
Gorlinov-Goltzov syndróm
Gorlin-Goltz syndrome
Background:
Gorlin-Goltz syndrome is an autosomal dominant inherited disorder characterized by a predisposition to various cancers. Clinicopathological findings of syndrome are very diverse and many symptoms begin to manifest in a certain period of life.
Case:
The authors describe a case report of a man who, at the age of 34 years, presented to a dermatologist with multiple tumor lesions of the skin. The lesions started to develop when he was 30 years old and thereafter increased in number. Histology revealed superficial, superficial-nodular and nodular basal cell carcinomas. A total of 11 basal cell carcinomas were surgically removed and microscopically investigated. The others were treated locally with imiquimod cream and cryotherapy. In addition, he was found to have multiple odontogenic keratocysts in the jaw and mandible, as well as supernumerary and retinated teeth. Stomatologic and maxillofacial surgery interventions were performed. Further clinical and imaging examinations confirmed macrocephaly, hypertelorism, calcification of falx cerebri, and abnormalities of the cervical vertebrae. The spectrum of pathological findings met the diagnostic criteria of Gorlin-Goltz syndrome.
Conclusion:
Although Gorlin-Goltz syndrome is very rare in routine practice, it usually represents a serious disease with multiple organ system involvement. From a prognostic point of view, early diagnosis with adequate therapy is critical. If a diagnosis is confirmed, lifetime dispensary care with interdisciplinary medical cooperation is necessary.
The authors would like to thank all physicians who participated in the diagnostics and therapy of the presented patient.
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted: 30. 8. 2018
Accepted: 8. 1. 2019
Keywords:
Gorlin-Goltz syndrome – basal cell carcinoma – odontogenic cysts
Authors:
V. Bartoš 1; M. Kullová 2; K. Adamicová 3; I. Paučinová 4
Authors‘ workplace:
Oddelenie patologickej anatómie, FNsP Žilina
1; Dermatovenerologický stacionár, FNsP Žilina
2; Ústav patologickej anatómie, JLF UK a UN Martin
3; Oddelenie lekárskej genetiky, FNsP Žilina
4
Published in:
Klin Onkol 2019; 32(2): 124-128
Category:
Case Report
doi:
https://doi.org/10.14735/amko2019124
Overview
Východiská:
Gorlinov-Goltzov syndróm je autozómovo dominantne dedičné ochorenie charakteristické predispozíciou k rôznym typom nádorov. Klinicko-patologické nálezy syndrómu sú veľmi pestré, pričom mnohé symptómy sa začínajú prejavovať až v určitom období života.
Prípad:
Autori opisujú prípad muža, ktorý sa vo veku 34 rokov dostavil na dermatologické vyšetrenie s mnohopočetnými tumoróznymi léziami kože. Ich vývoj začal pozorovať približne od 30. roku života a odvtedy sa ich počet zvyšoval. Histologicky išlo o bazocelulárne karcinómy superficiálneho, superficiálno-nodulárneho a nodulárneho typu. Celkovo mal chirurgicky odstránených a mikroskopicky vyšetrených 11 primárnych bazocelulárnych karcinómov. Ostatné boli liečené lokálne imiquimodom a kryoterapiou. Okrem toho mal z čeľuste a sánky exstirpované viacpočetné odontogénne keratocysty a extrahované retinované a nadpočetné zuby. Ďalšie klinické a zobrazovacie vyšetrenia potvrdili makrocefáliu, hypertelorizmus, kalcifikáciu falx cerebri a abnormality krčných stavcov. Spektrum chorobných zmien spĺňalo diagnostické kritériá Gorlinovho-Goltzovho syndrómu.
Záver:
Hoci je Gorlinov-Goltzov syndróm v bežnej praxi veľmi zriedkavý, väčšinou predstavuje závažnú chorobnú jednotku s multiorgánovým postihnutím. Z prognostického hľadiska je kľúčová jeho včasná diagnostika a zahájenie adekvátnej terapie. V prípade potvrdenia diagnózy je nevyhnutná celoživotná dispenzarizácia pacienta s medziodborovou lekárskou spoluprácou.
