
Molekulární podstata mnohočetného myelomu
Molecular basis of multiple myeloma
Background: Multiple myeloma (MM) is a heterogeneous hematological malignancy characterized by clonal expansion of malignant plasma cells in the bone marrow. The disease is accompanied by various clinical manifestations, such as bone lesions, anemia, hypercalcemia, and renal insufficiency. However, despite significant advances in treatment over the last two decades, the disease remains challenging to treat, and most patients relapse. Although its pathogenesis has not yet been elucidated, it is clear that genomic instability plays a key role in its development or resistance to treatment. In some instances, the cause of this instability is chromothripsis, a form of complex genomic rearrangement that involves shattering and subsequent haphazard reassembly of chromosomes within a single catastrophic event. The resulting rearrangements involve a variety of structural changes, including deletions, duplications, inversions, and translocations, that lead to genome disruption. Specifically, these changes may result in alteration or inactivation of tumor suppressor genes (TP53 and CDKN2C), activation of oncogenes (MAF, FGFR3, and CCND1) or genes involved in key cellular processes. Unraveling the mechanisms that result in chromothripsis provides opportunities to identify critical genes and pathways involved in MM pathogenesis. These findings may serve as a basis for improved diagnostic approaches. Purpose: The goal of this review is to summarize the common primary and secondary chromosomal aberrations in MM with a particular focus on introducing complex chromosomal aberrations, especially chromothripsis in MM.
Keywords:
chromothripsis – chromosome aberrations – Multiple myeloma
Authors:
D. Nižňanská 1; M. Vlachová 1; J. Gregorová 1; J. Kotašková 2; M. Jarošová 2; S. Ševčíková 1
Authors‘ workplace:
Babákova myelomová skupina, Ústav patologické fyziologie, LF MU Brno
1; Centrum molekulární biologie, a genetiky, Interní hematologická a onkologická klinika LF MU a FN Brno
2
Published in:
Klin Onkol 2024; 37(1): 27-33
Category:
Reviews
doi:
https://doi.org/10.48095/ccko202427
Overview
Východiska: Mnohočetný myelom (MM) je heterogenní hematoonkologické onemocnění charakterizované klonální expanzí maligních plazmatických buněk v kostní dřeni. Onemocnění je doprovázeno různými klinickými projevy, jedná se o kostní léze, anemii, hyperkalcemii a renální insuficienci. Navzdory značnému pokroku v léčbě v posledních dvou dekádách však zůstává onemocněním těžko léčitelným a většina pacientů relabuje. Ačkoliv dosud nebyla objasněna jeho patogeneze, je zřejmé, že genomová nestabilita hraje klíčovou roli v jeho rozvoji či v rezistenci na léčbu. V některých případech je příčinou této nestability chromotripse, tedy druh komplexní genomové přestavby, která zahrnuje rozsáhlou fragmentaci a opětovné náhodné spojení chromozomů během jediné katastrofické události. Výsledné přestavby zahrnují různé strukturální změny vč. delecí, duplikací, inverzí a translokací, což vede k narušení genomu, konkrétně např. k alteraci nebo inaktivaci tumor supresorových genů (TP53 a CDKN2C), aktivaci onkogenů (MAF, FGFR3 a CCND1) nebo genů zapojených do klíčových buněčných procesů. Odhalení mechanizmů, které vedou k chromotripsi, nabízí možnosti identifikace kritických genů a drah, které se podílejí na patogenezi MM. Tyto poznatky mohou být podkladem pro zlepšení diagnostických přístupů. Cíl: Cílem přehledového článku je shrnout časté primární a sekundární chromozomové aberace u MM a kromě toho představit komplexní chromozomové aberace s důrazem na chromotripsi u MM.
Klíčová slova:
chromotripse – mnohočetný myelom – chromozomové aberace
Úvod
Mnohočetný myelom (MM) je hematoonkologické onemocnění, k němuž dochází v důsledku maligní transformace plazmatických buněk, které nekontrolovatelně proliferují a akumulují se v kostní dřeni. Výchozím bodem hematopoézy plazmatických buněk jsou pluripotentní hematopoetické kmenové buňky, z nichž v tomto případě vznikají lymfoidní progenitory, které dále diferencují do stadia B lymfocytů. B lymfocyty v první fázi svého vývoje (stadium nezralého B lymfocytu), která probíhá v kostní dřeni bez setkání s antigenem, exprimují v procesu V(D)J rekombinace různé varianty B-buněčného receptoru schopného rozpoznávat konkrétní antigen. Pro konečný vývoj B lymfocytů je však nezbytné jejich setkání s antigenem v sekundárních lymfoidních orgánech, kdy dochází k jejich vývoji v paměťové B lymfocyty nebo plazmatické buňky. Plazmatické buňky (plasma cells – PC) jsou tedy specializované buňky hrající klíčovou roli v humorální imunitě díky produkci protilátek neboli imunoglobulinů [1–3].
