
Lymfangioleiomyomatóza
Lymphangioleiomyomatosis
Background: Lymphangioleiomyomatosis (LAM) is a rare systemic disease that occurs sporadically (S/LAM) or as part of tuberous sclerosis (TS/LAM). LAM is characterized by proliferation of abnormal smooth muscle cells. This disease clinically manifests as dyspnea on exertion and pneumothorax. Lymphadenopathy in the abdominal and pelvic region leading to lymphatic obstruction can also occur. LAM is associated with kidney angiomyolipoma and meningioma. The disease is diagnosed histologically and/or using typical high-resolution computed tomography findings and anamnestic information. In histopathological studies, the diagnosis is supported by detection of characteristic LAM cells. Mammalian target of rapamycin (mTOR) inhibitors are possible treatment options.
Material and methods: Ten consecutive patients diagnosed with LAM and pulmonary manifestation (eight with S/LAM and two with TS/LAM) in 2002–2018 were retrospectively analyzed. Their individual clinical characteristics and our treatment experience are described.
Results: The patients varied in terms of disease stage. The best predictors of prognosis were lung function parameters (forced vital capacity, forced expiratory volume in 1 second, and diffusing capacity for carbon monoxide). Four patients are currently being treated with mTOR inhibitors. This treatment stabilized lung functions in all four patients. The median follow-up was 48 months (12–132 months). Median survival was not achieved and only three patients died.
Conclusion: An interdisciplinary approach is required to care for LAM patients. Cooperation of pneumologists, surgeons, oncologists, and geneticists is needed. Treatment with mTOR inhibitors led to stabilization in our patients. The side effects were well managed.
Keywords:
sirolimus – lymphangioleiomyomatosis – rapamycin
Authors:
Martina. Doubková 1; Marianna Štefániková 1; Vladimír Čan 2; Zdeněk Merta 1; Marek Svoboda 3,4
Authors‘ workplace:
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
1; Chirurgická klinika LF MU a FN Brno
2; Klinika komplexní onkologické péče, Masarykův onkologický ústav, Brno
3; Oddělení epidemiologie a genetiky nádorů, Masarykův onkologický ústav, Brno
4
Published in:
Klin Onkol 2019; 32(5): 367-374
Category:
Original Articles
doi:
https://doi.org/10.14735/amko2019367
Overview
Východiska: Lymfangioleiomyomatóza (LAM) je vzácné systémové onemocnění, které se vyskytuje sporadicky (S/LAM) nebo je součástí tuberózní sklerózy (TS/LAM). Onemocnění je charakterizováno proliferací abnormálních buněk hladkého svalstva. LAM se klinicky manifestuje zhoršující se námahovou dušností a pneumotoraxy. Bývá zvětšení uzlin v abdominální a pánevní oblasti vedoucí k lymfatické obstrukci. LAM je asociována s angiomyolipomy v ledvinách. Diagnóza je stanovena histologicky a/nebo kombinací anamnézy a typického nálezu při vyšetření plic výpočetní tomografií s vysokým rozlišením. V histopatologickém nálezu je diagnóza podporována nálezem charakteristických LAM buněk. Je to onemocnění progredující, s možností kauzální léčby inhibitory mTOR. Materiál a metody: Na našem pracovišti bylo v letech 2002–2018 sledováno celkem deset pacientek s LAM (osm S/LAM, dvě TS/LAM) a plicním postižením. Popisujeme jednotlivé klinické charakteristiky a naše zkušenosti s léčbou.
Výsledky: LAM je prezentována na deseti případech s různým stupněm pokročilosti. Stanovení prognózy u konkrétního nemocného s plicní formou LAM je těžké. Nejlepším ukazatelem vývoje onemocnění je sledování hodnot plicních funkčních parametrů (usilovná vitální kapacita, usilovně vydechnutý objem za první sekundu, difuzní kapacita plic) v čase. V současné době jsou léčeni čtyři naši pacienti mTOR inhibitory, u všech vedla terapie ke stabilizaci plicních funkčních parametrů. Medián sledování byl 48 měsíců (12–132). Nebyl dosažen medián přežití, zemřeli jen tři pacienti.
Závěr: Společná mezioborová spolupráce pneumologa, chirurga, onkologa a genetika je základem naší péče o pacientky s LAM. Terapie inhibitory mTOR vedla ke stabilizaci LAM u našich nemocných. Nežádoucí účinky léčby byly dobře zvládnutelné.
Klíčová slova:
lymfangioleiomyomatóza – rapamycin – sirolimus
Úvod
Lymfangioleiomyomatóza (LAM) je vzácné onemocnění, které bylo poprvé popsáno v lékařské literatuře v roce 1918 Lauterbacherem [1]. Druhé pozorování publikoval von Stössel v roce 1937 [2]. Názvosloví nemoci se postupně vyvíjelo ze slova lymfangiomyom k lymfangiomyomatóze a následně lymfangioleiomyomatóze. Jako samostatná nosologická jednotka byla LAM vymezena až v roce 1958 [3]. Přestože je LAM vzácné onemocnění, bylo v naší a slovenské literatuře zaznamenáno hned několik případů nemoci. Jako první u nás LAM diagnostikoval Dvořáček v roce 1974. Tento první případ nemoci byl zmíněn v práci Miřejovského et al [4] a v práci Halíkové et al [5]. Dvořáček svůj případ časopisecky nepublikoval.
Známe dvě formy LAM – sporadická forma a forma v rámci tuberózní sklerózy (TS) [6]. Příčina nemoci je pravděpodobně genetická. Patologické nálezy u LAM jsou identické s těmi, které vidíme u TS, autozomálně dominantně dědičného onemocnění, u nějž jsou mutované tumor supresorové geny TSC1 a TSC2 na 9. a 16. chromozomu (9q34, 16p13) [7,8]. Sporadická LAM je zapříčiněna mutací TSC2 genu [9–11]. Komplex tuberózní sklerózy (tuberous sclerosis complex – TSC) je zapříčiněn mutací TSC1 nebo TSC2 [9,11]. Vzhledem k predilekčnímu postižení žen v reprodukčním věku a přítomnosti hormonálních receptorů v LAM tkáni byla za možnou příčinu nemoci považována nerovnováha mezi hladinou estrogenů a gestagenů v cirkulaci nebo ve tkáních a mezi počtem tkáňových receptorů pro tyto hormony [12,13]. Úloha hormonální regulace v patogenezi nemoci není stále jasná. Progresi nemoci je možné vidět během těhotenství, menstruace a užívání estrogenů [14,15].