Klíčová slova:
Gorlinov-Goltzov syndróm – bazocelulárny karcinóm – odontogénne cysty
Úvod
Gorlinov-Goltzov syndróm (G-G syndróm), nazývaný aj syndróm névoidných bazocelulárnych karcinómov (basal cell carcinoma – BCC), je autozómovo dominantne dedičné ochorenie charakteristické predispozíciou k rôznym typom nádorov [1–3]. Zapríčinený je mutáciami tumor supresorového génu PTCH1 (lokalizovaný na chromozóme 9q22.3-q31), ktorý kóduje transmembránový glykoproteín fungujúci ako antagonista Hedgehog signálnej dráhy [1]. V 20–30 % prípadov ide o de novo mutácie [1]. Okrem toho bol opísaný aj ďalší predisponujúci gén SUFU [1]. Ako samostatnú jednotku ho prvýkrát definovali v roku 1960 patológ a genetik Robert J. Gorlin a dermatopatológ Robert W. Goltz [4], podľa ktorých je pomenovaný. Z klinických príznakov syndrómu kládli pôvodne dôraz na mnohopočetné BCC (epiteliómy), keratocysty čeľuste a anomálie rebier. V súčasnosti je známe, že spektrum klinicko-patologických nálezov je pri tomto ochorení omnoho pestrejšie. Z doposiaľ zaužívaných diagnostických kritérií pre G-G syndróm sú považované za najrelevantnejšie kritériá navrhnuté Kimonisom et al [5] z roku 1997. Táto schéma zahrňuje šesť veľkých a šesť malých diagnostických znakov (tab. 1). Na potvrdenie G-G syndrómu sa vyžaduje prítomnosť dvoch veľkých, alebo jedného veľkého a dvoch malých diagnostických kritérií. V časopise Klinická onkologie bolo doteraz publikovaných niekoľko prehľadových prác, ktoré opisujú, resp. sa zmieňujú o G-G syndróme [1–3]. Žiadna z nich však neuvádza vlastný prípad z praxe. Preto sme sa rozhodli prezentovať kazuistiku mladého muža s týmto zriedkavým ochorením, ktorého sme diagnostikovali a liečili na našich pracoviskách.
![Diagnostické kritériá Gorlinov-
Goltzova syndrómu podľa Kimonisa
et al [5].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/ec9894c68d48ce37cffcc033149f578e.jpeg)
Kazuistika
Muž vo veku 34 rokov (fototyp III) bol začiatkom roka 2013 prijatý na dermatologické oddelenie FNsP v Žiline s mnohopočetnými tumoróznymi léziami kože. Ich vývoj začal pozorovať približne od 30. roku života a odvtedy sa ich počet zvyšoval. Pri vstupnom dermatologickom vyšetrení dominoval nález početných, prevažne ohraničených erytematóznych papúl a nodulov, ktoré sa vyskytovali najmä na chrbáte, pleciach a pažiach, menej na hlave a krku (obr. 1–3). Niektoré boli ulcerované s adherovanou krustou na povrchu. Klinicky a dermatoskopicky imponovali ako BCC. Palmárne a plantárne jamky neboli viditeľné, ale na dlani ľavej ruky sa vyskytoval drobný hyperkeratotický čap a na ploske ľavej nohy dve malé hyperkeratotické ložiská. V prvom slede boli chirurgicky excidované tumory lokalizované v lumbálnej oblasti vľavo, na pravej lopatke, na pravom pleci a na ľavej strane krku. Histologické vyšetrenie vo všetkých prípadoch potvrdilo BCC superficiálneho alebo superficiálno-nodulárneho typu. Postupne boli excidované aj ďalšie tumory vyrastajúce na chrbáte, na ramene, na čele a v parietálnej oblasti hlavy (obr. 4). Celkovo mal exstirpovaných a mikroskopicky vyšetrených 11 samostatných primárnych BCC, ktoré pozostávali zo superficiálneho (obr. 5), zmiešaného superficiálno-nodulárneho a nodulárneho typu (obr. 6). Ani v jednom prípade nemali histologicky infiltratívny rastový charakter a okolitá koža nevykazovala známky solárnej degenerácie.





