
Novinky z genetiky, molekulární biologie a klinické onkologie sarkomů
Novel Aspects of Genetics, Molecular Biology and Clinical Oncology of Sarcomas
The Connective Tissue Oncology Group Annual Meeting 2018 (CTOS 2018) took place in Rome from 4 to 17 November 2018, and the 39th Plenary Meeting of the Scandinavian Sarcoma Group (SSGM 2019) was held in Bergen from 8 to 10 May 2019. These two large international conferences brought together an overwhelming majority of molecular and clinical specialists in the sarcoma field, especially those working on soft tissue sarcoma. Topics discussed on the conferences included, among others, sarcoma genetics, clinical and molecular subclassification, targeted therapy, clinical prognostication, and new experimental sarcoma models. A large ongoing international study on germinal sarcoma genetics was presented, the interim results of which revealed the extremely complex nature of genetic disposition to sarcoma, and, surprisingly, a rather prominent place among predisposing genes for those coding for structural telomere constituents. Fusion oncogenes dominate somatic sarcoma genetics, especially because of their origin and impact on sarcoma clinical behaviour, and are especially relevant for karyotypically simple paediatric sarcomas. A crucial issue in karyotypically complex sarcomas are the efforts being made to obtain a subclassification of sarcoma, other than those based on pathology, using either the clinical characteristics of sarcomas (uterine leiomyosarcoma vs. soft tissue leiomyosarcoma) or specific gene expression profiles (molecular subtypes in undifferentiated pleiomorphic sarcoma), which showed that molecular characterization can open the way for subtype specific therapies. Other examples of where this type of strategy can be applied include gastrointestinal stromal tumours, infantile fibrosarcoma, and inflammatory myofibroblastic tumours, where targeted therapy could be conceived based on the actionable mutations identified. Attempts in this direction have been made also for clear cell sarcoma and dedifferentiated liposarcoma, albeit the effectiveness of molecular-targeted treatments for these sarcomas is still poor, and progress in the treatment of osteosarcoma is still rather slow. Actually, the platelet-derived growth factor signalling system holds a prominent position in searches for targeted therapies, not only against rare sarcoma types, where are activated by mutations (some gastrointestinal stromal tumours, infantile hereditary myofibromatosis, and dermatofibrosarcoma protuberans), but also against other more usual sarcoma types, where the blocking anti-PDGFRα-antibody olaratumab has been successfully integrated into combinatorial chemotherapeutic regimens. In the field of clinical prognostication, remarkable progress in sarcoma nomograms was reported. Interesting results were also presented in the area of new experimental sarcoma models.
Participation on both scientifi c conferences and all the experimental work leading to the presented sarcoma models were supported by the Czech Science Foundation project No. 17-17636S.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted: 20. 9. 2019
Accepted: 6. 11. 2019
Keywords:
osteosarcoma – soft tissue sarcomas – chondrosarcoma – genetic predisposition – molecular subtypes – targeted therapy – prognostic nomograms – experimental sarcoma models
Authors:
K. Houfková; J. Hatina
Authors‘ workplace:
Ústav biologie, LF UK v Plzni
Published in:
Klin Onkol 2020; 33(1): 66-78
Category:
Short Communication
doi:
https://doi.org/10.14735/amko202066
Overview
Ve dnech 14.–17. listopadu 2018 se v Římě uskutečnila velká mezinárodní konference Connective Tissue Oncology Group Annual Meeting 2018 (CTOS 2018), která svedla dohromady naprostou většinu nejvýznamnějších specialistů jak na molekulární biologii, tak i klinickou onkologii sarkomů, především sarkomů měkkých tkání. Ve dnech 8.–10. května 2019 se potom v Bergenu konala regionální konference The 39th Plenary Meeting of the Scandinavian Sarcoma Group (SSGM 2019). Soubory otázek, které byly na obou konferencích diskutovány formou přednáškových či posterových sdělení, lze rozdělit do několika zastřešujících okruhů. Byl představen velký projekt zaměřený na objasnění germinální genetiky sarkomů; dosavadní výsledky ukazují na překvapivě velký význam genů kódujících proteiny, které zajišťují integritu telomer, jakož i existenci komplexních mechanizmů genetické predispozice zahrnující polygenní dědičnost či modifikační geny. Problematice somatické genetiky sarkomů dominuje analýza vzniku a mechanizmu účinku fúzních onkogenů odpovídajících za iniciaci značné části především pediatrických sarkomů. U karyotypicky komplexních sarkomů je evidentní snaha po patobiologické specifikaci do subtypů, ať už na základě specifické klinické charakterizace (uterinní leiomyosarkom vs. leiomyosarkom měkkých tkání), či specifických expresních profilů genů (molekulární subtypy nediferencovaného pleiomorfního sarkomu). Molekulární charakterizace může být vodítkem pro koncipování subtypově specifických terapeutických protokolů, jak je tomu v různém měřítku u obou shora zmíněných příkladů. Mezi další prominentní typy sarkomů, u nichž se podařilo transformovat základní molekulárně biologické poznatky do podoby úspěšné cílené terapie, patří např. gastrointestinální stromální tumor, infantilní fibrosarkom či inflamatorní myofibroblastický tumor, a rovněž u světlobuněčného sarkomu a dediferencovaného liposarkomu byly obdobné koncepty prezentovány, byť jejich dosavadní účinnost zůstává za očekáváním, a podobná situace zatím přetrvává u osteosarkomu. Z hlediska molekulárních struktur, na které lze zaměřit cílenou terapii, zaujímá v oblasti sarkomů měkkých tkání zcela zásadní postavení signální systém destičkového růstového faktoru, a to jak u vzácných typů založených na přímé iniciační mutační aktivaci (relativně malá část gastrointestinálních stromálních tumorů, infantilní hereditární myofibromatóza, dermatofibrosarcoma protuberans), u kterých cílená terapie využívá především nízkomolekulárních tyrozinkinázových inhibitorů, tak rovněž u širokého spektra obvyklých typů sarkomů, kde se součástí kombinační chemoterapie stává specifická blokující anti-PDGFRα-protilátka olaratumab. V oblasti klinické prognózy byl pozoruhodný vývoj zaznamenán zejména u tzv. prognostických nomogramů. Zajímavé výsledky byly rovněž prezentovány v oblasti odvození nových experimentálních modelů vzniku a progrese sarkomů.
Klíčová slova:
sarkomy měkkých tkání – osteosarkom – genetická predispozice – molekulární subtypy – cílená terapie – prognostické nomogramy – experimentální modely sarkomatogeneze – chondrosarcoma
Genetika sarkomu
Germinální genetika sarkomů, tj. dědičná nádorová predispozice k sarkomu, představuje neobyčejně komplexní problematiku, a to nejen vzhledem k šíři různých konkrétních diagnóz, které lze v současnosti uvnitř rodiny mezenchymálních nádorů rozlišit (více než 70 různých typů [1]). Nejnovější vývoj v rámci projektu zaměřeného na zmapování genů zodpovědných za dědičnou nádorovou predispozici k sarkomu na konferencích Connective Tissue Oncology Group Annual Meeting 2018 (CTOS 2018) a The 39th Plenary Meeting of the Scandinavian Sarcoma Group (SSGM 2019) představila dr. M. Ballinger. Tento projekt nese označení International Sarcoma Kindred Study (ISKS) a ve své aktuální podobě zahrnuje 2 933 probandů postižených různými typy sarkomů, 2 623 jejich příbuzných 1. stupně, od nichž je k dispozici celkem 4 328 vzorků DNA a cca 600 histologických vzorků nádorové tkáně, a rovněž rodokmenovou informaci zhruba 105 000 příslušníků těchto rodin. Od 1 109 probandů již byla získána úplná genetická informace metodikou celogenomového sekvenování nové generace. Tyto impozantní rozměry studie si vynutila řada komplikujících faktorů genetiky sarkomu. Především, až na jednu výjimku, o které se zmíníme později, nebyl dosud popsán žádný mendelisticky dědičný syndrom nádorové predispozice, který by se týkal výlučně či převážně sarkomu (ani jakožto široké skupiny mezenchymálních nádorů, ani žádného z jednotlivých konkrétních typů), jinými slovy neexistuje prakticky žádná sarkomová obdoba např. syndromu dědičné nádorové predispozice k nádorům prsu a vaječníku, familiární adenomatózní polypózy nebo rodinného melanomu. Na druhé straně jsou ovšem různé sarkomy stabilní součástí řady různých syndromů dědičné nádorové predispozice, vč. tří výše zmíněných, a rovněž např. Lynchova syndromu, Li-Fraumeniho syndromu či rodinného retinoblastomu. Jedním z průběžných výsledků projektu ISKS je stanovení relativních rizik pro vývoj sarkomu podmíněných evidentními či pravděpodobnými mutantními alelami (označovanými jako C3, C4 a C5 alelické varianty Mezinárodní agenturou pro výzkum rakoviny (International Agency for Research on Cancer)) celé řady známých „nádorových“ genů (tab. 1).