Ve světové literatuře je možné nalézt více než 1 500 prací pojednávajících o LAM. Jejich počet stále narůstá v souladu s poznáním nemoci a její přesnější diagnostikou. LAM dominantně postihuje ženy ve fertilním věku [7,16,17], i když byly popsány případy žen po menopauze [18], u dětí a také u mužů [19]. LAM se může vyskytovat ve formě sporadické nebo ve spojení s TS. Některé případy LAM, které začaly již v reprodukčním věku ženy, mohou být při pomalé progresi diagnostikovány až postmenopauzálně. Dosud nebyl popsán familiární výskyt sporadické LAM a vyšší výskyt u některého z etnik [19]. Z velké části jsou pacienty nekuřáci nebo bývalí kuřáci [20]. Přesná data o výskytu nemoci v populaci nejsou známa. Sporadická LAM se vyskytuje u 3,3–7,7 žen na 1 milion obyvatel [21]. Odhadovaný medián přežití bez plicní transplantace je 29 let od začátku symptomů, 10leté přežití je 86 % [22]. LAM prevalence u TSC je většinou u žen. Mezi 67 japonskými ženami s TSC bylo 54 % s LAM. Frekvence u mužů je 1/28. LAM frekvence narůstá po 20. roku věku [23]. V jiné studii se LAM u TS vyskytovala až u 80 % žen [7,24]. Epidemiologická data v ČR chybí.
Materiál a metody
Provedli jsme retrospektivní analýzu pacientů sledovaných a léčených na našem pracovišti pro lymfangioleiomyomatózu v letech 2002–2018. V roce 2015 jsme vytvořili jako reakci na nová doporučení léčby LAM inhibitory mTOR (target of rapamycin), pracovní skupinu sestávající ze společné mezioborové spolupráce pneumologa, dětského onkologa, genetika a chirurga. Tato spolupráce má za cíl zlepšit diagnostiku a péči o pacienty a zajistit lepší dostupnost léčby inhibitory mTOR. Cílem práce bylo provést charakteristiku souboru našich pacientů s LAM, analyzovat přirozený vývoj onemocnění, příčiny úmrtí, léčebné postupy a jejich efekt a porovnat naše údaje s údaji publikovanými v české a světové lékařské literatuře.
Diagnóza LAM byla stanovena standardními postupy (klinický nález, sérologický biomarker vaskulární endoteliální růstový faktor D (vascular endothelial growth factor – VEGF-D), funkční vyšetření plic, zobrazovací metody, bronchoskopie s bronchoalveolární laváží k vyloučení jiných intersticiálních plicních procesů, histologický průkaz). Pro stanovení diagnózy jsme vyžadovali korelaci histologického nálezu, klinického nálezu, zobrazovacích a laboratorních vyšetření [6]. V případě absence histopatologického nálezu byla diagnóza stanovena na základě shody kliniky s výsledky zobrazovacích a laboratorních vyšetření (VEGF-D), vyšetření broncho-alveolární tekutiny, observací a vyloučením jiných intersticiálních plicních onemocnění.
Výsledky
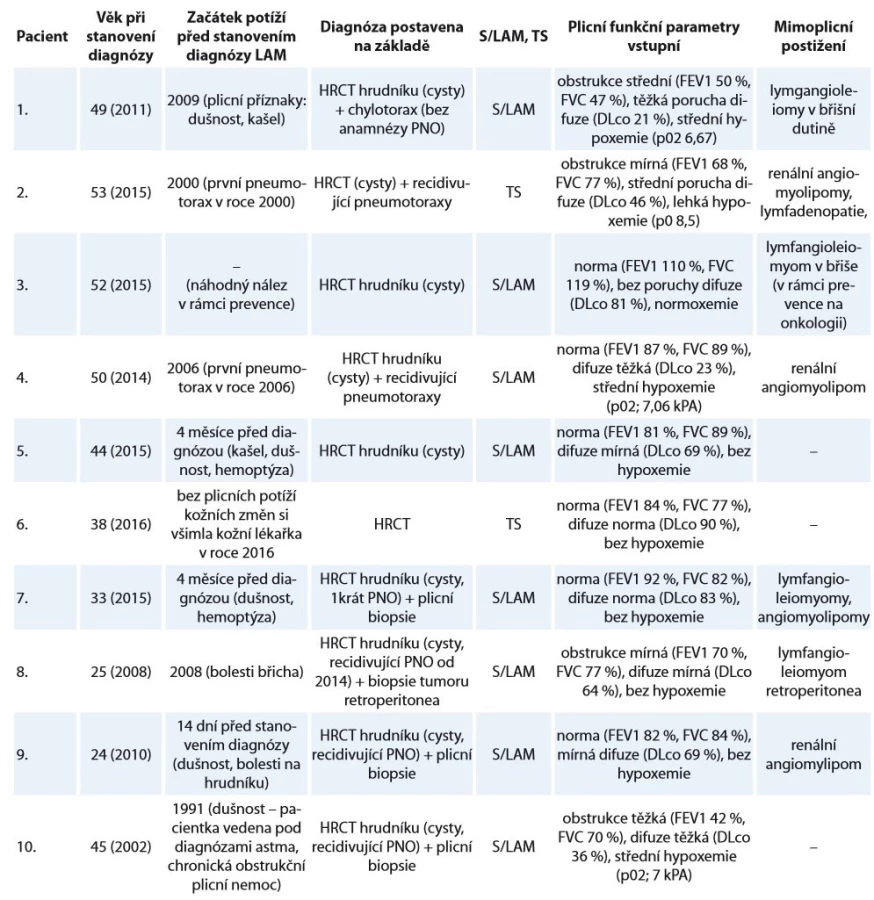
V letech 2002–2018 bylo na našem pracovišti sledováno deset pacientů (žen) s diagnózou LAM a plicním postižením, dvě S/LAM a osm TS/LAM. Dvě pacientky byly kuřačky, jeden pacient byl bývalý kuřák. Medián věku při stanovení diagnózy byl 44,5 roku, průměrný věk 41 let. Hodnocením klinických nálezů našich pacientů jsme zjistili, že v době stanovení diagnózy byla u čtyř (40 %) přítomna dušnost, u šesti (60 %) pneumotorax, u jednoho (10 %) chylotorax, u pěti (50 %) angiomyolipom a u čtyř (40 %) lymfangioleiomyom. Histologický průkaz LAM byl potvrzen u čtyř nemocných. U zbylých nemocných byla diagnóza stanovena na základě korelace klinického nálezu, zobrazovacích metod, laboratorních vyšetření s odběrem VEGF-D a vyloučením jiných intersticiálních plicních procesů.