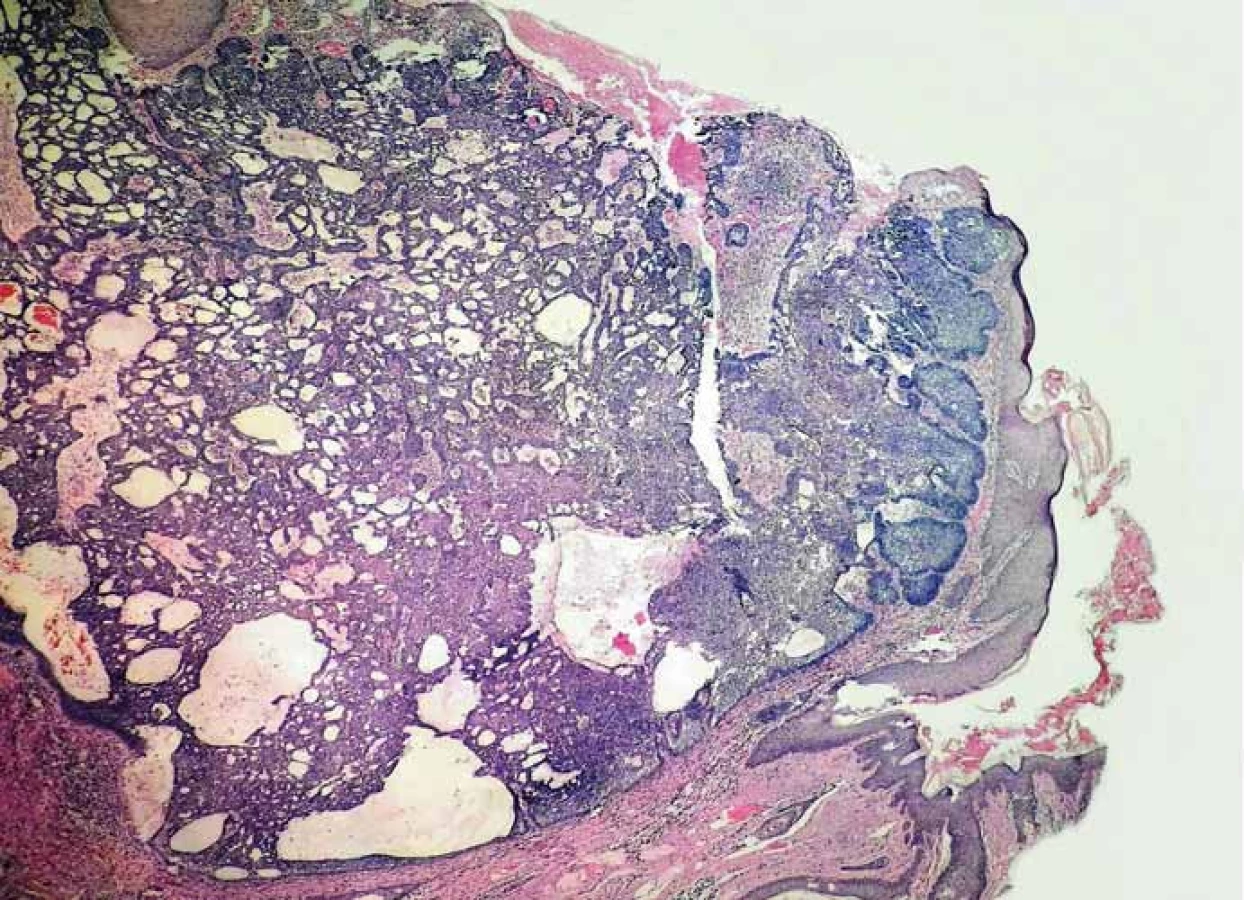
Okrem uvedených dermatologických ťažkostí sa pacient sťažoval aj na bolesti prednej časti tváre, pre ktoré absolvoval vyšetrenia na Klinike stomatológie a maxilofaciálnej chirurgie Jesseniovej lekárskej fakulty Univerzity Komenského a Univerzitnej nemocnice Martin. Röntgenové (RTG) vyšetrenie zobrazilo v maxile aj mandibule viacpočetné rôzne veľké cystické útvary, ktoré boli resekované. Histologicky išlo o odontogénne a epidermoidné cysty vyplnených keratínovým obsahom, ktorých výstelku tvoril vrstevnatý dlaždicový epitel bez dysplázie (obr. 7). Pacient mal zároveň extrahované retinované a nadpočetné zuby 18, 19, 28, 29, 38 a koreň zuba 54.