MM byl poprvé popsán v roce 1844 lékařem Samuelem Sollym, v té době byl nazývaný mollities ossium. Článek pojednával o devětatřicetileté ženě Sarah Newbury, u které se onemocnění projevovalo prudkou bolestí v zádech a následně i v jiných částech těla. Pacientka zůstala připoutaná na lůžko, na pohled měla mnohé deformity skeletu, které vznikly zlomením, popř. ohnutím kostí. Během jejího pobytu v nemocnici jí byly podávány rebarborové pilulky, pomerančová kůra a opiáty. Po její náhlé smrti byla odebrána moč, ve které se ve vysoké míře nacházel fosforečnan vápenatý. Posmrtný popis se věnoval hlavně charakteristice kostí, které byly měkké a vyplněné dutinami obsahujícími červenou želatinovou hmotu. V některých částech stehenní kosti nebyla kostní tkáň přítomna vůbec, zůstala jen okostice. Mikroskopické pozorování odhalilo přítomnost nepravidelných buněk se zřetelným jádrem, tehdy označovaných jako jaderné buňky maligního onemocnění. Fosforečnan vápenatý byl nalezen v částech ledvin [4].
Analýzu moči provedl Henry Bence Jones, který identifikoval specifický protein pro mollities ossium [5], přičemž bylo později zjištěno, že se jedná o lehký řetězec imunoglobulinu G (IgG) [6]. Charakteristika myelomových buněk vycházela z podrobného popisu histologických preparátů zasažených míst v hrudní kosti, žebrech, obratlech a lebce. Myelomové buňky byly klasifikovány jako různě velké buňky s excentrickým jedním nebo více jádry, vyplněné kondenzovaným chromatinem rozděleným do více menších oblastí a homogenní cytoplazmou. V tomto článku byla uvedena první zmínka o podobnosti mezi myelomovými a plazmatickými buňkami a také o tom, že myelomové buňky mohly vzniknout z plazmatických [7].
Příčiny vzniku MM nejsou stále objasněny [8]. Téměř všechny případy MM se vyvinou z asymptomatického premaligního stadia nazývaného monoklonální gamapatie nejistého významu (monoclonal gamapathy of undetermined significance – MGUS) [9,10]. MGUS je přítomna přibližně u 5 % populace ve věku nad 50 let. Vzhledem k tomu, že MGUS je asymptomatické onemocnění, více než 50 % jedinců, u kterých je diagnostikována MGUS, mělo tento stav více než 10 let před klinickou diagnózou [11]. U některých pacientů lze klinicky rozpoznat asymptomatické, ale pokročilejší premaligní stadium označované jako doutnající mnohočetný myelom (smoldering multiple myeloma – SMM). Během prvních 5 let od diagnózy SMM je roční riziko progrese do MM cca 10 %. Rychlost progrese je ovlivněna cytogenetickými změnami: pacienti s translokací t(4;14), del(17p) a ziskem na chromozomu 1 (1q) mají vyšší riziko progrese z MGUS nebo SMM do MM [12].
Epidemiologie v ČR
MM patří mezi nejčastější hematoonkologická onemocnění, ročně je v ČR diagnostikováno přibližně 500 nových případů. Data z roku 2018 uvádějí incidenci 5,3/ 100 000, nejčastěji jsou zastoupeni pacienti ve věkové kategorii 65–74 let. Medián věku při diagnóze je 70 let. Prevalence onemocnění podobně jako jeho incidence v posledních letech roste. Pětileté přežití léčených pacientů se však zvýšilo na 44 %, což je nárůst o více než 8 % od minulého statistického období (2009–2013) [13].