Plicní funkční vyšetření neprokázalo při zjištění diagnózy ventilační poruchu u šesti pacientů (60 %). Obstrukční ventilační porucha byla u čtyř (40 %) nemocných. Bez poruchy difuze byli tři (30 %) pacienti, ostatní měli poruchu difuze od mírné po těžkou. Vyšetřením krevních plynů (arterializovaná krev z ušního lalůčku) v klidu v době diagnózy jsme u šesti (60 %) neprokázali respirační nedostatečnost.
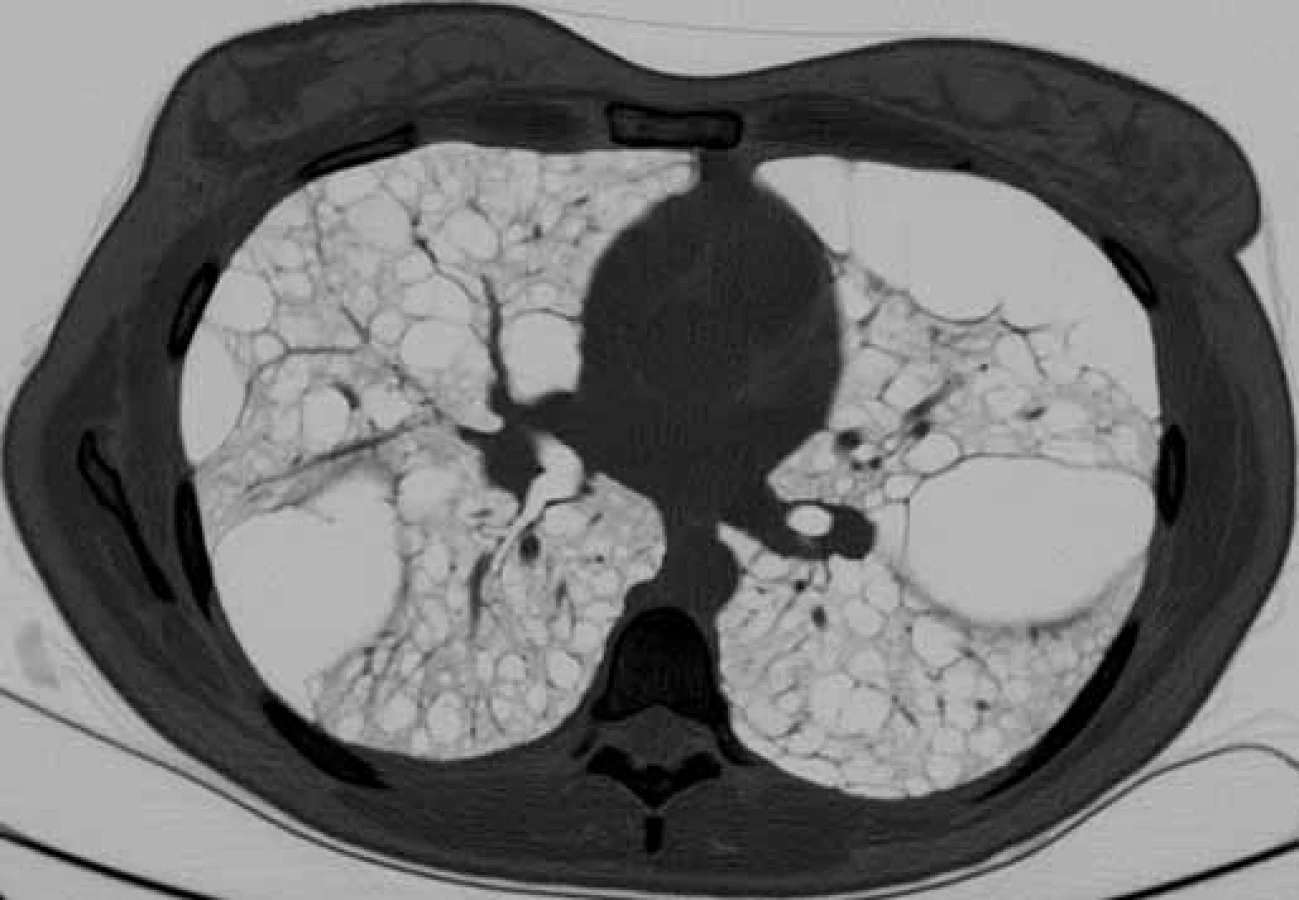
Bronchoskopie s bronchoalveolárním výplachem byla provedena u všech pacientů k vyloučení jiných intersticiálních plicních procesů s cystickou plicní přestavbou. Vyšetření výpočetní tomografií s vysokou rozlišovací schopností (high-resolution computed tomography – HRCT) hrudníku, ultrazvuk nebo CT břicha bylo provedeno u všech nemocných. Charakteristické nálezy na HRCT hrudníku u našich pacientek ukazují obr. 1, 2. K vyloučení TS pacienti podstoupili neurologické, kožní a oční vyšetření a při podezření na TS bylo provedeno i vyšetření genetické.