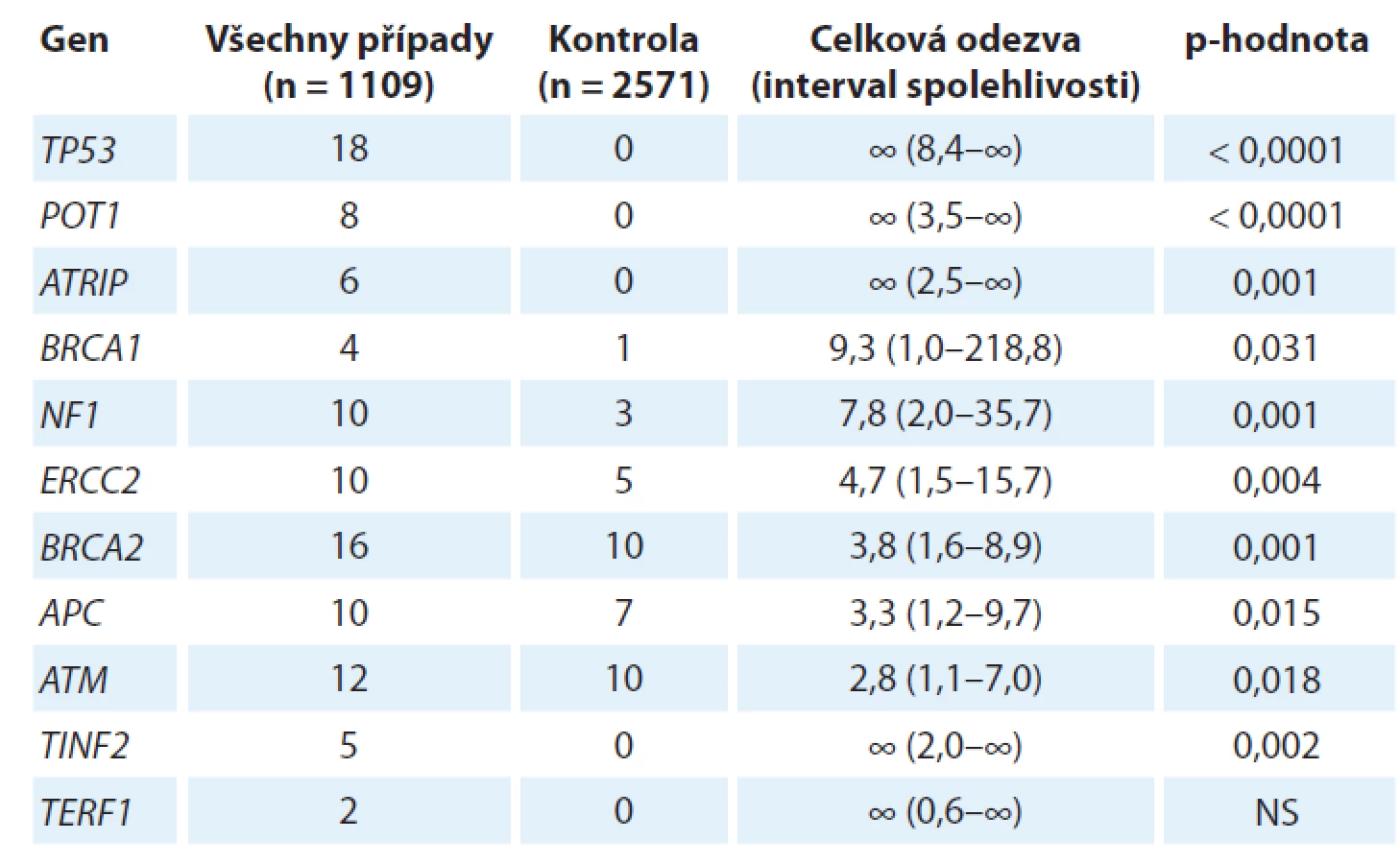
Jeden aspekt se v uvedené studii ukázal jako docela specifický pro různé typy sarkomů, a to velmi významné postavení genů kódujících proteiny zodpovědné za integritu telomery (POT1, TIN2, TRF1). Tyto proteiny spolu navzájem interagují a spolu s dalšími proteiny (TRF2, TPP1 a RAP1) tvoří tzv. „shelterin complex“, který chrání terminální části telomery. POT1 má klíčové postavení, protože přímo interaguje jednak s telomerovou jednořetězcovou DNA, jednak prostřednictvím TPP1 i s celým ochranným komplexem. POT1 blokuje přístup telomerázy, a je-li tato funkce porušena, dochází k aberantnímu prodloužení telomer, což má za následek jejich destabilizaci, která může být důležitou součástí, či dokonce iniciátorem destabilizace karyotypu. Samotný POT1 byl nezávisle identifikován jakožto kauzální mutovaný gen u čtyř španělských rodin segregujících celé spektrum nádorů vč. četných sarkomů, i velmi vzácných, jako je angiosarkom srdce či prsu [2]. Identifikovaná mutace (R117C) vede ke ztrátě afinity jak vůči telomeře, tak vůči TPP1 a u nositelů mutace skutečně dochází jak k signifikantnímu prodloužení telomery, tak k nárůstu ohnisek poškozené DNA v oblasti telomery. Dosavadní výsledky projektu ISKS tento předchozí raritní nález potvrzují a rozšiřují, zejména se ukazuje, že mutace v POT1 genu predisponují obzvláště ke vzniku častých karyotypicky komplexních sarkomů, jako je osteosarkom nebo leiomyosarkom. V této souvislosti stojí ovšem za to poukázat na výsledky, které prokázaly, že např. u liposarkomu dochází naopak ke stabilizaci telomer aktivací telomerázy, a to buď bodovou mutací v promotoru genu pro katalytickou podjednotku telomerázy hTERT, jako je tomu u myxoidního liposarkomu [3], nebo v důsledku chromozomální translokace a zvýšení exprese hTERT genu, která byla prokázána u dediferencovaného liposarkomu [4]. Tato oblast tedy ještě zřejmě přinese řadu překvapivých výsledků.
Jinou komplikací dědičné nádorové predispozice k sarkomu je existence polygenní dědičné složky. Již předchozí publikované dílčí výsledky projektu ISKS ukázaly, že C3 varianty mají výraznou tendenci chovat se aditivně, tj. současné zdědění několika C3 variant v různých genech proporcionálně zvyšuje riziko vzniku sarkomu [5]. Přechodem mezi monogenním a polygenním způsobem dědičnosti je uplatnění tzv. genů modifikátorů, jejichž patologické alely samy nemusejí nutně vést ke zvýšenému riziku, popř. mohou mít svůj specifický fenotypový dopad, ale v kombinaci s patologickou variantou tumorového supresoru či onkogenu výrazně modifikují jejich fenotypový projev, penetranci a expresivitu. Také v tomto ohledu mohou sarkomy nabídnout výborný příklad – mutace v genech EXT1 a EXT2 predisponují ke vzniku mnohočetného benigního osteochondromu (mnohočetné hereditární exostózy), v kombinaci s patologickými variantami celé řady jiných genů (PMS2, WRN, RECQ4, MLH3, FANCA, FANCL, BUB1B, XPC, TP53, BRCA1, BRCA2, ERC, WT1) ovšem signifikantně zvyšují riziko vzniku osteosarkomu a chondrosarkomu [5].
Somatické genetice sarkomů dominuje především téma fúzních onkogenů; současné představy o molekulární biologii a patologii sarkomů založených na fúzních onkogenech prezentovala na konferenci CTOS 2018 prof. Cristina R. Antonescu. Onkogeny aktivované genovou fúzí se pokládají za typickou vlastnost celé jedné skupiny sarkomů, které se vyznačují relativně stabilním karyotypem a relativně nízkým věkem nástupu, který ovšem vykazuje značné rozpětí u různých konkrétních sarkomů, od novorozeneckých až po sarkomy vyskytující se v časné dospělosti. Biologickým důsledkem genové fúze je vznik chimérických onkoproteinů, které typicky kombinují specifické domény dvou různých proteinů; v této formě nejsou genové fúze nikterak specifické pro sarkomy, tento mutační mechanizmus je velmi obvyklý i u značné části leukemií a lymfomů a rovněž u některých karcinomů (např. karcinomu prostaty). Dlouhou dobu se myslelo, že hlavním mechanizmem vzniku fúzních onkogenů jsou reciproké translokace, u nichž dochází k translokačním zlomům uvnitř zúčastněných onkogenů, popř. méně často inverze či intersticiální delece. V době velmi nedávné byl objeven rozsáhlejší mechanizmus strukturální přestavby karyotypů vedoucí k tvorbě fúzních onkogenů, tzv. chromoplexe, představující soubor chromozomálních přestaveb zahrnujících několik chromozomů, které jsou translokovány v jakési uzavřené „translokační smyčce“, tj. první chromozom s druhým, druhý s třetím, třetí se čtvrtým a čtvrtý zase s prvním (schéma 1) [6]. Zdá se, že se jedná o široce používaný mechanizmus, jenž byl zatím prokázán u Ewingova sarkomu, který tvoří historicky, biologicky i klinicky jakýsi prototyp sarkomu závislého na fúzním onkogenu, a dále u synoviálního sarkomu, chondromyxoidního fibromu a fosfaturického mezenchymálního tumoru a již dříve také v komplexnější podobě u karcinomu prostaty. Odhaduje se, že o něco méně než polovina případů Ewingova sarkomu připadá na vrub chromoplexii, zbytek připadá na reciproké translokace, přičemž první skupina vykazovala skoro dvojnásobnou četnost relapsu – komplexní způsob chromozomálních přestaveb v podobě chromoplexického translokačního cyklu lze tudíž považovat za jeden ze znaků agresivity nádoru [6].
![Schéma 1. Fúzní onkogeny jakožto výsledek komplexní chromozomální přestavby – chromoplexie. Upraveno podle [6].](https://pl-master.mdcdn.cz/media/image_pdf/a43fdc4e88857e502217d0edc0a80730.png?version=1582031972)
Ewingův sarkom představuje paradigmatický typ sarkomu závislého na fúzním onkogenu ještě z několika hledisek. Hlavními dvěma charakteristikami této skupiny sarkomů a genů zúčastněných na tvorbě fúzních onkogenů jsou promiskuita a heterogenita. Podkladem klasického Ewingova sarkomu je genová fúze mezi genem EWSR1 a jedním z genů kódujících transkripční faktory Ets-rodiny, nejčastěji FLI1 (90 %), méně často ERG (5–8 %) a výjimečně ETV1, FEV či E1AF (souhrnně 1–2 %). EWSR1 kóduje RNA-vazebný protein s kryptickou transkripčně aktivační doménou, která je mnohem silnější než přirozené transkripčně aktivační domény Ets-faktorů, fúzní onkogen tedy kombinuje tuto silnou transkripčně aktivační doménu původem EWSR1 s DNA-vazebnou doménou Ets-faktorů a tento aberantní transkripční faktor vede k rozsáhlé deregulaci transkripce v buňce, která je podkladem maligní transformace.
Jedním z možných vysvětlení těchto dramaticky různých četností vzniku různých fúzních onkogenů jsou vzájemné orientace transkripce zúčastněných genů; zatímco EWSR1 a FLI1 jsou transkribovány v témže směru (oba od centromery k telomeře), ERG je transkribován v opačném směru. Výsledkem chromozomální přestavby musí být kontinuální biologicky smysluplná kódující sekvence. Jsou-li oba zúčastněné geny orientovány na svých chromozomech opačně, je složitější tuto souvislou „správnou“ kódující sekvenci v důsledku chromozomální přestavby získat, a skutečně zatímco EWSR1-FLI1 může být výsledkem jak reciproké translokace, tak chromoplexie, EWSR1-ERG vzniká výhradně na podkladě chromoplexie [6,7].