![Revidovaný mezinárodní prognostický index mnohočetného myelomu. Upraveno podle [7].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/6ab21aa2104ad81a22f74873b9067bcb.png)
Klasifikace
MM je heterogenní onemocnění, délka přežití se pohybuje od několika měsíců až po ≥ 10 let. Tato variabilita se odvíjí od míry poškození genomu myelomových buněk a individuálních faktorů [14]. V současnosti se využívá Revidovaný mezinárodní klasifikační systém (Revised International Staging System – R-ISS), který je založen na koncentraci sérového albuminu a β2-mikroglobulinu v periferní krvi. Kromě toho jsou hodnoceny koncentrace laktátdehydrogenázy (LDH) a cytogenetické aberace (CA) myelomových buněk (tab. 1). Za vysoce rizikové CA jsou považovány del(17p), translokace t(4;14) a translokace t(14;16). Ostatní nálezy jsou spojeny se standardním rizikem [14].
Diagnostika
V minulosti bylo stanovení diagnózy MM možné až po klinickém projevu poškození koncových orgánů charakterizovaných tzv. CRAB symptomy (C – hyperkalcemie, R – renální poškození, A – anemie, B – kostní léze) [15]. S pokroky v diagnostických technikách došlo k časnějšímu stanovení diagnózy a zahájení terapie; kromě toho byly identifikovány biomarkery, které se vyskytují již ve dřívějších stadiích onemocnění. Současná diagnostická kritéria pro MM jsou: počet PC v kostní dřeni ≥ 10 % nebo biopticky potvrzený kostní nebo extramedulární plazmocytom a jeden nebo více nálezů definujících myelom (tab. 2) [16].
![Diagnostická kritéria mnohočetného myelomu. Upraveno podle [14].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/e96efbc76e4cb7c85301f954d9e87426.png)
Cytogenetické aberace u MM
MM je charakterizován rozsáhlou genetickou heterogenitou. K detekci genetických aberací se standardně využívají klasická cytogenetická vyšetření kostní dřeně, metoda interfázní fluorescenční in situ hybridizace (i-FISH) na separovaných PC nebo FISH na PC značených protilátkami proti lehkým řetězcům Ig (cIg FISH). Doplňkovou metodou může být komparativní genomová hybridizace na DNA mikročipech [17]. Genetické aberace jsou rozděleny na primární a sekundární.
Primární chromozomové aberace MM
Primární klonální aberace jsou rozděleny do dvou podskupin: hyperdiploidní (48–74 chromozomů) a non-hyperdiploidní (< 48 nebo > 78 chromozomů). Jako primární aberace jsou označovány počáteční události, které jsou přítomny již ve stadiu MGUS [18]. Tyto aberace se vyskytují v celé populaci klonálních maligních plazmatických buněk na rozdíl od sekundárních aberací, které se často vyskytují v subklonech [19,20].
Hyperdiploidní karyotyp je přítomný u 45–50 % případů MM. Zahrnuje trizomii lichých chromozomů, nejčastěji se jedná o chromozomy 3, 5, 7, 9, 11, 15, 19 a 21. Obecně jsou hyperdiploidie považovány za znak spojený s dobrou prognózou, nicméně příznivá prognóza byla prokázána pouze pro trizomie chromozomu 3 a 5, naopak nepříznivá prognóza byla potvrzena u trizomie chromozomu 21 [21].
Non-hyperdiploidní karyotyp se vyskytuje u 55 % pacientů s MM. Patří sem translokace chromozomu 14 zahrnující lokus pro těžký řetězec imunoglobulinu (IGH) lokalizovaný v oblasti 14q32. Výsledkem translokace je přeskupení eventuálních onkogenů do oblasti zesilovače IGH, což způsobí zvýšenou expresi těchto genů [22]. Nejčastěji je pozorována translokace t(11;14)(q13;q32), která se vyskytuje u 13–20 % pacientů s MM [23]. Méně frekventovaná je t(4;14)(p16;q32), kterou má 10–15 % pacientů. Ostatní translokace, vč. t(14;16)(q32;q23), t(14;20)(q32;q12) a t(6;14)(p21;q32), se vyskytují s prevalencí jen 2–5 % [24].
Translokace t(11;14) indukuje zvýšenou expresi genu pro cyklin D1 (CCND1) [25], což urychluje přechod plazmatických buněk z G1 fáze do S fáze [26]. Několik studií potvrdilo koexistenci této aberace spolu s aberacemi chromozomu 13 u více než 30 % pacientů [27,28].