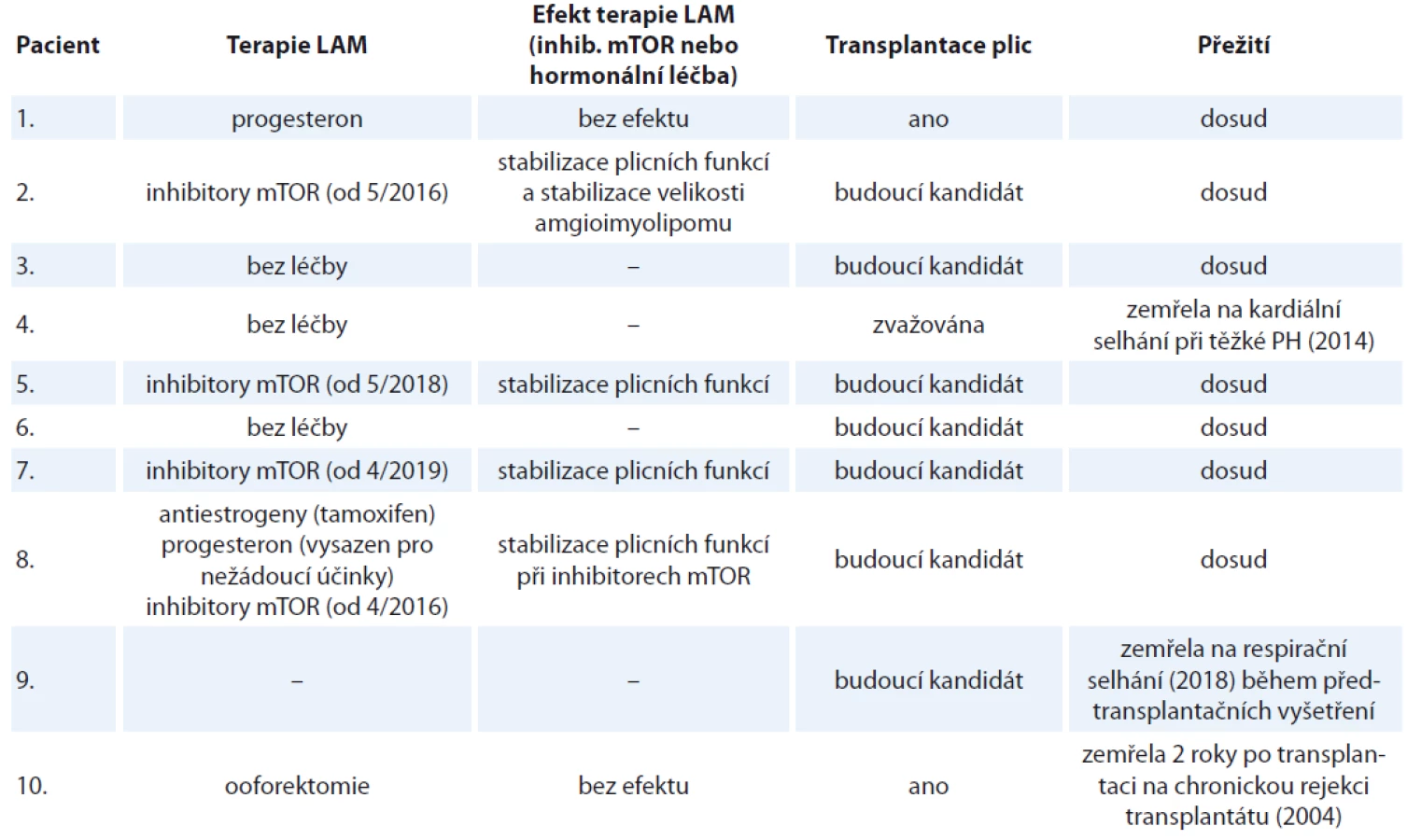
Kromě plicní rehabilitace, bronchodilatační léčby u nemocných s bronchiální obstrukcí, chemické a chirurgické pleurodézy (ošetření viscerální a parietální pleury za účelem srůstu obou listů a zamezení recidivě pneumotoraxu a pleurálních výpotků) terapie sestávala z hormonální léčby a z léčby inhibitory mTOR. Progesteron byl podáván u dvou pacientek, antiestrogen (tamoxifen) u jedné pacientky, radiační kastrace (ooforektomie) u jedné pacientky. Hormonální léčba nevedla ani u jedné z nich ke stabilizaci plicních nálezů a ke stabilizaci plicních funkčních parametrů v čase. U jedné pacientky ale měla antiestrogenová terapie vliv na redukci velikosti lymfangioleiomyomů v břiše. Čtyři pacienti jsou v současnosti léčeni inhibitory mTOR, snášenlivost je dobrá. Jen u jedné pacientky musela být léčba krátce přerušena z důvodu mírných kožních změn (purpura) a nevysvětlitelných pocitů chladu. U nemocných s mTOR inhibitory je pravidelně kontrolována hladina sirolimu, aby se pohybovala mezi doporučovanými hodnotami 4–10 ng/ml. U všech čtyř pacientek vedly inhibitory mTOR ke stabilizaci plicních funkčních parametrů v čase nebo mírnému zlepšení. Hodnoceny byly intervaly 6, 12, 18 měsíců a porovnávaly se parametry usilovné vitální kapacity (forced vital capacity – FVC), usilovně vydechnutého objemu za první sekundu (forced expiratory volume in one second – FEV1) a difuzní kapacity plic (diffusing capacity of carbon monoxide – DLco) v čase oproti předešlé kontrole.
Za dobu sledování tři pacienti zemřeli. Příčinou smrti bylo respirační selhání u nemocné s pokročilým plicním postižením na domácí dlouhodobé kyslíkové léčbě a v přípravě na plicní transplantaci; další příčinou bylo multiorgánové selhání u pacientky po plicní transplantaci s rejekcí transplantátu; v posledním případě se jednalo o kardiální selhání u nemocné s diagnózou LAM v menopauze, se třemi dětmi a pokročilým plicním postižením s těžkou plicní hypertenzí. Medián sledování (follow-up) byl 48 (12–132) měsíců. Nebylo dosaženo mediánu přežití, zemřeli jen tři pacienti. Shrnutí klinických charakteristik viz tab. 1, 2.
Diskuze
Lymfangioleimyomatóza je méně běžné multisystémové onemocnění vyskytující se samostatně nebo s TS. LAM je nově klasifikována jako nádor s nízkým stupněm metastazování (nízkým stupněm malignity) a je řazena do kategorie perivaskulárních epiteloidních buněčných tumorů [7]. Původ nemoci je genetický a LAM buňky obsahují abnormality (mutace a epigenetické modifikace) v TSC1 a TSC2 genech kódujících tuberin a hamartin, dva proteiny kontrolující signální dráhu kinázy mTOR. Nemoc je charakterizována: 1) proliferací abnormálních buněk hladké svaloviny v plicním intersticiu a podél axiálních lymfatických cest hrudníku a břicha a kolem mediastinálních, abdominálních a dolních krčních uzlin; 2) tenkostěnnými plicními cystami; 3) vyšší incidencí angiomyolipomů (abdominálních) [6,25]. Proliferující LAM buňky mající charakteristiky buněk hladké svaloviny a melanocytů pronikají do tkání plic, formují se do svazků a prorůstají do stěn dýchacích cest, krevních a lymfatických cév, čímž vedou k jejich poškození a obstrukci. Proto při vyšetření plicních funkčních parametrů nejčastěji zjišťujeme obstrukční ventilační poruchu se známkami plicní hyperinflace, současně bývá snížená difuzní kapacita plic. Obstrukce plicních krevních cév může vést k plicní hemoragii, hemoptýze a hemosideróze. Obstrukce lymfatických cest vede k chylotoraxům a chylózním ascitům [7].