Vzhľadom na veľmi suspektné syndromologické ochorenie bol pacient odoslaný na konzultáciu do genetickej ambulancie a absolvoval ďalšie cielené vyšetrenia. Rodinná anamnéza bola negatívna a v rodokmeni sa nepotvrdil výskyt klasických monogénových ochorení, vrodených vývojových chýb či reprodukčných strát. Somatické vyšetrenie pacienta potvrdilo robustný habitus s makrocefáliou (obvod hlavy 63 cm) a miernym hypertelorizmom, a klinodaktýliu 4. a 5. prsta na obidvoch rukách. Kostra bola súmerná, tvár bez dysmorfie.
RTG vyšetrenie hlavy a chrbtice zobrazilo makrocefalickú lebku s výraznou maxilárnou a etmoidálnou pneumatizáciou, vysoko postavenú masívnu sánku s cystickými prejasneniami, lamelárne kalcifikovaný falx cerebri a lakunárne výpadky kalcifikácie v parietálnej oblasti. Na krčnej chrbtici bola viditeľná synostóza stavcov C3 a C4 v oblasti třňových výbežkov s parciálne zaniknutou medzistavcovou štrbinou.
Pacient mal odobratú krv na molekulárno-genetické vyšetrenie. V analyzovanej DNA sa v géne PTCH1 metódou priameho sekvenovania jednotlivých exónov a priľahlých oblastí nedetekovala žiadna dovtedy známa patogénna kauzálna mutácia. Taktiež neboli metódou MLPA (Multiplex Ligation-dependent Probe Amplification) potvrdené delécie alebo duplikácie génu PTCH1. V géne PTCH1 sa však zistil variant rs141085821: c.-5-4dupGGC, ktorý je podľa prediktívneho algoritmu (Mutation Taster) považovaný za potenciálne kauzálny variant.
Bez ohľadu na výsledky genetického vyšetrenia však bola v našom prípade klinická diagnóza G-G syndrómu jednoznačná a podporená nálezom troch veľkých (mnohopočetné BCC kože, odontogénne keratocysty, kalcifikácia falx cerebri) a troch malých (makrocefália, hypertelorizmus, anomálie stavcov) diagnostických kritérií. Pacient bol poučený o charaktere ochorenia a jeho dedičnosti, ako aj o nutnosti komplexnej dispenzárnej starostlivosti. Zdôraznená mu bola ochrana pred solárnym a RTG žiarením.