Translokace t(4;14) způsobuje přeskupení genů pro methyltransferázu (MMSET) a receptor fibroblastového růstového faktoru 3 (FGFR3) do blízkosti zesilovače IGH, což způsobí jejich simultánní nadměrnou expresi. Onkogenní charakter MMSET byl prokázán pomocí snížené exprese MMSET, kdy v buňkách s t(4;14) nedocházelo k proliferaci, tumorigenezi ani ke klonogennímu růstu [29]. Přítomnost translokace t(4;14) predikuje horší prognózu [30,31].
Translokace t(14;16) a t(14;20) vedou k nadměrné expresi genu pro transkripční faktor MAF, který nepřímo ovlivňuje gen pro cyklin D2 (CCND2) [22]. Přímo cyklin D2 je pak partnerem druhé translokace. Bylo zjištěno, že uvedené translokace jsou doprovázeny mutacemi APOBEC genů, v důsledku čehož jsou asociovány s nepříznivou
prognózou [32].
Následkem t(6;14) je nadměrná exprese genu pro cyklin D3 (CCND3) [33]. Všechny výše uvedené translokace, které vedou k deregulaci cyklinů, jsou podle nově navržené mezinárodní klasifikace International Consensus Classification (ICC) zahrnuty do jedné podskupiny [34].
Sekundární chromozomové aberace MM
Sekundární aberace jsou zodpovědné za přechod onemocnění do stadia MM. Jejich frekvence se postupně zvyšuje spolu s rozvojem onemocnění [35]. Mezi nejběžnější patří deregulace genu MYC, která je přítomna až u 55 % pacientů s MM [22]. Jednou z častých subklonálních aberací je translokace t(8;22) genu pro λ IgL ke genu MYC. Přítomnost této translokace má výrazně horší prognózu než ostatní IgL amplifikace a translokace [36].
Dále jsou do této skupiny řazeny strukturní aberace chromozomů – delece dlouhého ramene chromozomu 13 (del(13q)), zisk dlouhého ramene chromozomu 1 (amp1q)) a delece krátkého ramene chromozomu 17 (del(17p)). Ztráta 13q vede ke snížení exprese několika genů, zejména genu RB1, což je důležitý tumor supresorový gen [37]. Důsledkem zisku 1q jsou nadbytečné kopie genu CKS1B zodpovědné za aktivaci růstu myelomových buněk i rezistenci vůči některým lékům [38]. Zisk 1q je proto popisován jako nepříznivý genetický prognostický faktor [39]. Delece krátkého ramene chromozomu 1 zahrnující oblast 1p32 je považována za deleci s negativním prediktivním významem. V oblasti jsou lokalizovány geny jako FAF1 zahrnutý do iniciace apoptózy nebo CDKN2C, který brání progresi buněčného cyklu. Delece těchto genů by mohla podporovat tumorigenezi a analýza uskutečněná na souboru 2 551 pacientů s MM ukázala, že delece se vyskytuje v 11 % a je negativním, nezávislým prognostickým faktorem [40].
Del(17p) způsobuje snížení aktivity tumor supresorového genu TP53. Často se vyskytuje jako double hit mutace, kdy je na jedné alele chromozomu 17 přítomna delece genu TP53 a na druhé mutace, což způsobí úplnou deaktivaci genu TP53, přičemž bialelická inaktivace je spojena s horší prognózou [41,42].
Dalším typem sekundárních aberací jsou rekurentní mutace genů KRAS, NRAS, BRAF aktivujících signální dráhy MAPK a NF-κB. Tyto mutace v myelomových buňkách způsobují zvýšení proliferace, diferenciace a progresi onemocnění [43].
Komplexní genetické aberace – chromoanageneze
Pomocí metody celogenomového sekvenování (whole-genome sequencing – WGS) je možné analyzovat aberace v celém genomu [44]. WGS u pacientů s MM odhalilo, že některé rekurentní translokace, které byly původně považovány za jednoduché a reciproké, jsou ve skutečnosti často komplexní události zahrnující více než dvě chromozomové oblasti [44].
Obecně jsou rozeznávány tři hlavní komplexní události – chromoanasyntéza, chromoplexe a chromotripse – které jsou souhrnně označovány pojmem chromoanageneze (obr. 1) [45,46].