Průměrný věk stanovení diagnózy je 35 let [26]. V naší skupině byl průměrný věk 41 let. Dvěma nejčastějšími klinickými manifestacemi jsou dušnost a pneumotorax [7]. Klinické příznaky se různí dle studií, zahrnují dušnost (73 % u S/LAM, 71 % u TS/LAM), spontánní pneumotorax (57 % u S/LAM, 47 % u TS/LAM), hemoptýzu (10 % u S/LAM, 3 % u TS/LAM), abdominální angiomyolipomy (41 % u S/LAM, 96 % u TS/LAM), lymfangioleiomyom (38 % u S/LAM, 13 % u TS/LAM), chylotorax (20 % u S/LAM, 14 % u TS/LAM), méně často chylózní ascites, chylurie, chyloptysis a abdominální krvácení způsobené renálními angiolipomy [7,8]. Uvedené údaje korespondují s našimi výsledky. LAM vede obvykle k progresivní destrukci plicní tkáně a respiračnímu selhání. Doba od začátku symptomů ke stanovení diagnózy je podle zahraniční literatury 3–4 roky [20]. U některých našich pacientek to bylo i déle. LAM se může projevovat i jako mimoplicní postižení, což jsme pozorovali i v našem souboru. Matsui et al popisují ve své práci 22 nemocných s postižením lymfatických uzlin mediastina a retroperitonea. U většiny těchto pacientů diagnóza extrapulmonální LAM předcházela plicní projevy obvykle o 1–2 roky [27]. Častým nálezem u 20–54 % pacientů s LAM je benigní angiomyolipom. Jde většinou o malý, bilaterální, často bezpříznakový útvar složený z tuku, hladké svaloviny a krevních cév [28].
Diagnostická kritéria jsou následující (spolehlivá diagnóza LAM): charakteristický nález cyst na HRCT hrudníku + jeden z následujících klinických příznaků: TSC, renální angiomyolipomy, cystický lymfangioleiomyom, chylózní pleurální výpotek nebo chylózní tekutina v břišní dutině. V případě, že není diagnóza jistá a některé z následujících příznaků nejsou přítomny, pak je indikována plicní biopsie [25]. V našem souboru pacientů byla histologická verifikace provedena u čtyř nemocných. Vyšetření sérové hladiny VEGF-D je vhodné v případě kontraindikace invazivního odběru vzorku. VEGF-D je lymfangiogenní růstový faktor (přispívá k lymfangiogenezi, invazi a metastazování); je zvýšen v séru pacientů s LAM [7,29] a pomáhá při stanovení diagnózy u pacientů s LAM bez mimoplicního postižení nebo TSC a k vyloučení jiných cystických plicních postižení [7,25]. Hladiny VEGF-D v séru nad 800 pg/ml jsou diagnostické pro LAM [7,25,30]. Vyšetření VEGF-D jsme využívali i my pro diagnostiku LAM v případě, že nebyla provedena histologická verifikace. Sérové koncentrace VEGF-D mají i prognostický význam, hladiny klesají u nemocných léčených inhibitory mTOR oproti placebu [25].
Na HRCT hrudníku je nález homogenně difuzně rozložených tenkostěnných cyst různých velikosti (od mm po cm) (obr. 1, 2). Na diagnózu LAM je nutno pomýšlet v diferenciální diagnostice plicních intersticiálních procesů, zvláště pokud jsou spjaty s recidivujícími pneumotoraxy, chylotoraxem a obstrukční ventilační poruchou u mladých žen. U pacientů s početnými cystami na CT skenech hrudníku by mohlo v diagnostické rozvaze pomoci i provedení abdominálního CT vyšetření k ověření extrapulmonální LAM. U pacientů s lymfatickým postižením vidíme pleurální tekutinu charakteru chylotoraxu a plicní infiltráty způsobené kumulací chylu v intersticiu. Další nálezy zahrnují již zmiňované angiomyolipomy, lymfadenopatie, lymfangioleiomyomy a ascites [7]. Diferenciálně diagnosticky je potřeba odlišit S/LAM od TS/LAM (nejeví sexuální predilekci, je charakterizována mnohočetnými hamartomy, epilepsií, mentální retardací a kožními změnami typu adenoma sebaceum).
Nejvíce specifickou metodou průkazu LAM je plicní biopsie demonstrující přítomnost LAM buněk, buď pro jejich typický histologický obraz, nebo pro jejich reakci s HMB-45 protilátkami (myší monoklonální protilátka reagující s extraktem z humánního maligního melanomu) způsobenou nekompletní formou melanogeneze v LAM buňkách. LAM buňky exprimují kromě markerů buněk melanozomů, estrogenových a progesteronových receptorů [7,31], chemokinových receptorů [32], CD44v6 (glykoproteinu spojeného s metastazováním) [33] a matrix metaloproteináz rovněž receptor pro vaskulární endoteliální růstový faktor (VEGF-D3) [7,29].
Současná terapie vychází z několika doporučení [25,34]. Obecně je doporučováno ženám udržovat normální tělesnou hmotnost (negativní vliv estrogenů tvořených v tukové tkáni) a nekouřit. Ve světě existuje sdružení na podporu žen (samy sobě si říkají „LAMies“), které poskytují informace o nemoci a doporučení pro život a podporu dalšího výzkumu (www.thelamfoundation.org). U nás zatím takové pacientské sdružení není. Vhodné je zvýšení rehabilitačního potenciálu, proto i většina našich pacientek podstupuje dechovou plicní rehabilitaci. Symptomaticky všem nemocným s bronchiální obstrukcí podáváme bronchodilatační léčbu, protože obstrukce může být částečně reverzibilní a může pomáhat ke zlepšení subjektivního vnímání dušnosti [6].
Gravidita představuje pro pacientku s LAM a jejího lékaře velké dilema. Je prokázáno, že estrogeny v průběhu těhotenství zhoršují průběh onemocnění. To byl i případ naší pacientky (případ č. 9). Nemocná ale byla pečlivě informována o rizicích, aby se mohla svobodně rozhodnout, zda je ochotna podstoupit riziko zhoršení nemoci. Hormonální faktory pravděpodobně hrají úlohu v patogenezi nemoci, a to proto, že onemocní prakticky výlučně jen ženy [25], stabilizace nemoci bývá vidět v postmenopauzálním období [35], zhoršení LAM je spojené s menstruačním cyklem [36], těhotenstvím [37,38], hormonální forma antikoncepce obsahující estrogeny zhoršuje průběh onemocnění [39]. I přes tyto skutečnosti (viz výše) užívání progesteronu u žen s LAM neposkytlo jasný důkaz o jeho účinnosti. Obdobně nejsou v léčbě LAM doporučovány antiestrogenová léčba ve formě tamoxifenu, ooforektomie nebo podávání agonistů gonadotropinů uvolňujících hormony [40–42]. My jsme podali hormonální léčbu dvěma pacientkám, ani u jedné nebylo dosaženo efektu na průběh nemoci a plicní funkce (případ 1 a 8). Třetí podstoupila v roce 2002 radiační kastraci, opět bez pozitivního efektu na plicní funkce. Nemocné byla posléze provedena plicní transplantace (případ č. 10).