Řada dalších typů sarkomů na podkladě fúzních onkogenů vykazuje tuto molekulární heterogenitu, přičemž jeden z konkrétních fúzních onkogenů obvykle vysoce převažuje. Například inflamatorní myofibroblastický tumor zahrnuje více než deset alternativních fúzních onkogenů obsahujících gen pro tyrozinovou kinázu ALK, synoviální sarkom je iniciován fúzí genu SS18 a jednoho z genů SSX lokalizovaných na X chromozomu (SSX1, SSX2 nebo SSX4; je zajímavé, že SSX3, který je jako jediný transkribován v opačném směru, zatím jakožto součást fúzního onkogenu identifikován nebyl [7]) a u alveolárního rhabdomyosarkomu existuje heterogenita v obou genech (PAX3/PAX7 a FOXO1/ NCOA1 [7]). Na druhou stranu EWSR1 se účastní tvorby fúzních onkogenů u podstatně širšího souboru sarkomů než je Ewingův sarkom (tab. 2) a současně Ewingův sarkom vykazuje nápadné morfologické i klinické podobnosti se sarkomy, jejichž podkladem jsou jiné fúzní onkogeny, např. CIC-DUX4 (ve srovnání s klasickým Ewingovým sarkomem prognosticky horší a s výraznou preferencí pro měkké tkáně) či BCOR-CCNB3 (prognosticky srovnatelný s klasickým Ewingovým sarkomem, s výraznou tkáňovou preferencí pro kost); všechny tyto tři typy jsou v současnosti řazeny do rodiny Ewingova sarkomu (Ewing-and Ewing-like tumours [8]).

V některých případech může dokonce prakticky jeden a tentýž fúzní onkogen tvořit podklad různých typů sarkomů, např. EWSR1 fúzovaný s geny pro transkripční faktory ATF/CREB (tab. 3). Jak je možné vysvětlit tuto fenotypovou variabilitu? Hlavním možným vysvětlením jsou odlišné sekundární mutace typické pro každý jednotlivý typ nebo rozdíly v buněčných typech, u nichž dochází k prvotní chromozomální přestavbě a iniciaci sarkomageneze.

Je zajímavé, že použití celogenomového sekvenování nové generace umožnilo detekovat fúzní onkogeny i u typů sarkomů s komplexním karyotypem, jako leiomyosarkom, dediferencovaný liposarkom [4] nebo nediferencovaný pleiomorfní sarkom, není ovšem dosud jasné, jaká je jejich patobiologická funkce v iniciaci sarkomageneze těchto komplexních sarkomů, na rozdíl od předešle diskutované skupiny převážně pediatrických sarkomů se stabilním karyotypem, kde všechno hovoří pro to, že fúzní onkogeny hrají naprosto klíčovou úlohu, představují tzv. „driver-mutace“. Rovněž vzhledem ke značné nestabilitě karyotypicky komplexních sarkomů není jasné, jestli fúzní onkogeny přetrvávají např. v jistém klonu sarkomových buněk, nebo jestli jsou kontinuálně vytvářeny a ztráceny.
Molekulární subtypy sarkomů
Odhlédneme-li od zajímavých molekulárně genetických aspektů zmíněných výše, je nepochybné, že stanovení kauzální mutace, např. ve formě kritického fúzního onkogenu, je klíčové pro stanovení diagnózy. Se stále se prohlubujícím porozuměním molekulární biologii i klinickému chování sarkomů přitom pozorujeme stále jemnější a podrobnější diagnostické členění, kdy se dříve jednotná diagnostická skupina ukazuje jako heterogenní a rozpadá se do jemnějších, homogennějších a molekulárně i klinicky lépe definovaných podskupin. Zajímavý vývoj v tomto směru byl pozorován např. u embryonálního rhabdomyosarkomu (prof. C. R. Antonescu, CTOS 2018). Na rozdíl od alveolárního rhabdomyosarkomu s jeho dobře definovanými fúzními onkogeny PAX3/PAX7-FOXO1/NCOA1 u embryonálního rhabdomyosarkomu doposud žádné molekulárně či patologicky definované podskupiny známy nebyly, obecnou genetickou charakteristikou byla absence chromozomálních přestaveb a zásadní onkogenní funkce bodových mutací v klíčových onkogenech a tumorových supresorových genech. V poslední době byla definována nová patologická skupina označená jako vřetenobuněčný/sklerozující rhabdomyosarkom, jejíž molekulární charakteristika opět prozradila značnou úroveň heterogenity. Jedna část těchto převážně pediatrických nádorů je opět iniciována fúzními onkogeny mezi geny kódujícími transkripční faktory podílející se významně na diferenciaci kosterního svalu (SRF, TEAD1, VGLL2) a transkripčními korepresory CITED a NCOA2 – výsledkem je transkripční represe genů, které by normálně měly být v průběhu diferenciace kosterního svalu aktivovány, tedy dediferenciace a sarkomageneze. Druhá skupina vřetenobuněčného/sklerozujícího rhabdomyosarkomu má svůj molekulární podklad v inaktivujících bodových mutacích v genu kódujícím klíčový a hierarchicky nadřazený transkripční faktor diferenciace kosterního svalu MyoD1 (nejčastější je bodová mutace změny smyslu L122R v DNA-vazebné doméně). Podstatné je, že se tyto dva typy vřetenobuněčného/sklerozujícího rhabdomyosarkomu podstatně liší prognózou – zatímco subtyp založený na fúzních onkogenech je prognosticky příznivý, u MyoD1-mutovaných nádorů se jedná o velice agresivní typ spojený s vysokou a časnou mortalitou.
Z nádorů s komplexním karyotypem se už dlouhou dobu spekuluje o jisté molekulární dichotomii u leiomyosarkomu, přesněji opakovaně se objevují názory, že uterinní leiomyosarkom není biologicky ani klinicky totožný s leiomyosarkomem měkkých tkání. V době velmi nedávné byla provedena komplexní transkriptomická a genomická analýza v rámci projektu The Cancer Genome Anatomy (TCGA), která poskytla těmto hypotézám částečnou oporu [9]. Přestože uterinní leiomyosarkom a leiomyosarkom měkkých tkání si jsou svým expresním profilem a mutačním vzorem navzájem bližší než každý z nich vůči jakémukoliv jinému typu sarkomu, přece se v segregační analýze řadí do odlišných skupin, a bylo možné identifikovat molekulární dráhy, které jsou typické pro každý z nich. U uterinního leiomyosarkomu tak byla významně aktivnější dráha aktivovaná poškozenou DNA (DNA damage response pathway), zatímco leiomyosakrom měkkých tkání měl signifikantně aktivnější signální dráhu aktivovanou hypoxií a transkripčním faktorem HIF1α. Je také už delší dobu známá rozdílná aktivita steroidních receptorů; výše uvedená studie TCGA prokázala specifickou hypometylaci cílových genů pro estrogenový receptor (ERα) specificky u uterinního leiomyosarkomu a zvýšená exprese a aktivita samotného estrogenového, jakož i progesteronového receptoru u uterinního leiomyosarkomu vedla již k několika malým klinickým studiím testujícím prospěšnost integrace inhibitorů aromatáz do různých léčebných protokolů [10]. Tyto výsledky byly na konferenci CTOS 2018 rozšířeny o imuno-histochemickou analýzu velkého souboru nádorů shromážděných v podobě „tissue microarrays“ (TMA) (přednáška dr. I. M. Schaefer) (tab. 4), která potvrdila signifikantní rozdíly v četnosti mutací klíčových tumorových supresorových genů a v expresi steroidních receptorů mezi oběma subtypy leiomyosarkomu. Za povšimnutí stojí jeden rozpor mezi uváděnou studií TCGA a výsledky imunohistochemické analýzy TMA – zatímco první z nich identifikovala značnou frekvenci delecí TP53 genu, v imunohistochemické studii TMA prezentované na konferenci byl naopak nalezen ve velké většině případů aberantně exprimovaný p53 prozrazující jeho stabilizaci specifickými bodovými mutacemi. Zatím není jasné, co by mohlo být příčinou tohoto rozdílu, jedná se ovšem o otázku, která je svými důsledky, zejména s ohledem na možnou perspektivní terapii, docela důležitá, poněvadž stabilizovaný bodově mutovaný p53 může být přístupný navrácení do nativní konformace specifickými malými molekulami [11] (autoři výše uvedené přednášky dokonce spekulovali o budoucí možnosti této léčby), což samozřejmě u deletovaného p53 nepřichází v úvahu.