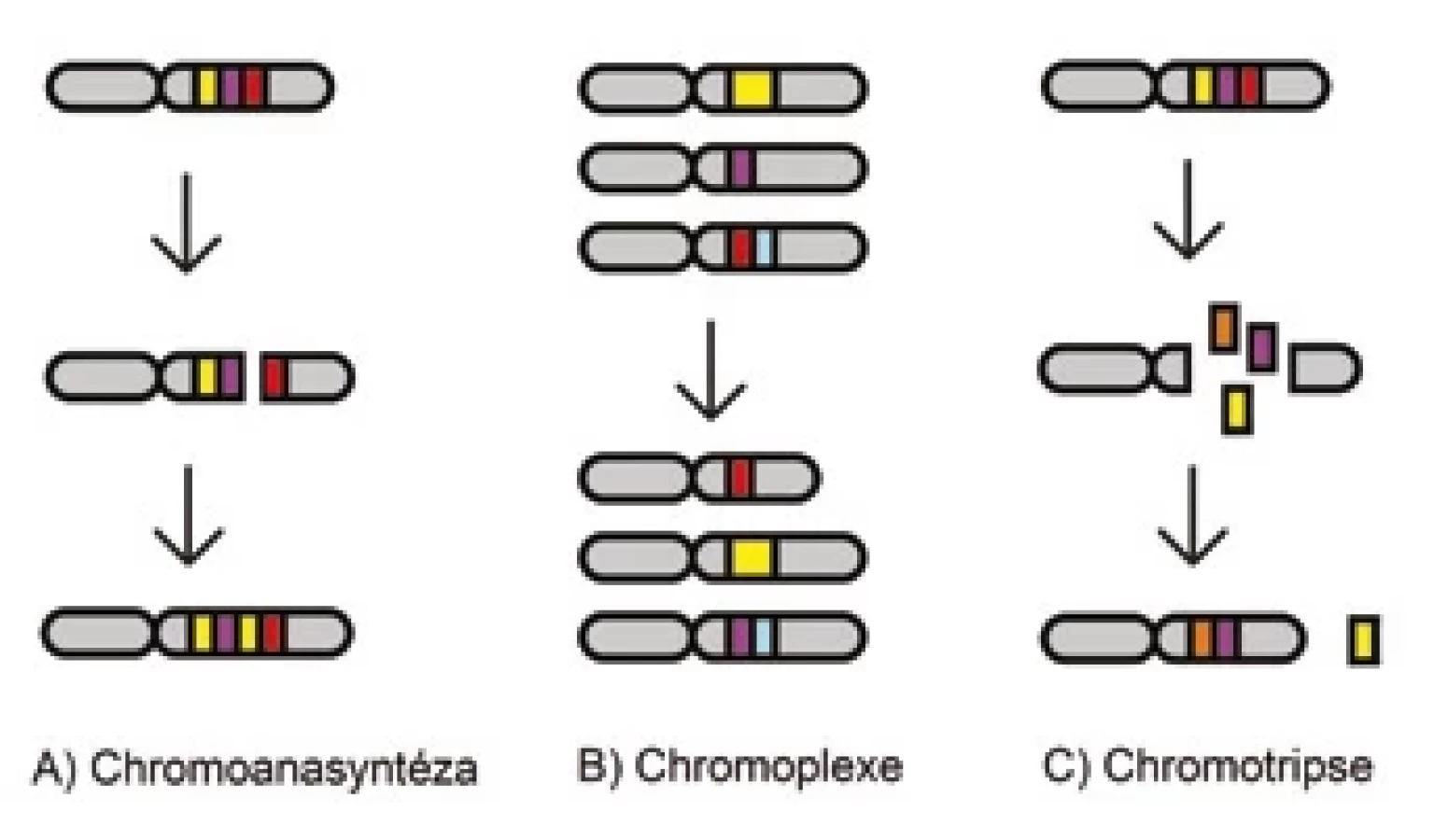
Chromoanasyntéza
Chromoanasyntéza je komplexní přeskupení závislé na poruše v replikačním procesu (obr. 1A). Během chromoanasyntézy dochází k oddělení zaostávajícího vlákna z defektní replikační vidlice a k jejímu připojení k jiné prostorově blízké intrachromozomové nebo interchromozomové oblasti pomocí mikrohomologických úseků a obnovení replikační vidlice. Tento mechanizmus vede většinou k tvorbě inzercí, duplikací a triplikací v kombinaci s delecemi i početně neutrálními segmenty [47,48].
Chromoplexe
Chromoplexe je další typ komplexního přeskupení genomu, do kterého bývá zapojeno několik chromozomů (obr. 1B). Je charakteristická akumulací translokací někdy až šesti chromozomů současně, které mohou způsobit deleci některých úseků. Nicméně výsledné přeskupení vykazuje jen mírné nebo žádné změny v počtu kopií [49,50].