V rokoch 2013–2016 pravidelne absolvoval vyšetrenia u kožného lekára. Ďalšie tumorózne lézie kože vzhľadu BCC dermatológ liečil lokálnou aplikáciou imiquimodu (Aldara) a kryoterapiou. Počas tohto obdobia mal pacient vyšetrením nukleárnou magnetickou rezonanciou opäť potvrdené recidivujúce cystické lézie v mandibule, ako aj cystu v pravom maxilárnom sínuse, ktorá zapríčiňovala sťažené dýchanie cez pravý nosový priechod. Vyšetrenie hlavy nukleárnou magnetickou rezonanciou navyše zobrazilo ložisko nešpecifickej gliózy mozgu v paraventrikulárnej oblasti, ktoré však ostalo v ďalšom sledovaní bez známok progresie a nevyžadovalo neurochirurgickú intervenciu. V období spracovania tohto príspevku (august 2018) nám aktuálny zdravotný stav pacienta nebol známy, nakoľko od novembra 2016 nemal v elektronickom databázovom systéme našej nemocnice evidované žiadne nové lekárske záznamy.
Diskusia
G-G syndróm je multisystémové dedičné ochorenie s takmer úplnou penetranciou a variabilnou expresivitou [3,6,7]. Keďže jeho klinické prejavy sú veľmi pestré a mnohé vznikajú, resp. dominujú až v určitom období života, odhaliť sa môže prakticky v každom veku. V jednej štúdii analyzujúcej 105 pacientov s G-G syndrómom [5] varírovalo vekové rozpätie v čase stanovenia diagnózy od 4 mesiacov do 87 rokov. Ochorenie sa niekedy prejavuje už po narodení makrocefáliou, rázštepmi pery či podnebia, anomáliami rebier alebo inými malformáciami skeletu [5]. Okolo druhého roka života má viac ako 5 % detí diagnostikovaný zhubný nádor mozočka – meduloblastóm [1–3]. Až u 90 % osôb vznikajú do 40. roku života odontogénne keratocysty (keratocystický odontogénny tumor) čeľuste [2,3] ktoré sú často prvým klinickým príznakom dovtedy nerozpoznaného ochorenia [7]. Hoci ide o benígne cystické lézie, zvyčajne sa správajú agresívne, rastú lokálne deštruktívne a recidivujú. Mnohokrát si vyžadujú opakované stomatochirurgické intervencie. Takmer u všetkých pacientov s G-G syndrómom vznikajú v priebehu života BCC kože, ktoré sú jeho typickým príznakom. Progredovať začínajú od 3. dekády a ich počet sa zvyšuje bez ohľadu na mieru expozície slnečnému žiareniu. Do 20. roku života má BCC kože približne 75 % a vo veku 40 rokov až 90 % osôb s G-G syndrómom [1,3]. Druhým charakteristickým kožným prejavom sú viacpočetné malé kožné jamky na dlaniach a ploskách nôh. Ďalším významným patologickým nálezom je kalcifikácia falx cerebri, ktorá sa odhalí len zobrazovacím vyšetrením. Približne u štvrtiny postihnutých žien vznikajú vo fertilnom veku fibrómy vaječníkov [3].
Hoci je spektrum klinicko-patologických nálezov pri G-G syndrómu veľmi rôznorodé, mnohé z nich nie sú konštantné, v dôsledku čoho je manifestácia a tým aj prognóza ochorenia u pacientov individuálna. V tab. 2 uvádzame prehľad percentuálneho zastúpenia základných diagnostických znakov u osôb postihnutých G-G syndrómom, ktoré sme získali z troch pôvodných štúdií z USA [5], Japonska [8] a Austrálie [9]. Viditeľné sú odlišnosti vo frekvencii niektorých nálezov, pričom zjavné rozdiely sú najmä vo výskyte BCC kože. Zatiaľ čo min. jeden BCC sa u Austrálčanov a Američanov svetlej pleti vyskytoval v 75–78,9 % prípadov, u Japoncov a amerických černochov to bolo len v 37,8–38,4 % prípadov. Tieto výsledky napovedajú, že výskyt BCC kože pri G-G syndróme závisí aj od etnicity a/alebo individuálneho genetického pozadia jednotlivcov. Fenotypová rôznorodosť sa dá vysvetliť expresiou rozdielnych (a v mnohých prípadoch zatiaľ neobjasnených) mutácií génu PTCH, ako aj vplyvu environmentálnych faktorov a iných modifikovaných génov. Veľmi nekonzistentné údaje sú aj ohľadom samotnej prevalencie G-G syndrómu. Napríklad v Spojenom kráľovstve je odhadovaná prevalencia 1 prípad na 30 827 obyvateľov [10], ale v japonskej populácii iba 1 prípad na 235 800 obyvateľov [8]. Keďže klinické prejavy G-G syndrómu závisia aj od veku a pri ľahších formách ochorenia môžu ostať dlho nerozpoznané, niektorí autori polemizujú [6,7], či nie je reálne poddiagnostikovaný a jeho skutočná prevalencia môže byť omnoho vyššia.