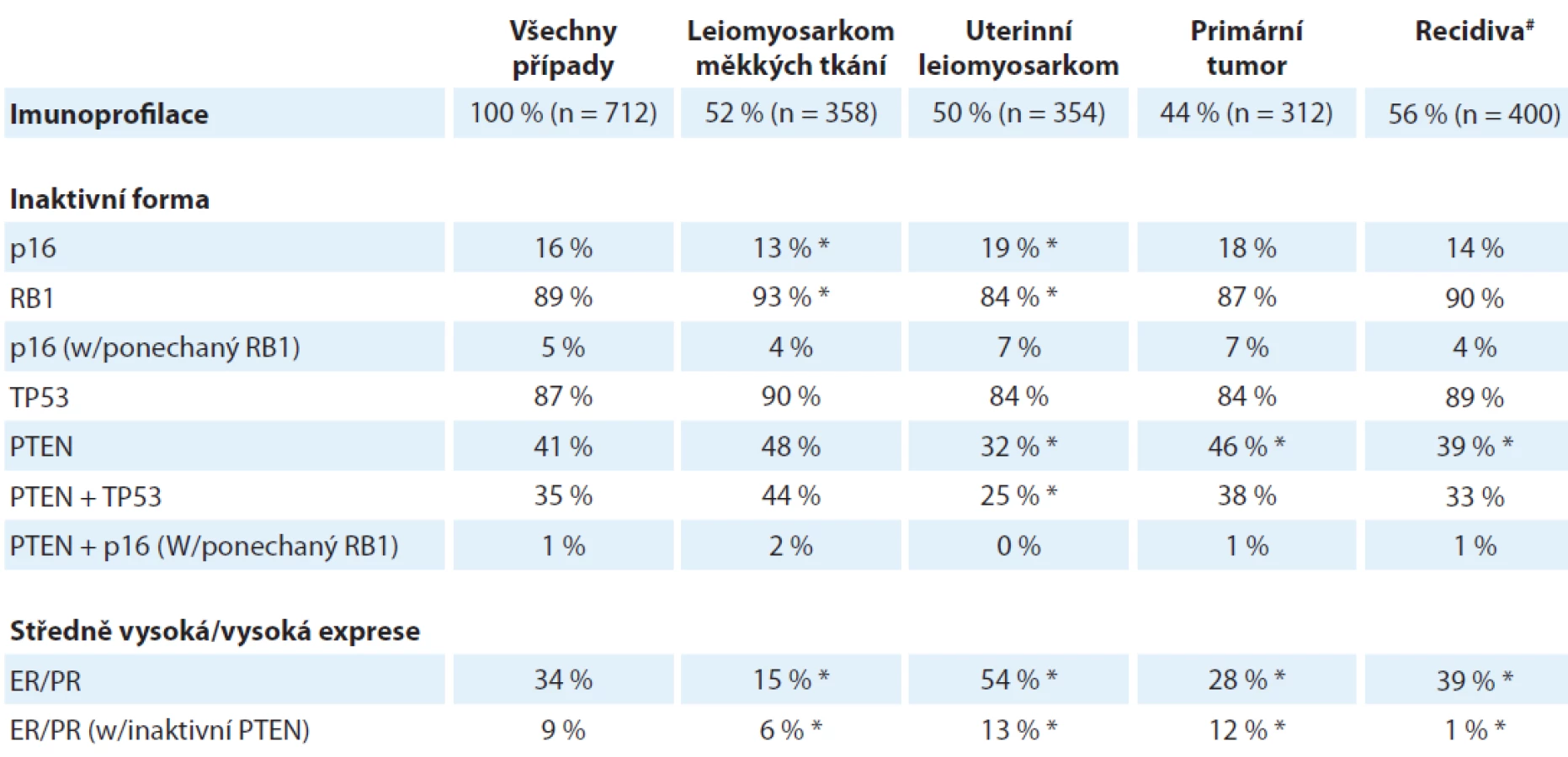
ER – estrogenový receptor, PR – progesteronový receptor
Není asi nikterak překvapující, že jednou ze sarkomových diagnóz, která přímo volala po identifikaci molekulárních subtypů, je nediferencovaný pleiomorfní sarkom (undifferentiated pleomorphic sarcoma – UPS), diagnóza, která je často přijímána vylučovacím způsobem a je tudíž ze své podstaty heterogenní. Ostatně už výše citovaná komplexní studie TCGA prokázala kontinuum mezi nediferencovaným pleiomorfním sarkomem a myxofibrosarkomem. Problematiku heterogenity UPS objasnila ve své přednášce na konferenci CTOS 2018 dr. M. Toulmondé, která podrobila sérii 26 primárních nádorových vzorků diagnostikovaných s tímto typem sarkomu komplexní expresní (RNA sekvenování) i genomové (celoexomové sekvenování) a proteomické analýze. Vzorky nediferencovaného pleiomorfního sarkomu se na základě zejména expresní analýzy rozpadly do tří skupin, dvou vyhraněně odlišných a mezi nimi jedné intermediární. Jedna ze skupin v sobě nesla část expresního profilu prozrazujícího aktivní zánětlivou reakci (pojmenovaná jako „hot-nádory“). Tato skupina měla signifikantně vyšší hladinu bodových mutací; není vyloučeno, že zde existuje oboustranná příčinná souvislost s aktivní zánětlivou reakcí – kyslíkové a dusíkové radikály vylučované zánětlivými buňkami mají prokazatelnou mutagenní aktivitu, současně vysoká úroveň bodových mutací a z toho vyplývající prezentace nádorových antigenů může přilákat do prostředí nádoru imunokompetentní buňky. Druhá, vyhraněně odlišná skupina nádorů, nazvaná případně „cold“, vykazovala výrazně vyšší strukturální chromozomální nestabilitu (copy number alteration, tj. rozsáhlejší delece a amplifikace) a byla prognosticky signifikantně méně příznivá. Jednou ze signálních molekul specificky aktivovaných v této skupině je FGFR2, receptorová tyrozinová kináza, pro niž jsou k dispozici specifické nízkomolekulární inhibitory v pokročilé fázi vývoje, a skutečně jeden z těchto inhibitorů (JNJ-42756493) významně inhiboval proliferaci „cold-UPS“ buněčných linií i primárního xenotransplantačního modelu; tento efekt byl vysoce specifický, u „hot-UPS“ experimentálních modelů jej nebylo možné detekovat. Naopak signální dráhy typické pro „hot“ podskupinu zahrnovaly transkripční faktor Myc a dále faktory asociované s epiteliálně-mezenchymální tranzicí. Vzhledem k vysoké imunitní infiltraci by se v tomto případě nabízel imunoterapeutický postup, např. formou protilátek proti klíčovým inhibitorům T lymfocytů (tzv. checkpoint inhibitory – CTLA-4, PD-1 či PD-1L [12]), o nichž se dnes diskutuje v souvislosti s většinou nádorů, vč. sarkomů (např. právě probíhající studie IMMUNOSARC [13] či možné uplatnění imunomodulátoru mifamutridu, které se pozvolna začíná stále více prosazovat u osteosarkomu – viz dále).
Molekulární biologie sarkomů jakožto cesta k identifikaci racionální personalizované terapie
Příklad molekulárních subtypů nediferencovaného pleiomorfního sarkomu ukazuje možnou hlavní aplikaci molekulární biologie sarkomageneze, totiž možnou identifikaci optimální protinádorové terapie, která by byla přesně přizpůsobena molekulárním změnám v nádorových buňkách a přesně na tyto změny zacílena [14]. Bohužel už výše uvedené příklady sarkomů založených na fúzních onkogenech naznačily, že pro většinu z nich je podobná strategie zatím nedostupná, poněvadž hlavní funkční kategorií proteinů, kterých se vznik fúzních onkogenů týká, jsou aberantní transkripční faktory a v současné době není k dispozici žádná klinicky zralá strategie jejich inhibice. Již současné použití cílené terapie u karcinomů a hematologických malignit prozrazuje, že naopak v zásadě dobře zvládnutá je farmakologická inhibice různých onkogenních kináz. Bohužel jen nemnoho sarkomů je molekulárně založeno právě na aberantní kinázové aktivitě. Nicméně takové příklady také existují a u některých z nich byl na konferenci prezentován pozoruhodný terapeutický pokrok.
Poměrně velká pozornost byla na konferenci CTOS 2018 věnována infantilnímu fibrosarkomu (satelitní sympozium vedené dr. G. S. Demetrim, dr. S. DuBoisem a prof. J. Y. Blayem, přednáška pronesená dr. N. Federmanem). Jedná se o vzácný novorozenecký až dětský nádor, jehož metastatická kapacita je sice omezená, který ale roste rychle a lokálně invazivně, což při radikálním operativním zásahu s sebou často nese trvalou invalidizaci dítěte, a pro nějž nebyl k dispozici spolehlivý chemoterapeutický režim, nehledě na obecnou problematičnost klasické chemoterapie u dětí takto nízkého věku. Již koncem 90. let 20. století byla u infantilního fibrosarkomu odhalena fúze genu pro transkripční faktor ETV6 a genu NTRK3 kódujícího tyrozinovou kinázu TRKC [15]; v tomto případě, přestože se genové fúze účastní gen pro transkripční faktor, je její dopad odlišný než u příkladů citovaných výše, poněvadž ETV6 přispívá do výsledného fúzního onkogenu, resp. onkoproteinu toliko svým promotorem a dimerizační doménou, a NTRK3 dodává intracelulární doménu vč. katalytické domény receptorové tyrozinkinázy TRKC. Výsledkem fúze tedy není globální deregulace transkripce, ale konstitutivně aktivní kináza. TRKC je jednou z trojice receptorových kináz pro neurotrofní růstové faktory, zbylé dvě jsou TRKA a TRKB, kódované geny NTRK1 a NTRK2 [16], jejichž genové fúze byly v nádorech rovněž identifikovány, vč. infantilního fibrosarkomu (např. TMP3-NTRK1, PDE4DPI-NTRK1, SQSTM1-NTRK1 nebo LMNA-NTRK1 [17,18]; 5’-fúzní partner vždy musí dodávat promotor aktivní v mezenchymálních buňkách, jelikož uvedené kinázy sloužící jako receptory pro neurotrofní růstové faktory jsou fyziologicky exprimovány v nervové tkáni, a nějakou formu dimerizační či oligomerizační domény – dimerizace jakékoliv receptorové kinázy ligandem je nezbytnou součástí jejich fyziologické aktivace [16]. Fúzní onkoproteiny zahrnující TRK-receptorové kinázy jsou nalézány v celém spektru nádorů, vč. velmi obvyklých karcinomů (např. plic, prsu, kolorekta, pankreatu aj.), melanomu či akutní lymfoblastické a myeloidní leukemie či gastrointestinálního stromálního tumoru (GIST), vždy ovšem tvoří pouze výraznou menšinu případů, typicky kolem 5 %. Naproti tomu u infantilního fibrosarkomu a několika dalších dětských nádorů (jako dětský sekretující karcinom prsu či mezoblastický nefrom) jsou NTRK-fúzní onkogeny prakticky definiční a vyskytují se ve více než 90 % případů [16]. Podstatné je rovněž, že máme k dispozici velmi dobré nízkomolekulární inhibitory entrectinib a larotrectinib. U infantilního fibrosarkomu znamenalo nasazení těchto inhibitorů opravdový průlom v léčbě, především v neoadjuvantním a částečně i adjuvantním režimu ve spojení se šetrnou a neinvalidizující chirurgickou léčbou, při velice příznivém profilu vedlejších účinků.
Existuje několik dalších typů mezenchymálních nádorů, u kterých léčba zahrnuje či je přímo postavena na využití cílených kinázových inhibitorů. Příkladem, který není možné opominout, je samozřejmě GIST, jeden z paradigmatických příkladů cílené protinádorové terapie [14]. Přestože GIST lze rovněž zařadit ke spektru nádorů, u nichž je malá část případů způsobena shora uvedenými NTRK-fúzními onkogeny, a tudíž i citlivá vůči entrectinibu nebo larotrectinibu, v naprosté většině případů je mutační mechanizmus odlišný a spočívá v bodových aktivačních mutacích genů pro receptorové tyrozinkinázy c-Kit (80–85 % případů) a PDGFRα (10 % případů), a bylo šťastnou shodou okolností, že se velice záhy zjistilo, že většina těchto nádorů je citlivá vůči jednomu z prvních dostupných kinázových inhibitorů imatinibu. V současné době existují s touto léčbou již bohaté zkušenosti a do popředí se tak dostává otázka terapeutické rezistence a jejího adekvátního řešení, ať se už jedná o primárně rezistentní nádory (např. nádory nesoucí specifickou mutaci PDGFRα D842V), nebo pokud se týče vývoje sekundární rezistence v důsledku selekce klonů nesoucích specifické mutace v ATP-vazebné doméně nebo aktivační smyčce c-Kit nebo PDGFRα. Novou nadějí pro tyto pacienty jsou kinázové inhibitory nové generace, jednak avapritinib (BLU-285 – přednáška na konferenci CTOS 2018, dr. M. Heinrich), jednak DCC-2618 (přednáška na konferenci CTOS 2018, dr. S. George). Obě přednášky prezentovaly výsledky klinických studií fáze I zahrnující pacienty s pokročilým GIST s historií několika linií předchozí standardní terapie a obě prokázaly akceptovatelný profil toxicity a velmi povzbudivou klinickou účinnost.