Chromotripse
Chromotripse je definována jako rozpad jednoho nebo více chromozomů na desítky až stovky částí během jedné katastrofické události (obr. 1C) [51]. Následná reparace probíhá v náhodném pořadí a orientaci, většinou pomocí nehomologního nebo alternativního spojování konců [52]. Chromotripse zpravidla způsobuje četné zlomy jen v určitých sousedících oblastech na jedné chromatidě; ostatní části chromozomu nejsou zasaženy. Vznikají komplexní změny jako ztráta heterozygotnosti, vytvoření fúzních genů nebo snížení exprese nádorových supresorů. Výsledkem může být také delece některých fragmentů nebo vytvoření tzv. double-minute chromozomů, což jsou extrachromozomové kruhové struktury DNA, které mohou obsahovat onkogeny (např. MYC), a tím poskytovat nádorovým buňkám proliferační výhodu [53,54].
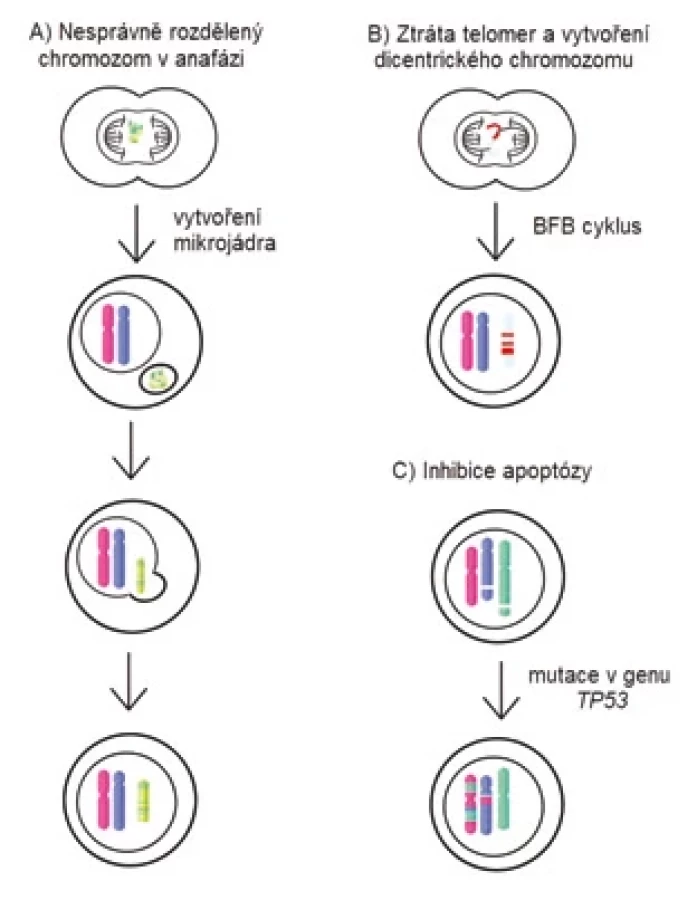
Příčina vzniku chromotripse nebyla dosud objasněna. Zpočátku se předpokládalo, že vzhledem k mnohočetnosti zlomů ve dvouřetězcové DNA by příčinou mohly být exogenní stresové faktory [55]. V současnosti je však nejvíce uznávaný model, který je založen na rozpadu chromozomů v mikrojádru (obr. 2A). Mikrojádra se vytvářejí jako důsledek zaostání některých chromozomů nebo jejich částí při přechodu buňky z metafáze do anafáze. Tyto chromozomy jsou odděleny od vzniklých dceřiných jader. Jsou obaleny jadernou membránou a existují jako mimojaderné struktury [56]. Některé procesy v mikrojádrech probíhají pomaleji nebo neprobíhají vůbec. Chromozomy v mikrojádrech se nedostatečně replikují a vykazují značné známky poškození kvůli nedostatku potřebných proteinů na replikaci a opravu DNA [57]. DNA replikace v mikrojádře je vůči replikaci v primárním jádře asynchronní. Buňka tedy může zahájit dělení, ačkoliv replikace v mikrojádře ještě není dokončena, což v něm způsobí rozsáhlé poškození DNA [58]. Osud mikrojaderné DNA může být různý. Buď se tato poškozená DNA během následujících dělení začlení zpět do jednoho z dceřiných jader, nebo během S fáze buněčného cyklu dojde ke zničení membrány mikrojádra, a tím k ztrátě veškeré DNA, kterou obsahovala [56].