V roce 2015 byla americkým Úřadem pro kontrolu potravin a léčiv (U.S. Food and Drug Administration – FDA) schválena kauzální léčba inhibitory mTOR (tato terapie ale není v ČR pro indikaci LAM dosud schválena a musí se žádat pojišťovnu o úhradu). Patologická proliferace LAM buněk je totiž zapříčiněna dysregulací mTOR dráhy, která přes hamartin a tuberin, proteiny kódované TSC1 a TSC2 geny, reguluje intracelulární serin/threonin kinázu mTOR signalizační cesty. Ta pak ve výsledku kontroluje velikost, proliferaci, dělení a přežití buněk. mTOR obsahuje dva komplexy (mTORC1, mTORC2). mTORC1 je citlivý na mTOR inhibitory (sirolimus, everolimus) (schéma 1). Sirolimus stabilizuje nebo zlepšuje plicní funkce (FEV1 a FVC), kvalitu života, redukuje velikost angiomyolipomů, lymfangioleiomyomů a množství chylózní tekutiny [25,43]. Sirolimus je na základě uvedených studií indikován pro LAM pacienty s FEV1 menší než 70 % [25,43]. Indikací u našich nemocných byla rovněž obstrukční ventilační porucha s FEV1 menší než 70 % a progrese plicních funkčních parametrů v čase nebo progrese velikosti angiomyolipomů. Doporučená dávka pro pacienty s LAM/TSC je 2 mg/den s případnou korekcí, aby hladina sirolimu v krvi byla mezi 4–10 ng/ml, max. 15 ng/ml [25,43]. Nejběžnější nežádoucí účinky jsou stejné jako u jiných indikací, např. mukozitidy, zažívací potíže (průjmy, zvracení), hypercholesterolemie, kožní změny (purpura), otoky dolních končetin, elevace jaterních testů, infekce, dysmenorhea, proteinurie, cysty ovaria [25,43,44]. U našich pacientek jsme se setkali s mírnými nežádoucími účinky většinou typu mukozitidy. U žádné ze čtyř pacientek jsme nemuseli z důvodů nežádoucích účinků zcela vysadit terapii.
![Schéma 1. Signalizační dráha mTOR a působení inhibitoru mTOR sirolimu. Podle
Taveira-DaSilva [7].](https://pl-master.mdcdn.cz/media/image_pdf/4c45c1bce06fa78894fdecbf231deb0b.png?version=1570999645)
Dalším lékem, který byl klinicky studován, je doxycyklin. Doxycyklin je tetracyklinové antibiotikum, které inhibuje produkci a aktivitu matrix metaloproteináz (MMP-2, MMP-9). Degradace extracelulární matrix proteolytickými enzymy, jako jsou MMP, přispívá k tvorbě cyst [45]. Bohužel terapie doxycyklinem neprokázala žádnou účinnost na plicní funkce [46]. Jiné léčebné možnosti se nadále hledají, zejména inhibitory autofagie, hydroxychlorochin a simvastatin jsou nyní pod klinickým testováním, protože inhibice mTORC1 sirolimem vede ke zvýšení autofagie a přežití buněk [7]. Kombinace mTOR s dalšími léčebnými modalitami tedy čeká na výsledky dalších studií.
Symptomatická léčba se zaměřuje na ovlivnění recidivujících pneumotoraxů, pleurálních výpotků a chylózních ascitů. Pleurodéza, plerektomie, pleurální abraze se mohou použít k minimalizování rizika opakujících se pneumotoraxů, zároveň ale mohou komplikovat provedení plicní transplantace [6,34,47]. V našem souboru pacientů byla chirurgická pleurodéza pro recidivující pneumotorax provedena u čtyř nemocných. V případě, že dochází k progresi, je indikována plicní transplantace, která obvykle přináší nemocným dlouhodobý benefit. V naší kohortě byla plicní transplantace provedena u dvou nemocných. Transplantace plic bývá zvažována v případě, že se pacientka funkčně posune do stadia dušnosti NYHA III–IV s klidovou hypoxemií a těžkým postižením plicních funkcí. Komplikací pro pacienty s LAM jsou pneumotoraxy v nativní plíci po jednostranné plicní transplantaci, chylotorax a v literatuře zřídkavé popisy rekurence nemoci v transplantované plíci, které bývají často asymptomatické [6,48–51]. Jsou známy zkušenosti s retransplantací plic pro selhání štěpu nebo při recidivě plicního postižení v transplantátu [50,51].
Klinický průběh je variabilní a nemoc většinou pomalu progreduje a vede k respiračnímu selhání a ke smrti v horizontu let. Prognostické faktory jsou závislé na plicních funkčních parametrech a histopatologickém zastoupení obou složek: cystické degenerace a proliferace abnormálních buněk hladké svaloviny. Mladší, premenopauzální ženy mají obvykle rychlejší pokles plicních funkčních parametrů nežli ženy starší, postmenopauzální [6,7]. Předpokládá se, že pacienti s TS/LAM mají ve srovnání se S/LAM mírnější a méně progresivní onemocnění [52,53]. V našem souboru máme jen dvě pacientky s TS/LAM. První je ve sledování od roku 2000 a je stabilní na léčbě inhibitory mTOR, druhá je diagnostikována v roce 2016 a nemá respirační potíže ani plicní funkční poruchu. Mladší pacienti, kteří se prezentují pneumotoraxem jako první manifestací LAM, mají lepší prognózu než ti, kteří trpí progredující dušností bez anamnézy pneumotoraxu [7,54]. S tímto závěrem se plně neshodujeme, protože pacienti, kteří se prezentují dlouhodobou dušností, mohou být často vedeni pod jinými diagnózami (viz pacientka 10, která byla 11 let léčena pro astma, pak pro chronickou obstrukční plicní nemoc) a LAM je pak diagnostikována až v pokročilém stadiu. Pacientka č. 9 se prezentovala jen recidivujícími pneumotoraxy, bez subjektivně udávané dušnosti, byla premenopauzální a u ní i vzhledem ke dvěma graviditám nemoc rychle progredovala. Ve studii Hayashida et al pacienti s poklesem plicních funkčních parametrů v čase (FEV1) a DLco pod 40 % projevující se dušností bez pneumotoraxu měli horší prognózu [54]. U našich pacientek byla také deklinace FEV1, DLco v čase spojena s progresí a závažností plicního procesu, a funkční parametry tak byly citlivé ukazatele pokročilosti plicního postižení.