V léčbě sarkomů nachází uplatnění rovněž crizotinib, duální inhibitor kináz ALK a MET. V případě ALK je zjevnou aplikací inflamatorní myofibroblastický tumor (viz výše) a v případě MET je to světlobuněčný sarkom. Ten je sice založen na translokacích t (12; 22) (q13; q12) či t (2; 22) (q32.3; q12), které vedou k vytvoření fúzních onkogenů EWSR1-ATF, resp. EWSR1-CREB1 (tab. 3), nedochází tedy k přímé ligand-nezávislé aktivaci kinázy jako např. u infantilního fibrosarkomu, nicméně tento aberantní transkripční faktor aktivuje, pravděpodobně s výrazným přispěním jiného transkripčního faktoru MITF, gen kódující tyrozinovou kinázu MET. Výsledky prezentované malé klinické studie (přednáška na konferenci CTOS 2018, dr. P. Schöffski) jsou ovšem dosti skromné, jen u necelých 4 % pacientů došlo k objektivní částečné klinické odpovědi, nicméně u značné části (60 %) došlo ke stabilizaci onemocnění. Tato studie ovšem trpěla některými koncepčními nedostatky, jednak u pacientů nebyla analyzována a použita jako vstupní kritérium exprese/aktivita MET, jednak jsou dnes již k dispozici účinnější inhibitory MET-kinázy než crizotinib, takže definitivní závěr ještě není možné učinit. Poslední ze skupiny kinázových inhibitorů, které byly na konferenci diskutovány, jsou inhibitory CDK4 (abemaciclib a ribociclib) u dediferencovaného liposarkomu; podkladem je zde notorická amplifikace části chromozomu 12 (12q13-q15), která zahrnuje CDK4 gen a rovněž gen pro inhibitor p53 MDM2, vůči němuž jsou také k dispozici specifické farmakologické inhibitory (např. nutliny nebo SAR405838). Obě dvě skupiny inhibitorů byly nezávisle samostatně testovány (přednášky na konferenci CTOS 2018, dr. M. Gounder a dr. M. A. Dickson) a výsledky obou studií prezentovaných na konferenci jsou opět relativně střízlivé, s nejčastější odpovědí ve formě stabilizace onemocnění. Vyvstává proto myšlenka kombinace obou inhibitorů, otázkou ovšem je, jestli taková terapie nebude zatížena příliš vysokou toxicitou.
Systém destičkového růstového faktoru jakožto terapeutický cíl sarkomů měkkých tkání
Zcela specifické a do jisté míry výjimečné postavení v sarkomagenezi má signální systém destičkového růstového faktoru (platelet-derived growth factor – PDGF). Lidský genom nese celkem čtyři PDGF geny, označované jako A, B, C a D. Signálně aktivní ligand je dimer, a to buď homodimer, nebo heterodimer PDGFAB. K dispozici jsou dva geny pro receptory, PDGFRA a PDGFRB (receptory samotné se označují jako α a β), přičemž tak jako u všech tyrozinkinázových receptorů prvním krokem transdukce signálu je vazba ligandu a ligandem zprostředkovaná dimerizace receptoru, jejímž výsledkem tedy mohou být tři formy aktivovaného receptoru (αα, αβ a ββ). Existují přitom dosti komplexní preference mezi dostupnými pěti ligandy a třemi receptorovými komplexy [19].
PDGF je jedním z nejsilnějších mitogenů pro mezenchymální buňky a má klíčové kauzální postavení u několika typů karyotypicky jednoduchých sarkomů. Již byla řeč o aktivačních somatických mutacích PDGFRA u asi 10 % GIST. Germinální aktivační mutace u PDGFRB jsou podkladem vzácného syndromu dědičné nádorové predispozice (zřejmě jediného, který se manifestuje výlučně ve formě sarkomu – viz výše) – hereditární infantilní myofibromatózy [20,21]. Jedná se o nádory tvořené aktivovanými fibroblasty, které mohou vznikat v jakémkoli věku, ovšem často jsou pediatrické, a jejichž klinické chování velmi závisí na výchozí tkáni. Pokud vznikají v kůži, podkoží, svalu či kosti, je jejich chování často indolentní a dochází u nich často dokonce ke spontánní regresi, pokud se vyvíjejí ve vnitřních orgánech, jsou často agresivní a spojené s vysokou mortalitou. Žádná standardní terapie nebyla pro tyto nádory formulována, na konferenci CTOS 2018 byly prezentovány výsledky izraelské skupiny s klasickou chemoterapeutickou léčbou (poster dr. M. Manisterski) spočívající v kombinaci nízkodávkovaného metotrexátu a vinblastinu, přičemž ze čtyř pediatrických pacientů s postižením vnitřních orgánů tři přežili (jeden ovšem zaznamenal relaps onemocnění po vysazení chemoterapie) a jeden zemřel. Možná je rovněž také cílená léčba kinázovými inhibitory imatinibem nebo sunitinibem, s nímž byla u pozoruhodné kazuistiky úspěšná brněnská skupina [22].
Samotný ligand PDGFB má kauzální postavení v případě dermatofibrosarcoma protuberans, kožního sarkomu iniciovaného translokací t (17; 22) (q22; q13), jejímž výsledkem je fúzní gen mezi genem pro kolagen 1A1 a právě PDGFB genem. Translokační zlom je u kolagenového genu velmi variabilní, podstatné je, že jeho promotor řídí transkripci fúzního onkogenu. U PDGFB je naproti tomu translokační zlom lokalizován vždy v prvním intronu. Kolagenový promotor je samozřejmě v mezenchymálních buňkách velmi aktivní, výsledkem je tedy masivní nadprodukce PDGFB-faktoru. Také v tomto případě je u lokálně pokročilých nebo metastatických nádorů, které se ovšem vyskytují velmi vzácně, indikován imatinib [23].
Zatímco výše uvedené příklady se vždy týkají jen zcela specifické, geneticky přesně definované a početně zpravidla malé skupiny pacientů, signální systém PDGF se v nedávné době stal cílovou strukturou terapie sarkomů měkkých tkání v této široké specifikaci, tj. napříč různými typy a vč. obvyklých karyotypicky komplexních typů. Stalo se tak v podobě zavedení terapeutické protilátky proti PDGFRα olaratumabu. Jedná se o lidskou monoklonální protilátku třídy IgG1, která specificky a s vysokou afinitou rozeznává extracelulární ligand-vazebnou doménu PDGFRα, čímž zabraňuje vazbě ligandu a aktivaci receptoru. V in vitro a in vivo podmínkách olaratumab signifikantně inhiboval růst řady sarkomových buněčných linií [24] a překvapivě dobrého výsledku bylo dosaženo v kombinované klinické studii fáze I/II (studie JGDG), u které kombinace olaratumabu a doxorubicinu prakticky zdvojnásobila celkovou dobu přežití oproti monoterapii doxorubicinem u souboru pacientů s pokročilými sarkomy měkkých tkání [25]. Na konferenci CTOS 2018 byla představena návazná studie (přednáška dr. S. Bauer), u které byl kombinován olaratumab s kombinační chemoterapií doxorubicinem a ifosfamidem. Studie fáze I, která byla předmětem přednášky, zahrnovala 15 pacientů (3 se synoviálním sarkomem, 3 s maligními tumory z pochvy periferního nervu (malignant peripheral nerve sheath tumours – MPNST), 3 s liposarkomem, 2 s leiomyosarkomem, 2 se světlobuněčným sarkomem a 2 s nediferencovaným pleomorfním sarkomem). Plánovaná kombinační chemoterapie zahrnovala šest cyklů s podáním olaratumabu (15 mg/kg) 1. a 8. den, doxorubicinu (75 mg/m2) 1., 2. a 3. den a ifosfamidu (20 g/m2) 1., 2., 3. a 4. den, následovanými monoterapií olaratumabem až do progrese. V rámci výsledků studie fáze I nebyla přídavkem olaratumabu zaznamenána zvýšená toxicita oproti publikované toxicitě kombinační chemoterapie samotné [26]. Z pacientů, kteří dokončili plánovaný režim, zaznamenal pouze 1 progresi, u 5 došlo k částečné odpovědi (z čehož ovšem 1 progredoval při monoterapii olaratumabem), u 6 došlo ke stabilizaci onemocnění (klinická odpověď 25 %, kontrola onemocnění 69 %). U 3 pacientů umožnila dosažená klinická odpověď následnou operaci.
Bude nepochybně zajímavé sledovat tuto studii v delším časovém horizontu, aby bylo možné vyhodnotit důsledky pro celkové přežití. Lze rovněž doufat, že tato nová kombinace pomůže osvětlit některé z ne zcela jednoznačně interpretovatelných závěrů studie JGDG, např. ne zcela snadno vysvětlitelný nesoulad mezi jen poměrně skromným zvýšením doby do progrese a poměrně razantním zvýšením celkového přežití, jakož i nemožnost prokázat jakýkoliv vztah mezi expresí PDGFRα v nádoru a léčebnou odpovědí [25].
Otevře molekulární biologie osteosarkomu dveře k nové terapii?
Jedním z častých typů sarkomů, u kterých by zavedení nových terapeutických postupů bylo skutečně na místě, je osteosarkom. Pro tyto pacienty zůstává základní léčebný protokol prakticky nezměněn od 70. let, kdy byla formulována základní kombinační chemoterapie metotrexátem, doxorubicinem a cisplatinou a u refrakterních a relapsujících nádorů gemcitabinem a docetaxelem. Jedinou inovací, která ovšem stále zůstává pojímána jako experimentální léčba, je doplnění této polychemoterapie imunomodulační látkou mifamurtidem, který pravděpodobně stimuluje tumoricidní aktivitu alveolárních makrofágů a snižuje tak pravděpodobnost růstu a progrese plicních metastáz [26]; přestože se jedná o experimentální léčbu, většina na osteosarkom specializovaných účastníků konference SSGM 2019 se spíše přiklání k názoru, že je tato imunomodulační léčba klinicky přínosná.
