hypoxic enviroment. Oncol Lett 2013; 6 (5): 1201–1206.
79. Nusse R, Fuerer C, Ching W et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol 2008; 73 : 59–66. doi: 10.1101/sqb.2008.73.035.
80. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012; 149 (6): 1192–1205. doi: 10.1016/j.cell.2012.05.012.
81. Guo J, Fu Z, Wei J et al. PRRX1 promotes epithelial-mezenchymal transition through the Wnt/beta-catenin pathways in gastric cancer. Med Oncol 2015; 32 (1): 393. doi: 10.1007/s12032-014-0393-x.
82. Beachy PA, Karhadkar SS, Berman DM et al. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004; 432 (7015): 324–331.
83. Lei J, Ma J, Ma Q et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manne. Mol Cancer 2013; 12 : 66. doi: 10.1186/1476-4598-12-66.
84. Cano A, Nieto MA. Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol 2008; 18 (8): 357–359. doi: 10.1016/j.tcb.2008.05.005.
85. Christoffersen NR, Silahtaroglu A, Orom UA et al. MiR-200b mediates post-transcriptional repression of ZFHX1B. RNA 2007; 13 (8): 1172–1178.
86. Park SM, Gaur AB, Lengyel E et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008; 22 (7): 894–907. doi: 10.1101/gad.1640 608.
87. Gregory PA, Bert AG, Paterson EL et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10 (5): 593–601. doi: 10.1038/ncb1722.
88. Burk U, Schubert J, Wellner U et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008; 9 (6): 582–589. doi: 10.1038/embor.2008.74.
89. Bracken CP, Gregory PA, Kolesnikoff N et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008; 68 (19): 7846–7854. doi: 10.1158/0008-5472.CAN-08-1942.
90. Camps C, Buffa FM, Colella S et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 2008; 14 (5): 1340–1348. doi: 10.1158/1078-0432.CCR-07-1755.
91. Fu X, Han Y, Wu Y et al. Prognostic role of microRNA-21 in various carcinomas: a systematic review and meta-analysis. Eur J Clin Invest 2011; 41 (11): 1245–1253. doi: 10.1111/j.1365-2362.2011.02535.x.
92. Mei M, Ren Y, Zhou X et al. Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat 2010; 9 (1): 77–86.
93. Iorio MV, Casalini P, Piovan C et al. MicroRNA-205 regulates HER3 in human breast cancer. Cancer Res 2009; 69 (6): 2195–2200. doi: 10.1158/0008-5472.CAN-08-2920.
94. Kumarswamy R, Mudduluru G, Ceppi P et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer 2012; 130 (9): 2044–2053. doi: 10.1002/ijc.26218.
95. Yang AD, Fan F, Camp ER. Chronic oxaliplatin resistence induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12 (14 Pt 1): 4147–4753.
96. Papageorgis P, Cheng K, Ozturk S et al. Smad4 inactivation promotes malignancy and drug resistence of colon cance. Cancer Res 2011; 71 (3): 998–1008. doi: 10.1158/0008-5472.CAN-09-3269.
97. Hoshinono H, Miyoshi N, Nagai K et al. Epithelial-mesenchymal transition with expression of SNAIl-induced chemoresistance in colorectal cancer. Biochem Biophys Res Commun 2009; 390 (3): 1061–1065. doi: 10.1016/j.bbrc.2009.10.117.
98. Chen X, Wang Y, Xia H et al. Loss of E-cadherin promotes the growth, invasion and drug resistence of colorectal cancer cells and is associated with liver metastasis. Mol Biol Rep 2012; 39 (6): 6707–6714. doi: 10.1007/s11033-012-1494-2.
99. Buck E, Eyzaguirre A, Barr S et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther 2007; 6 (2): 532–541.
100. Trumpp A, Wiestler OD. Mechanisms of disease: cancer stem cells – targeting the evil twin. Nat Clin Pact Oncol 2008; 5 (6): 337–347. doi: 10.1038/ncponc1110.
101. Fan CW, Chen T, Shang YN et al. Cancer-initiating cells derived from human rectal adenocarcinoma tissues carry mesenchymal phenotypes and resist drug therapies. Cell Death Dis 2013; 4: e828. doi: 10.1038/cddis.2013.337.
102. Hirano T, Satow R, Kato A et al. Identification of novel small compounds that restore E-cadherin expression and inhibit tumor cell motility and invasiveness. Biochem Pharmacol 2013; 86 (10): 1419–1429. doi: 10.1016/j.bcp.2013.09.001.
103. Fernando RI, Litzinger M, Trono P et al. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest 2010; 120 (2): 533–544. doi: 10.1172/JCI38379.
104. Roselli M, Fernando RI, Guadagni F et al. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res 2012; 18 (14): 3868–3879. doi: 10.1158/1078-0432.CCR-11-3211.
105. Chua KN, Sim WJ, Racine V et al. A cell-based small molecule screening method for identifying inhibitors of epithelial-mesenchymal ransition in carcinoma. PLoS One 2012; 7 (3): e33183. doi: 10.1371/journal.pone.0033183.
106. Flanigan SA, Pitts TM, Newton TP et al. Overcoming IGF1R/1R resistence through inhibiton of MEK signaling in colorectal cance models. Clin Cancer Res 2013; 19 : 6219–6129. doi: 10.1158/1078-0432.CCR-13-0145.
107. Bocca C, Bozzo F, Cannito S et al. Celecoxib inactivates epithelial-mesenchymal transition stimulated by hypoxia and/or epidermal growth factor in colon cancer cells. Mol Carcinog 2012; 51 (10): 783–795.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2017 Issue 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Lymfedém po operacích na spádových lymfatických uzlinách pro karcinom prsu
- Daratumumab – naděje pro myelomové pacienty, výzva pro klinické laboratoře
- Epitelo-mezenchymální tranzice v nádorové tkáni a její role při metastatickém šíření karcinomů
- Možnosti chemoterapie v léčbě karcinomu prostaty