Závěr
LAM je pomalu progresivní, metastatické onemocnění způsobené TSC1/TSC2 mutací. Negativními prognostickými faktory jsou gravidita a pokles plicních funkčních parametrů v čase. Předpovědět průběh onemocnění neumíme, ale základem správné péče o naše pacienty je dobrá mezioborová spolupráce s cílem časné diagnostiky a léčby, která je v současné době již kauzální. mTOR inhibitory jsou indikovány u progredujícího onemocnění a u tří našich pacientek vedly ke stabilizaci plicních funkcí, u jedné pacientky ke stabilizaci plicních funkcí a stabilizaci velikosti angiomyolipomu. Nežádoucí účinky asociované s mTOR u LAM jsou v našem souboru shodné s těmi u jiných indikací a jsou dobře terapeuticky zvládnutelné.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato/Accepted: 5. 8. 2019
MUDr. Martina Doubková, Ph.D.
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: doubkovamartina@seznam.cz
Sources
1. Lauterbacher R. Dysembriomes metotypiques des reins carcinnose submiliere aique poumon avec emphysemae generalise et double pneumothorax. Ann Med Interne (Paris) 1918; 5 : 435–450.
2. Van Stössel E. Uber muskuläre Cirrhose der Lunge. Beitr Klin Tuberk 1937; 90 : 432–442.
3. Laipply TC, Sherrick JC. Intrathoracic angiomyomatous hyperplasia associated with chronic chylothorax. Lab Incest 1958; 7 (4): 387–400.
4. Miřejovský P, Kulhánková J. Plicní lymfangioleiomyomatóza. Česk Patol 1980; 16 (3): 132–138.
5. Halíková J, Miřejovský P, Fučík J et al. Plicní lymfangioleiomyomatóza. Stud Pneumol Phtiseol Cechoslov 1990; 50 (4): 239–243.
6. Johnson SR, Cordier JF, Lazor R et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J 2010; 35 (1): 14–26. doi: 10.1183/09031936.00076209.
7. Taveira-DaSilva AM, Moss J. Epidemiology, pathogenesis and diagnosis of lymphangioleiomyomatosis. Expert Opin Ophran Drugs 2016; 4 (4): 369–378. doi: 10.1517/21678707.2016.1148597.
8. Ryu JH, Moss J, Beck GJ et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med 2006; 173 (1): 105–111. doi: 10.1164/rccm.200409-1298OC.
9. Smolarek TA, Wessner LL, McCormack FX et al. Evidence that lymphangiomyomatosis is caused by TSC2 mutations, chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet 1998; 62 (4): 810–815. doi: 10.1086/301804.
10. Carsillo T, Astrinidis A, Henske EP. Mutation in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 2000; 97 (11): 6085–6090. doi: 10.1073/pnas.97.11.6085.
11. Yu J, Astrinidis A, Henske EP. Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med 2001; 164 (8Pt1): 1537–1540. doi: 10.1164/ajrccm.164.8.2104095.
12. Colley MH, Geppert E, Franklin WA. Immunohistochemical detection of steroid receptors in case of pulmonary lymphangioleiomyomatosis. Am J Surg Pathol 1989; 13 (9): 803–807. doi: 10.1097/00000478-198909000-00011.
13. Matsui K, Takeda K, Yu ZX et al. Down regulation of estrogen and progesterone receptors in the abnormal smooth muscle cells in pulmonary lymmphangioleiomyomatosis following therapy: an immunohistochemical study. Am J Respir Crit Care Med 2000; 161 (3Pt1): 1002–1009. doi: 10.1164/ajrccm.161.3.9904009.
14. Oberstein EM, Fleming LE, Gomez-Marin O et al. Pulmonary lymphangioleiomyomatosis (LAM): examining oral contraceptive pills and the onset of disease. J Women Health 2003; 12 (1): 81–85. doi: 10.1089/154099903 321154176.
15. Silerstein EF, Ellis K, Wolff M et al. Pulmonary lymphangioleiomyomatosis. Am J Roentgenol 1974; 120 (4): 832–850.
16. Doubková M, Turčáni P, Pokojová E et al. Lymfangioleiomyomatóza (kazuistika a přehled případů popsaných v české a slovenské lékařské literatuře). Stud Pneumol Phtiseol 2005; 656 (4): 154–159.
17. Svatoň M, Pešek M, Ferda J. Lymfangioleiomyomatóza – cesta do nitra buněk. Kazuistiky v alergologii, pneumologii a ORL 2013; 10 (3): 3–8.
18. Homolka J, Svobodová L, Slováková A. Lymfangioleiomyomatóza u postmenopauzální ženy. Cas Lek Cesk 2003; 142 (2): 117–119.
19. Aubry MC, Myers JL, Ryu JH et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med 2000 (2Pt1); 162 : 749–752. doi: 10.1164/ajrccm. 162.2.9911006.
20. Urban T, Lazor R, Lancronique J et al. Pulmonary lymphangioleiomyomatosis: a study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine (Baltimore) 1999; 78 (5): 321–337. doi: 10.1097/00005792-199909000-00004.
21. Harknett EC, Chang WY, Byrnes S et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM 2011; 104 (11): 971–979. doi: 10.1093/qjmed/hcr116.
22. Oprescu N, McCormack FX, Byrnes S et al. Clinical predictors of mortality and cause of death in lymphangioleilomyomatosis: a population-based registry. Lung 2013; 191 (1): 35–42. doi: 10.1007/s00408-012-9419-3.
23. Wataya-Kaneda M, Tanaka M, Hamasaki T et al. Trends in the prevalence of tuberous sclerosis complex manifestations: an epidemiological study of 1666 Japanese patients. PLoS One 2013; 8 (5): e63910. doi: 10.1371/journal.pone.0063910.
24. Cudzilo CJ, Szczesniak RD, Brody AS et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest 2013; 144 (2): 578–585. doi: 10.1378/chest.12-2813.
25. McCormack FX, Gupta N, Finlay GR et al. Official American Thoracic Society/Japanese Respiratory Society clinical practice guidelines: lymphangioleiomyomatosis diagnosis and management. Am J Respir Crit Care Med 2016; 194 (6): 748–761. doi: 10.1164/rccm.201607-1384ST.
26. Gupta N, Vassallo R, Wikenheiser-Brokamp KA et al. Diffuse cystic lung disease: part I. Am J Respir Crit Care Med 2015; 191 (12): 1354–1366. doi: 10.1164/rccm.201411-2094CI.
27. Matsui K, Tatsuguchi A, Valencia J et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol 2000; 31 (10): 1242–1248. doi: 10.1053/hupa.2000.18500.