Neblahou vlastností osteosarkomu je náhlý explozivní růst a poměrně rychlá progrese, takže naprostá většina nádorů je diagnostikována jakožto „high grade“ a u většiny pravděpodobně již v době diagnózy byly založeny mikrometastázy. Jedná se rovněž o sarkom s vyhraněně nestabilními karyotypy s desítkami, možná stovkami numerických a strukturálních chromozomových aberací a polyklonálním vývojem [27]. Svými vlastnostmi osteosarkom docela překvapivě připomíná serózní „high grade“ ovariální karcinom, a možná právě to bylo impulzem k tomu, že v nedávné době byl osteosarkom podrobně geneticky analyzován na aktivitu enzymatických komplexů DNA-reparační dráhy homologní rekombinací (tzv. BRCAness, podle BRCA-1 a BRCA-2 genů) a skutečně se zjistilo, že tato reparační dráha je porušena u značné části osteosarkomů, což samozřejmě hned vyvolalo naději na možné léčebné uplatnění PARP-inhibitorů [28]. Přestože se zprvu zdálo, že se jedná o skoro univerzální vlastnost osteosarkomu, situace asi bude o mnoho složitější a zejména heterogennější. Na konferenci SSGM 2019 prezentoval poměrně rozsáhlou experimentální studii citlivosti etablovaných osteosarkomových buněčných linií vůči PARP-inhibitoru BMN 673 prof. O. Myklebost a zjistil jednak značnou heterogenitu, jednak, aspoň prozatím, nemožnost předpovědět citlivost na základě jakékoli molekulárně biologické charakteristiky. Studie právě publikovaná na primárních osteosarkomových liniích udržovaných ve formě xenotransplantátů (patient derived xenografts – PDX), viz dále, tento nález potvrdila [29]. Bez identifikace spolehlivého prediktivního markeru lze tudíž o této terapii uvažovat jen obtížně.
Prognostické nomogramy sarkomů měkkých tkání
Viděli jsme již, že jak v případě Ewingova sarkomu, rhabdomyosarkomu i nediferencovaného pleomorfního sarkomu nese v sobě molekulárně genetická charakterizace, ať už v jakékoliv podobě (mutační mechanizmus, konkrétní „driver“ mutace, molekulární subtyp charakterizovaný specifickým expresním profilem), také významnou prognostickou informaci. Stanovení prognózy onemocnění na základě takové molekulárně genetické informace je v oblasti sarkomu měkkých tkání ovšem přinejlepším záležitostí klinických studií a pro standardní odhad prognózy onemocnění jsou určující klinické a patologické charakteristiky. Ty je možné kombinovat v podobě tzv. prognostických nomogramů. Klasický prognostický nomogram představuje grafický prognostický nástroj, kdy je každá nezávislá prognosticky významná charakteristika vynesena na své podélné ose a její přímá či konvertovaná kvantitativní hodnota je převedena do podoby bodového skóre [30]. Pravděpodobnost, že k definované klinické události (celkové přežití, sarkom-specifické přežití, přežití bez metastatického rozsevu apod.) dojde v předem stanoveném časovém okamžiku, je odvozena od celkového bodového součtu po sečtení bodového příspěvku všech zohledněných klinických charakteristik [31,32]. Některé z klinických ukazatelů vstupují do prognostických nomogramů jakožto kvantitativní veličiny (typicky např. věk pacienta), některé kvantitativní proměnné mohou být někdy naopak dichotomizovány (např. velikost nádorů může do prognostických nomogramů vstupovat jako kvantitativní veličina nebo nádory mohou být např. diferencovány a odlišně bodově ohodnoceny podle toho, jestli nedosahují určité arbitrárně stanovené hodnoty, např. 15 cm, nebo ji naopak přesahují [33]). Některé ze vstupních proměnných mají duální charakter samy o sobě (např. fokalita – unifokální vs. multifokální nádory), některé jsou ze své podstaty kvalitativní a bodové ohodnocení je jim přiděleno specificky v rámci daného nomogramu (u sarkomů typicky histopatologický typ). Rovněž záleží na tom, jestli je konstruován obecný nomogram, nebo nomogram specializovaný pouze na určitou podskupinu. První sarkomový nomogram [34] měl obecný charakter, novější nomogramy jsou už specializované. K této specializaci může u sarkomů docházet ve dvou ohledech – jednak jsou k dispozici nomogramy omezené jen na specifický histopatologický typ (např. synoviální sarkom [35] či uterinní leiomyosarkom [36]) či histopatologickou rodinu (liposarkom [37]), jednak byly konstruovány nomogramy specializované na určité anatomické místo prezentace nádoru (retroperitoneální sarkomy [38] a končetinové sarkomy [39]). Důsledkem této specializace je možnost vzít při konstrukci nomogramu v úvahu specifické proměnné, které mají dominantní postavení jen u určité skupiny sarkomů. Např. u retroperitoneálního liposarkomu byly při konstrukci specializovaného liposarkomového nomogramu rozlišeny případy, kdy chirurgická resekce mohla být zaměřena jen na nádor jako takový, od případů, kdy předmětem resekce musel být také nějaký vnitřní orgán, s vyšším bodovým ohodnocením druhé z možností [37]. Někdy se dokonce může jednat o odlišnou formu distribuce společné kvantitativní proměnné. Například u současného nomogramu zaměřeného na končetinové sarkomy je velikost nádoru zohledněna jakožto kvantitativní proměnná s lineární distribucí [40], zatímco současný nomogram konstruovaný pro retroperitoneální sarkomy nahlíží na velikost nádoru jako na kvantitativní proměnnou s nelineální distribucí – extrémně velké nádory dosahují nižšího bodového ohodnocení než nádory s velikostí 30 cm, což je hraniční hodnota, do které je distribuce lineární [41]. Příčinou je skutečnost, že obzvláště velké retroperitoneální sarkomy jsou z velké většiny dobře diferencované liposarkomy s příznivějším klinickým průběhem než jiné typy sarkomů, jako např. dediferencovaný liposarkom či leiomyosarkom [42]. Tento poslední příklad rovněž ilustruje interakci různých proměnných, které, ač a priori stanovené jako nezávislé, se navzájem podmiňují. Konstrukce nomogramů je statisticky poměrně komplexní problém, kvalita každého nomogramu je určena především dvěma ukazateli – kalibrací a diskriminací [40,42]. Kalibrace je grafický výraz validace prognostické hodnoty nomogramu, kdy na jedné ose vynášíme vypočítanou předpokládanou hodnotu veličiny (např. celkové přežití), na druhé ose hodnotu reálnou – u ideálního nomogramu bychom tak měli získat přímku se směrnicí 45°. Diskriminace je pravděpodobnost, že z náhodně vybrané dvojice pacientů se předpokládaná událost, jejíž pravděpodobnost nomogram stanovuje (smrt, rekurence, metastáza apod.) objeví dříve u pacienta s vyšším bodovým součtem, a tudíž vyšší vypočítanou pravděpodobností. Ideální diskriminace dosahuje hodnoty 1 (tj. 100 %), diskriminace 0,5 (50 %) znamená nulovou prognostickou hodnotu nomogramu. Spolehlivost nomogramu je zejména dána jeho externí validací, tj. spolehlivý nomogram dosahuje podobné kalibrace a diskriminace u etnicky a geograficky různých populací pacientů ošetřovaných v různých specializovaných centrech [43].
Příspěvky na konferenci CTOS 2018 se týkaly tří aspektů vývoje prognostických nomogramů sarkomů měkkých tkání. Nevyhnutelná interakce různých proměnných byla zohledněna ve specifické modifikaci nomogramu pro končetinové sarkomy, tzv. dynamickém prognostickém nomogramu (přednáška prof. D. Callegaro). Jak toto označení již nechává tušit, tou hlavní proměnnou, jejíž interakce s ostatními je předmětem této modifikace, je čas; zatímco klasické nomogramy, o nichž jsme diskutovali výše, umožňují jednorázový odhad úmrtí, rekurence či metastatického rozsevu na základě hodnot jednotlivých proměnných zjištěných v okamžiku operace, tento dynamický nomogram umožňuje opakovanou kalkulaci po 12, 24 a 36 měsících, a to na základě jak vstupních klinických dat v okamžiku operace, tak i klinického vývoje od okamžiku operace po provedení opakované kalkulace. Druhým aspektem je integrace molekulárně genetické informace; v našem dosavadním textu jsme prognostické nomogramy dávali do jakéhosi protikladu k molekulárně genetickým a molekulárně biologickým faktorům odhadu prognózy, a to čistě z důvodu logického členění textu. Lze ovšem téměř s jistotou předpovědět, že tato hranice se bude v budoucnosti čím dále tím více stírat a molekulárně geneticky či biologicky založené charakteristiky individuálních nádorů se stanou jedněmi z proměnných prognostických nomogramů. Jakousi předzvěstí tohoto vývoje byla přednáška dr. I. Wei, která ukázala možnosti integrace konkrétní mutační informace (tři nejčastější mutace CTNNB1 genu či wt-alela či jiná mutace) do prognostického nomogramu rekurence desmoidní fibromatózy; podstatné přitom je, že začleněním této molekulární proměnné došlo ke zvýšení diskriminace z hodnoty 0,707, dosažené pouze s klinickými proměnnými, na 0,729 u všech pacientů a 0,744 u pacientů s primárně diagnostikovanou desmoidní fibromatózou. Konečně, třetí oblastí, ve které prognostické nomogramy sarkomů doznaly v poslední době významného rozvoje, je jejich aplikační forma. Cílem je co nejvíce rozšířit a usnadnit jejich praktické použití v každodenní klinické praxi. Sarkulátor je jedna z takových aplikačních inovací (www.sarculator.com). Jedná se o volně dostupnou aplikaci vyvinutou pro smartphony a tablety, která je určena dospělým pacientům po resekci retroperitoneálního a končetinového sarkomu. Poskytuje odhad onkologických výsledků na základě věku pacienta, stadia onemocnění a velikosti a histologie nádoru. Po vložení těchto specifických kovariátů do aplikace je uživateli vykalkulována pravděpodobnost celkového přežití, bezpříznakového přežití nebo výskytu vzdálených metastáz po resekci primárního sarkomu. Koncepčně podobná, i když v detailech odlišná, a nikoliv široce dostupná aplikační forma byla vyvinuta také specificky pro myxoidní liposarkom (poster dr. D. D. McMillan). Jedním z příkladů uplatnění Sarkulátoru je studie prezentovaná v přednášce dr. S. Pasqualiho, jejímž cílem bylo pokusit se redefinovat postavení adjuvantní chemoterapie v léčbě končetinových sarkomů měkkých tkání. Dosavadní klinické studie poskytly velmi rozporuplné výsledky; poslední velká randomizovaná klinická studie [41] neprokázala klinický benefit kombinační adjuvantní chemoterapie doxorubicinem a ifosfamidem. Jestliže ovšem totožní pacienti byli ex post stratifikováni na základě Sarkulátorem kalkulovaného odhadu pravděpodobnosti 8letého celkového přežití (jako hraniční hodnota bylo zvoleno 60 %), pak u vysoce rizikových pacientů (tzn. s kalkulovanou pravděpodobností celkového 8letého přežití pod 60 %) byl efekt adjuvantní terapie statisticky významný.