Dalším z možných mechanizmů vzniku chromotripse může být vytvoření tzv. breakage-fusion-bridge cyklu, kde dochází ke tvorbě chromozomového můstku (obr. 2B). Můstek vzniká spojením konců chromozomů po zlomu DNA, nekompletní DNA replikaci nebo při kriticky zkrácených telomerách [59]. Vytvoření chromozomového můstku způsobí vznik dicentrického chromozomu, který se v anafázi nedokáže normálně oddělit do dceřiných buněk. Chromozomový můstek mezi dceřinými buňkami je zachován až do interfáze a poté je zničen pomocí cytoplazmatické 3‘-exonukleázy TREX1 [60]. Toto může vést k četným ztrátám a inverzím chromozomových fragmentů a také ke tvorbě double-minute chromozomů [58].
Další zmiňovaný mechanizmus vzniku chromotripse je spojován s mutací v genu TP53 (obr. 2C). Koexistence mutace v TP53 a chromotripse byla prokázána u mnohých typů nádorů [61]. Ve studii z roku 2022 byla prevalence chromotripse u MM vyšší u pacientů s mutací v genu TP53, nicméně se vyskytovala i u pacientů s funkčním genem [62,63].
Chromotripse u MM
Nejrozšířenější komplexní událostí u MM je chromotripse [45]. Původně se předpokládalo, že se chromotripse objevuje jen u 2–3 % všech nádorových onemocnění [51]. Techniky jako WGS však detekovaly chromotripsi a jiná chromozomová přeskupení v celé řadě nádorů. Dvě nedávné studie prokázaly 30–50% prevalenci chromotripse napříč více typy nádorů, což naznačuje, že se může jednat o jeden z hlavních procesů, které jsou zodpovědné za rozvoj některých nádorových onemocnění [64,65]. Vyšší prevalence byla zjištěna u solidních a agresivních nádorů, např. u liposarkomu dosahovala až 100 %. U hematologických nádorů je obecně nízká prevalence chromotripse [64], avšak u MM byla potvrzena až u 24 % nově diagnostikovaných pacientů [44]. Chromotripse u MM, v porovnání se solidními nádory, se považuje za ranou událost, která zřejmě vznikla ještě v germinálním centru. Tato domněnka je podporována několika skutečnostmi. Jednak bývá chromotripse detekovatelná již v premaligních stadiích [66], dále bylo také zjištěno, že k nárůstu počtu kopií asociovaných s chromotripsí pravděpodobně dochází dříve během života pacientů [35]. Dále se zjistilo, že chromotripse bývá stabilní v průběhu času, což znamená, že po progresi z prekurzorového stadia do MM i při relapsu nejsou pozorovány významné strukturní nebo početní změny. Chromotripse se také zřídka objevuje při pozdních stadiích onemocnění [49]. Taktéž chromotripse u MM i jiných hematologických nádorů je obvykle jednodušší než u solidních nádorů. Dochází k menšímu počtu dvouvláknových zlomů, tedy celkový počet strukturních přestaveb i nárůst počtu kopií v oblasti, kde došlo ke chromotripsi, je nižší. Rovněž tvorba double-minute chromozomů je méně častá než u solidních nádorů [67,68].
Nepříznivý prognostický dopad chromotripse zjištěný u MM je podobný jako u jiných hematoonkologických onemocnění [69]. Přítomnost chromotripse je asociována se známými vysoce rizikovými markery MM, zahrnujícími vysokou mutační nálož APOBEC (z angl. apolipoprotein B mRNA editing enzyme, catalytic polypeptide) genů, deaktivaci genu TP53 a translokace genů MMSET a MAF, což naznačuje její využití jako silný a nezávislý prognostický faktor [62].
Moderní diagnostika MM se postupně přesouvá od vyšetření karyotypu a vybraného panelu chromozomových aberací pomocí FISH k pokročilým molekulárně biologickým technikám umožňujícím identifikovat strukturní a početní změny napříč celým genomem, vč. identifikace mutací v klíčových genech. Komplexní genetická analýza zahrnující určení přítomnosti a rozsahu chromotripse u nemocných s MM pak poskytuje nový pohled na mechanizmus vzniku a vývoje MM, ale i na mechanizmus vzniku amplifikace onkogenů a ztráty nádorových supresorů. Ukazuje, jak může být deregulováno mnoho genů jedinou genetickou událostí, která ve svém důsledku významně ovlivní prognózu a klinické chování onemocnění. Všechny tyto znalosti mohou také významným způsobem ovlivnit celkový pohled na onemocnění, stejně jako koncepci a strategii léčby.