28. Avilla NA, Kelly J, Chu S et al. Lymphangioleiomyomatosis: abdominopelvic CT and US findigs. Radiology 2000; 216 (1): 147–153. doi: 10.1148/radiology.216.1.r00jl42147.
29. Glasgow CG, Avila NA, Lin JP et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other disease. Chest 2009; 135 (5): 1293–1300. doi: 10.1378/chest.08-1160.
30. Young L, Lee HS, Inoue Y et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med 2013; 1 (6): 445–452. doi: 10.1016/S2213-2600 (13) 70090-0.
31. Grzegorek I, Lenze D, Chabowski M et al. Immunohistochemical evaluation of pulmonary lymphangioleiomyomatosis. Anticancer Res 2015; 35 (6): 3353–3360.
32. Pacheco-Rodriguez G, Kumaki F et al. Chemokine-enhanced chemotaxis of lymphangioleiomymatosis cells with mutation in the tumor suppressor TSC2 gene. J Immunol 2009; 182 (3): 1270–1277. doi: 10.4049/jimmunol.182.3.1270.
33. Pacheco-Rodriguez G, Steagall WK et al. TSC2 los in lymphangioleiomyomatosis cells correlated with expression of CD44v6, a molecular determinant of metastasis. Cancer Res 2007; 67 (21): 10573–10581. doi: 10.1158/0008-5472.CAN-07-1356.
34. Gupta N, Finlay GA, Kotloff RM et al. Lymphangioleiomyomatosis diagnosis and management: high-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An official American Thoracic Society/Japanese Respiratory Society clinical practice guideline. Am J Respir Crit Care Med 2017; 196 (10): 1337–1348. doi: 10.1164/rccm.201709-1965ST.
35. Johnson SR, Tattersfield AE. Decline in lung function in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med 1999; 160 (2): 628–633. doi: 10.1164/ajrccm.160.2.9901027.
36. Sandrini A, Silverstone E, Yates DH. Menstrual cycle variation of retroperitoneal lymphangioleiomyomas in lymphangioleiomyomatosis. Intern Med J 2011; 41 (12): 832–835. doi: 10.1111/j.1445-5994.2011.02593.x.
37. Yockey CC, Riepe RE, Ryan K. Pulmonary lymphangioleiomyomatosis complicated by pregnancy. Kans Med 1986; 87 (10): 277–278, 293.
38. Yano S. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous oestrogen used for infertility treatment. Thorax 2002; 57 (12): 1085–1086. doi: 10.1136/thorax.57.12.1085.
39. Shen A, Iseman MD, Waldron JA et al. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogens. Chest 1987; 91 (5): 782–785. doi: 10.1378/chest.91.5.782.
40. Taveira-Da Silva AM, Stylianou MP, Hedin CJ et al. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest 2004; 126 (6): 1867–1874. doi: 10.1378/chest.126.6.1867.
41. Banner AS, Carrington CB, Emory WB et al. Efficacy of oophorectomy in lymphangioleiomyomatosis and benign metastasizin leiomyoma. N Engl J Med 1981; 305 (4): 204–209. doi: 10.1056/NEJM198107233050406.
42. Harari S, Cassandro R, Chiodini I et al. Effect of a gonadotrophin-releasing homrone analogue on lung function in lymphangioleiomyomatosis. Chest 2008; 133 (2): 448–454. doi: 10.1378/chest.07-2277.
43. McCormack FX, Inoue Y, Moss J et al. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011 (17); 364 : 1595–1606. doi: 10.1056/NEJMoa1100391.
44. Gao N, Zhang T, Ji J et al. The efficacy and adverse events of mTOR inhibitors in lymphangioleiomyomatosis: systematic review and meta-analysis. Orphanet J Rare Dis 2018; 13 (1): 134. doi: 10.1186/s13023-018-0874-7.
45. Bendeck MP, Conte M, Zhang M et al. Doxycycline modulates smooth muscle cell grouwth, migration, and matrix remodeling after arterial injury. Am J Pathol 2002; 160 (3): 1089–1095. doi: 10.1016/S0002-9440 (10) 64929-2.
46. Chang WY, Cane JL, Kumaran M et al. A 2-year randomised placebo-controlled trial of doxycykline for lymphangioleiomyomatosis. Eur Respir J 2014; 43 (4): 1114–1123. doi: 10.1183/09031936.00167413.
47. Čan V, Doubková M, Hanke I, Penka I. Lymfangioleiomyomatóza z pohledu chirurga. Kazuistiky v alergologii, pneumologii a ORL 2016; 13 (2): 10–15.
48. Bittmann I, Dose TB, Muller C et al. Lymphangioleiomyomatosis: recurrence after single-lung transplantation. Hum Pathol 1997; 26 (12): 1420–1423. doi: 10.1016/s0046-8177 (97) 90233-1.
49. Zaki KS, Aryan Z, Mehta AC et al. Recurrence of lymphangioleiomyomatosis: nine years after a bilateral lung transplantacion. World J Transplant 2016; 6 (1): 249–254. doi: 10.5500/wjt.v6.i1.249.
50. Pechet TT, Meyers BF, Guthrie TJ et al. Lung transplantation for lymphangioleiomyomatosis. J Heart Lung Transplnat 2004; 23 (3): 301–308. doi: 10.1016/S1053-2498 (03) 00195-5.
51. Sakompani P, Kasemsam C, Yottasurodom C. Retransplantaton after single lung transplantation. Transplant Proc 2008; 40 (8): 2617–2619. doi: 10.1016/j.transproceed.2008.07.120.
52. Taveira-DaSilva AM, Pacheco-Rodriguez G, Moss J. The natural history of lymphangioleiomyomatosis: markers of severity, rate of progression and prognosis. Lymphat Res Biol 2010; 8 (1): 9–19. doi: 10.1089/lrb.2009.0024.
53. Gupta N, Lee HS, Young LR et al. Analysis of the MILES cohort reveals determinants of disease progression and treatment response in lymphangioleiomyomatosis. Eur Respir J 2019; 53 (4): 1802066. doi: 10. 1183/13993003.02066-2018.
54. Hayashida M, Seyama K, Inoue Y et al. The epidemiology of lymphangioleiomyomatosis in Japan: a nationwide cross-sectional study of presenting features and prognostic factors. Respirology 2007; 12 (4): 523–530. doi: 10.1111/j.1440-1843.2007.01101.x.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2019 Issue 5
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Alopecie a poškození vlasů indukované onkologickou terapií
- Pseudomyxom peritonea
- Maligní peritoneální mezoteliom a jeho léčba
- Maligní nádory peritonea – úvod do problematiky