Nové experimentální modely sarkomu měkkých tkání
Jedním z problémů experimentální onkologie sarkomů obecně je nedostatek vhodných experimentálních modelů; ostatně to bylo také téma našich příspěvků. Na konferenci CTOS 2018 jsme posterovou formou prezentovali naše dosavadní výsledky získané analýzou progresivní série fibrosarkomových buněčných linií odvozených z fibrosarkomu indukovaného hlubokým poraněním u v-jun transgenní myši [42,43], zejména výsledky transkriptomické analýzy. Tento experimentální systém zahrnuje relativně málo transformovanou sarkomovou linii JUN-2 s nízkou úrovní proliferace, motility a invazivity, z ní odvozenou dceřinou linii JUN-2fos3, která si zachovává nízkou úroveň proliferace, ale došlo u ní k výrazné manifestaci buněčné motility a invazivity, a vysoce transformovanou sarkomovou linii JUN-3 s vysokou úrovní proliferace, motility i invazivity. Tato unikátní sestava sarkomových buněčných linií nám umožnila identifikovat v jediné transkriptomické analýze jak geny zodpovědné za proliferaci (kontrastní expresní profil u JUN-3 oproti JUN-2 a JUN-2fos3), tak geny zodpovědné za motilitu a invazivitu (kontrastní expresní profil u JUN-3 a JUN-2fos3 oproti JUN-2). Ukázalo se tak, že JUN-3 sarkomové buňky disponují značnou autokrinní aktivací jak buněčné proliferace, tak motility; jeden z těchto autokrinních motilitních faktorů, chemokin Ccl8, může být farmakologicky inhibován, a skutečně tato inhibice signifikantně snižuje motilitu této sarkomové buněčné linie. Autokrinní regulace buněčné proliferace má zřejmě komplexnější charakter; JUN-3 buňky skutečně specificky nadměrně exprimují řadu růstových faktorů (amfiregulin, epiregulin, fibroblastové růstové faktory 10 a 13, hepatocytární růstový faktor) či receptorů (rovněž výše diskutovaný PDGFRα) a jejich proliferace není závislá na obsahu séra v médiu, na druhou stranu jimi kondiciované médium specificky inhibuje růst JUN-2 sarkomových buněk. To by mohlo otevřít cestu k objasnění interakce nezávislých nádorových klonů v komplexních nádorech vč. klasického fenoménu klonální dominance. Na konferenci SSGM 2019 jsme potom formou komentovaného posteru tento model rozšířili o další progresivní sarkomovou sérii založenou na velice obvyklé preadipocytární buněčné linii 3T3L1. Francouzská skupina publikovala spontánní transformaci této preadipocytární buněčné linie in vivo do dediferencovaného liposarkomu, ze kterého byla odvozena příslušná buněčné linie LM3D [44]. Opět jsme postupovali cestou celogenomové transkriptomické analýzy, která nám poskytla vhled do možných základních mechanizmů vzniku a progrese nádorů tukové tkáně. Obecnější význam by mohl mít bioinformaticky identifikovaný průnik transkriptomických profilů obou agresivních sarkomových linií, tj. JUN-3 a LM3D, který jsme označili jako „sarcoma progression signature“. Celkem se nám podařilo identifikovat 95 souhlasně transkripčně aktivovaných genů a 78 souhlasně transkripčně reprimovaných genů, které by mohly ukrývat obecnější informaci o biologických mechanizmech progrese sarkomu. Prvotní analýza aktivovaných genů odhalila přítomnost překvapivě různorodé skupiny genů, které byly v různém kontextu publikovány jakožto inhibitory klasické, tzv. konvenční Wnt/β-kateninové signální dráhy (Dickkopf-2 a -3, Apcdd1, Meg3, Fibulin-5, Ints6, Msx1), a současně též aktivaci exprese genu kódujícího receptor tyrosine kinase-like orphan receptor 2 (Ror-2); to by napovídalo, že během progrese sarkomů (na rozdíl od mnohých karcinomů) dochází k utlumení klasické Wnt/β-kateninové signální dráhy a k jejímu nahrazení tzv. nekonvenční Wnt5a/Ror-2 signální dráhou [45]. To by potvrzovala i přítomnost některých genů, které byly publikovány jakožto aktivované právě touto nekonvenční Wnt5a/Ror-2 signální dráhou, mezi aktivovanými geny našeho „sarcoma progression signature“, jako je gen pro onkoprotein c-jun anebo geny pro metaloproteinázy matrix, konkrétně MMP-16. Podle stejné logiky by farmakologické aktivátory konvenční Wnt/β-kateninové dráhy mohly představovat slibné kandidáty pro novou chemoterapii pokročilých sarkomů; za zmínku v tomto ohledu stojí např. bortezomib [46].
Obecným problémem klasických buněčných kultur jsou arteficiální podmínky růstu nádorových buněk, které nemohou napodobit komplexní mikroprostředí nádoru [47]. Jednou z možností, jak zmírnit tento problém, jsou xenotransplantační modely, kdy se nativní fragmenty nádorů přímo implantují přísně imunodeficientní myši, tzv. PDX (patient-derived xenografts). Tuto techniku využila i skupina, která popsala molekulární subtypy nediferencovaného pleomorfního sarkomu pro analýzu účinnosti FGFR2 – specifických inhibitorů u tzv. „cold-UPS“ nádorů, nebo nedávná studie hledající nové terapeutické možnosti u osteosarkomu (viz výše) [29]. Skupina z belgické univerzity v Leuvenu (projekt XenoSarc – poster na konferenci CTOS 2018, dr. A. Wozniak) se takto zaměřila na celou škálu sarkomů měkkých tkání zahrnující myxofibrosarkom, dediferencovaný liposarkom, synoviální sarkom, MPNST, leiomyosarkom, epitelioidní hemangioendoteliom, mezenchymální chondrosarkom, rhabdomyosarkom a nediferencovaný pleiomorfní sarkom. Celkem se v současnosti jedná o 30 xenotransplantačních modelů, a současně s nimi je připravována i odpovídající tissue microarray, takže by mělo být možné současně analyzovat jednotlivé sarkomy imunohistochemicky i funkčním testem. Samozřejmě že metodika PDX je o mnoho komplikovanější a zejména experimentálně méně přístupná než klasické buněčné kultury. Skupina z milánského Istituto Nazionale dei Tumori (poster na konferenci CTOS 2018, dr. C. Colombo) se pokusila tento problém zmírnit současným odvozením xenotransplantátů i klasických buněčných linií ze vzorků získaných po chirurgickém odstranění retroperitoneálně lokalizovaných dediferencovaných liposarkomů a podle prezentovaných výsledků se zdá, že úspěšně; ze tří dediferencovaných liposarkomů se podařilo odvodit paralelně oba modely a využít je pro zatím základní screening chemosenzitivity. Jinou možností, jak zachovat komplexní nádorové interakce a současně získat experimentálně přístupnější model, než jsou xenotransplantáty, je použít trojrozměrné buněčné kultivace. Skupina z německé kliniky Helios (poster na konferenci CTOS 2018, dr. M. Gaebler) se zaměřila právě tímto směrem. Jedná se o tzv. sarkomové organoidy, tedy trojrozměrné mikronádory kultivované v komplexní extracelulární matrix (tzv. Matrigel); tento systém se označuje PD3D (patient-derived 3D-culture). Smyslem je získat experimentálně schůdný systém, který bude např. přístupný dlouhodobé kultivaci s cílem identifikovat potenciálně terapeuticky využitelné mutace každého individuálního sarkomového pacienta a na základě toho formulovat individualizovanou terapii, nebo použít tento systém jakožto screeningovou platformu pro identifikaci nových farmakologicky zajímavých molekul.
Primární buněčné kultury klinických sarkomů představují metodicky poměrně nelehký úkol; vzhledem k histopatologické variabilitě je obtížné definovat kultivační podmínky tak, aby se podařilo ustanovit primární buněčné kultury s rozumnou úspěšností u různých pacientů se sarkomy. Pozoruhodného úspěchu, prezentovaného formou přednášky a komentovaného posteru na konferenci SSGM 2019, dosáhla v tomto ohledu švédská skupina z Karolinska Institutet [48], která s více než 50% úspěšností kultivovala primární nádorové kultury odvozené z aspirátů získaných tenkou jehlou nebo z chirurgicky získaných vzorků několika typů sarkomů (Ewingova sarkomu, embryonálního rhabdomyosarkomu, alveolárního sarkomu měkkých tkání, angiosarkomu). Primární buněčné kultury byly následně podrobeny expresní analýze a současně testovány na citlivost vůči farmakologické knihovně zahrnující 525 farmakologicky aktivních látek a následně konfrontovány s pozorovanou odpovědí pacientů na empiricky podanou chemoterapii. Dosavadní výsledky (rozsah studie je zatím malý – 14 primárních buněčných kultur) ukazují na velmi dobrou schopnost předpovědět klinickou rezistenci a v jednom případě rovněž senzitivitu. Prezentovaným cílem projektu je tento kultivační a screeningový systém v blízké budoucnosti použít jakožto platformu pro in vitro test chemosenzitivity a podle něj modifikovat, či dokonce formulovat individualizovanou terapii.
Účast na obou konferencích i veškerá experimentální práce vedoucí k prezentovaným sarkomovým modelům byla podpořena projektem Grantové agentury České republiky 17-17636S.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. Ing. Jiří Hatina, CSc.