Záver
V našom príspevku sme sa snažili poukázať, že hoci je G-G syndróm v bežnej praxi veľmi zriedkavý, väčšinou predstavuje závažnú chorobnú jednotku s multiorgánovým postihnutím. Z prognostického hľadiska je kľúčová jeho včasná diagnostika a zahájenie adekvátnej terapie. V prípade potvrdenia diagnózy je nevyhnutná celoživotná dispenzarizácia pacienta s medziodborovou lekárskou spoluprácou.
Autori ďakujú všetkým lekárom, ktorí sa podieľali na diagnosticko-terapeutickom procese prezentovaného pacienta.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr., PhDr. Vladimír Bartoš, PhD., MPH
Oddelenie patologickej anatómie Fakultná nemocnica s poliklinikou Žilina
Vojtecha Spanyola 43 012 07 Žilina
e-mail: vladim.bartos@gmail.com
Obdržané: 30. 8. 2018
Prijaté: 8. 1. 2019
Sources
1. Krutílková V. Genetické syndromy predisponující k dětským nádorům centrálního nervového systému. Klin Onkol 2016; 29 (Suppl 1): S71–S77. doi: 10.14735/amko2016S71.
2. Plevová P, Krutílková V, Puchmajerová A et al. Gorlinův syndrom. Klin Onkol 2009; 22 (Suppl 1): S34–S35.
3. Plevová P, Šilhánová E, Foretová L et al. Vzácné hereditární syndromy s vyšším rizikem vzniku nádorů. Klin Onkol 2006; 19 (Suppl 1): S68–S75.
4. Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Eng J Med 1960; 262 : 908–912. doi: 10.1056/NEJM196005052621803.
5. Kimonis VE, Goldstein AM, Pastakia B et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 1997; 69 (3): 299–308.
6. Thomas N, Vinod SV, George A et al. Gorlin–Goltz syndrome: an often missed diagnosis. Ann Maxillofac Surg 2016; 6 (1): 120–124. doi: 10.4103/ 2231-0746.186148.
7. Visioli F, Martins CA, Heitz C et al. Is nevoid basal cell carcinoma syndrome really so rare? Proposal for an investigative protocol based on a case series. J Oral Maxillofacial Surg 2010; 68 (4): 903–908. doi: 10.1016/j.joms.2009.03.032.
8. Endo M, Fujii K, Sugita K et al. Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am J Med Genet A 2012; 158A (2): 351–357. doi: 10.1002/ajmg.a.34421.
9. Shanley S, Ratcliffe J, Hockey A et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 1994; 50 (3): 282–290. doi: 10.1002/ajmg.1320500312.
10. Evans DG, Howard E, Giblin C et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic service. Am J Med Genet A 2010; 152A (2): 327–332. doi: 10.1002/ajmg.a.33139.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2019 Issue 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Chromotripse – rozsáhlé chromozomové přestavby a jejich význam u onkologických onemocnění
- Gorlinov-Goltzov syndróm
- Oligometastatický karcinom prostaty
- Postižení jater po jedné dávce nivolumabu – kazuistika a přehled literatury