Ústav biologie
LF UK v Plzni
alej Svobody 1655/76
326 00 Plzeň
e-mail: jiri.hatina@lfp.cuni.cz
Obdrženo: 20. 9. 2019
Přijato: 6. 11. 2019
Sources
1. Fletcher CD, Unni KK, Mertens F (eds). WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press 2013.
2. Calvete O, Martinez P, Garcia-Pavia P et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li–Fraumeni-like families. Nat Commun 2015; 6 : 8383. doi: 10.1038/ncomms9383.
3. Koelsche C, Renner M, Hartmann W et al. TERT promoter hotspot mutations are recurrent in myxoid liposarcomas but rare in other soft tissue sarcoma entities. J Exp Clin Cancer Res 2014; 33 (1): 33. doi: 10.1186/1756-9966-33-33.
4. Delespaul L, Lesluyes T, Pérot G et al. Recurrent TRIO fusion in nontranslocation–related sarcomas. Clin Cancer Res 2017; 23 (3): 857–867. doi: 10.1158/1078-0432.CCR-16-0290.
5. Ballinger ML, Goode DL, Ray-Coquard I et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol 2016; 17 (9): 1261–1271. doi: 10.1016/S1470-2045 (16) 30147-4.
6. Anderson ND, de Borja R, Young MD et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018; 361 (6405): 8419. doi: 10.1126/science.aam8419.
7. Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer 2016; 55 (4): 291–310. doi: 10.1002/gcc.22335.
8. Specht K, Hartmann W. Ewing-Sarkome und Ewing-artige Sarkome. Pathologe 2018; 39 (2): 154–163. doi: 10.1007/s00292-018-0421-2.
9. Cancer Genome Atlas Research Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017; 171 (4): 950–965. doi: 10.1016/j.cell.2017.10.014.
10. George S, Serrano C, Hensley ML et al. Soft tissue and uterine leiomyosarcoma. J Clin Oncol 2017; 36 (2): 144–150. doi: 10.1200/JCO.2017.75.9845.
11. Synnott NC, Bauer MR, Madden S et al. Mutant p53 as a therapeutic target for the treatment of triple-negative breast cancer: preclinical investigation with the anti-p53 drug, PK11007. Cancer Lett 2018; 414 : 99–106. doi: 10.1016/j.canlet.2017.09.053.
12. Darvin P, Toor SM, Sasidharan Nair V et al. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 2018; 50 (12): 165. doi: 10.1038/s12276-018-0191-1.
13. Broto JM, Hindi N, Redondo A et al. IMMUNOSARC: A collaborative Spanish (GEIS) and Italian (ISG) Sarcoma Groups phase I/II trial of sunitinib plus nivolumab in selected bone and soft tissue sarcoma subtypes – results of the phase I part. Ann Oncol 2019; 30 (Suppl 5): v683-v709. doi: 10.1093/annonc/mdz283.
14. Dufresne A, Brahmi M, Karanian M et al. Using biology to guide the treatment of sarcomas and aggressive connective-tissue tumours. Nat Rev Clin Oncol 2018; 15 (7): 443–458. doi: 10.1038/s41571-018-0012-4.
15. Knezevich SR, McFadden DE, Tao W et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998; 18 (2): 184–187. doi: 10.1038/ng0298-184.
16. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018; 15 (12): 731–747. doi: 10.1038/s41571-018-0113-0.
17. DuBois SG, Laetsch TW, Federman N et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer 2018; 124 (21): 4241–4247. doi: 10.1002/cncr.31701.
18. Davis JL, Lockwood CM, Albert CM et al. Infantile NTRK-associated mesenchymal tumors. Pediatr Dev Pathol 2018; 21 (1): 68–78. doi: 10.1177/1093526617712639.
19. Papadopoulos N, Lennartsson J. The PDGF/PDGFR pathway as a drug target. Mol Aspects Med 2018; 62 : 75–88. doi: 10.1016/j.mam.2017.11.007.
20. Cheung YH, Gayden T, Campeau PM et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 2013; 92 (6): 996–1000. doi: 10.1016/ j.ajhg.2013.04.026.
21. Martignetti JA, Tian L, Li D et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet 2013; 92 (6): 1001–1007. doi: 10.1016/ j.ajhg.2013.04.024.
22. Mudry P, Slaby O, Neradil J et al. Case report: rapid and durable response to PDGFR targeted therapy in a child with refractory multiple infantile myofibromatosis and a heterozygous germline mutation of the PDGFRB gene. BMC Cancer 2017; 17 (1): 119. doi: 10.1186/s12885-017-3115-x.
23. Noujaim J, Thway K, Fisher C et al. Dermatofibrosarcoma protuberans: from translocation to targeted therapy. Cancer Biol Med 2015; 12 (4): 375–384. doi: 10.7497/ j.issn.2095-3941.2015.0067.
24. Lowery CD, Blosser W, Dowless M et al. Olaratumab exerts antitumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor α. Clin Cancer Res 2018; 24 (4): 847–857. doi: 10.1158/1078-0432.CCR-17-1258.
25. Antoniou G, Lee AT, Huang PH et al. Olaratumab in soft tissue sarcoma – current status and future perspectives. Eur J Cancer 2018; 92 : 33–39. doi: 10.1016/ j.ejca.2017.12.026.
26. Meyers PA, Chou AJ. Muramyl tripeptide-phosphatidyl ethanolamine encapsulated in liposomes (L-MTP-PE) in the treatment of osteosarcoma. Adv Exp Med Biol 2014; 804 : 307–321. doi: 10.1007/978-3-319-04843-7_17.
27. Baumhoer D. Die klonale Evolution des Osteosarkoms. Pathologe 2016; 37 (Suppl 2): 163–168. doi: 10.1007/s00292-016-0200-x.
28. Kovac M, Blattmann C, Ribi S et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun 2015; 6 : 8940. doi: 10.1038/ncomms9940.
29. Loh AH, Stewart E, Bradley CL. Combinatorial screening using orthotopic patient derived xenograft-expanded early phase cultures of osteosarcoma identify novel therapeutic drug combinations. Cancer Letters 2019; 442 : 262–270. doi: 10.1016/j.canlet.2018.10.033.
30. Balachandran VP, Gonen M, Smith JJ et al. Nomograms in oncology: more than meets the eye. Lancet Oncol 2015; 16 (4): 173–180. doi: 10.1016/S1470-2045 (14) 71116-7.
31. Eilber FC, Kattan MW. Sarcoma nomogram: validation and a model to evaluate impact of therapy. J Am Coll Surg 2007; 205 (Suppl 4): S90–S95. doi: 10.1016/j.jamcollsurg.2007.06.335.
32. Tattersall HL, Callegaro D, Ford SJ et al. Staging, nomograms and other predictive tools in retroperitoneal soft tissue sarcoma. Chin Clin Oncol 2018; 7 (4): 36. doi: 10.21037/cco.2018.08.01.
33. Anaya DA, Lahat G, Wang X et al. Postoperative nomogram for survival of patients with retroperitoneal sarcoma treated with curative intent. Ann Oncol 2009; 21 (2): 397–402. doi: 10.1093/annonc/mdp298.
34. Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol 2002; 20 (3): 791–796. doi: 10.1200/JCO.2002.20.3.791.
35. Canter RJ, Qin LX, Maki RG et al. A synovial sarcoma-specific preoperative nomogram supports a survival benefit to ifosfamide-based chemotherapy and improves risk stratification for patients. Clin Cancer Res 2008; 14 (24): 8191–8197. doi: 10.1158/1078-0432.CCR-08-0843.
36. Zivanovic O, Jacks LM, Iasonos A. et al. A nomogram to predict postresection 5-year overall survival for patients with uterine leiomyosarcoma. Cancer 2012; 118 (3): 660–669. doi: 10.1002/cncr.26333.
37. Dalal KM, Kattan MW, Antonescu CR et al. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg 2006; 244 (3): 381–391. doi: 10.1097/01.sla.0000234795.98607.00.
38. Gronchi A, Miceli R, Shurell E et al. Outcome prediction in primary resected retroperitoneal soft tissue sarcoma: histology-specific overall survival and disease-free survival nomograms built on major sarcoma center data sets. J Clin Oncol 2013; 31 (13): 1649–1655. doi: 10.1200/JCO.2012.44.3747.
39. Callegaro D, Miceli R, Bonvalot S et al. Development and external validation of two nomograms to predict overall survival and occurrence of distant metastases in adults after surgical resection of localised soft-tissue sarcomas of the extremities: a retrospective analysis. Lancet Oncol 2016; 17 (5): 671–680. doi: 10.1016/S1470-2045 (16) 00010-3.
40. Raut CP, Miceli R, Strauss DC et al. External validation of a multi-institutional retroperitoneal sarcoma nomogram. Cancer 2016; 122 (9): 1417–1424. doi: 10.1002/cncr.29931.
41. Woll PJ, Reichardt P, Le Cesne A et al. Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 2012; 13 (10): 1045–1054. doi: 10.1016/S1470-2045 (12) 70346-7.
42. Hatina J, Hájková L, Peychl J et al. Establishment and characterization of clonal cell lines derived from a fibrosarcoma of the H2-K/v-jun transgenic mouse. Tumour Biol 2003; 24 (4): 176–184. doi: 10.1159/000074427.
43. Peychl J, Hatina J, Reischig J et al. Vztah motility a invazivity transformovaných buněk-model H2-K/V-JUN fibrosarkomových buněčných linií. Klin Onkol 2003; 16 (5): 223–226.
44. Mariani O, Brennetot C, Coindre JM et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer Cell 2007; 11 (4): 361–374. doi: 10.1016/j.ccr.2007. 02.007.
45. Endo M, Nishita M, Fujii M et al. Insight into the role of Wnt5a-induced signaling in normal and cancer cells. Int Rev Cell Mol Biol 2015; 314 : 117–148. doi: 10.1016/ bs.ircmb.2014.10.003.
46. Qiang YW, Hu B, Chen Y et al. Bortezomib induces osteoblast differentiation via Wnt-independent activation of beta-catenin/TCF signaling. Blood 2009; 113 (18): 4319–4330. doi: 10.1182/blood-2008-08-174300.
47. Witz IP. Tumor–microenvironment interactions: dangerous liaisons. Adv Cancer Res 2008; 100 : 203–229. doi: 10.1016/S0065-230X (08) 00007-9.
48. Brodin BA, Wennerberg K, Lidbrink E et al. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br J Cancer 2019; 120 (4): 435–443. doi: 10.1038/s41416-018-0359-4.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2020 Issue 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Nežádoucí účinky a efekt imunoterapie
- Neurotoxicita a imunoterapie
- Důležitá role STAT3 v biologii chronické lymfocytární leukemie
- Toxicita imunoonkologické léčby – myokarditida