Podporující agentury
Publikace vznikla díky podpoře grantu AZV NU21-03-00076 a LF MUNI/ A/ 1370/ 2022 a Projektem Národní ústav pro výzkum rakoviny (Program EXCELES, číslo projektu: LX22NPO5102) – Financováno Evropskou unií – Next Generation EU. Podpořeno MZ ČR – RVO (FNBr, 65269705).
doc. RNDr. Sabina Ševčíková, Ph.D.
Babákova myelomová skupina
Ústav patologické fyziologie,
LF MU Brno
Kamenice 5
625 00 Brno
e-mail: sevcik@med.muni.cz
Obdrženo/Submitted: 14. 8. 2023
Přijato/Accepted: 14. 12. 2023
Sources
1. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol 2013; 131(4): 959–971. doi: 10.1016/ j.jaci.2013.01.046.
2. Kurosaki T. B-lymphocyte biology. Immunol Rev 2010; 237(1): 5–9. doi: 10.1111/ j.1600-065X.2010.00946.x.
3. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol 2015; 15(3): 137–148. doi: 10.1038/ nri3804.
4. Solly S. Remarks on the pathology of mollities ossium; with cases. Med Chir Trans 1844; 27 : 435–498.8. doi: 10.1177/ 095952874402700129.
5. Jones HB. English: Original Paper Describing the Bence-Jones-Protein. 1848. [online]. Available from: https:/ / commons.wikimedia.org/ wiki/ File:Bence_Jones-On_a_New_Substance_Occurring_in_the_Urine.pdf.
6. Edelman GM, Gally JA. The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins. J Exp Med 1962; 116(2): 207–227. doi: 10.1084/ jem.116.2.
207.
7. Wright JH. A case of multiple myeloma. J Boston Soc Med Sci 1900; 4(8): 195–204.5.
8. Yang P, Qu Y, Wang M et al. Pathogenesis and treatment of multiple myeloma. MedComm 2022; 3(2): e146. doi: 10.1002/ mco2.146.
9. Landgren O, Kyle RA, Pfeiffer RM et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009; 113(22): 5412–5417. doi: 10.1182/ blood-2008-12-194241.
10. Abeykoon JP, Tawfiq RK, Kumar S et al. Monoclonal gammopathy of undetermined significance: evaluation, risk assessment, management, and beyond. Fac Rev 2022; 11 : 34. doi: 10.12703/ r/ 11-34.
11. Therneau TM, Kyle RA, Melton LJ et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc 2012; 87(11): 1071–1079. doi: 10.1016/ j.mayocp.2012.06.014.
12. Rajkumar SV, Gupta V, Fonseca R et al. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013; 27(8): 1738–1744. doi: 10.1038/ leu.2013.86.
13. Krejčí D, Pehalová L, Talábová A et al. Novotvary 2018 – Současné epidemiologické trendy novotvarů v České republice. [online]. Dostupné z: https:/ / www.uzis.cz/ index.php?pg=record&id=8352.
14. Palumbo A, Avet-Loiseau H, Oliva S et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol 2015; 33(26): 2863–2869. doi: 10.1200/ JCO.2015.61.2267.
15. Kumar SK, Rajkumar V, Kyle RA et al. Multiple myeloma. Nat Rev Dis Primers 2017; 3(1): 17046. doi: 10.1038/ nrdp.2017.46.
16. Rajkumar SV, Dimopoulos MA, Palumbo A et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12): e538–e548. doi: 10.1016/ S1470-2045(14)70442-5.
17. Crabtree M, Cai J, Qing X. Conventional karyotyping and fluorescence in situ hybridization for detection of chromosomal abnormalities in multiple myeloma. J Hematol 2022; 11(3): 87–91. doi: 10.14740/ jh1007.
18. Fonseca R, Bailey RJ, Ahmann GJ et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood 2002; 100(4): 1417–1424. doi: 10.1182/ blood.V100.4.1417.h81602001417_1417_1424.
19. Schmidt-Hieber M, Gutiérrez ML, Pérez-Andrés M et al. Cytogenetic profiles in multiple myeloma and monoclonal gammopathy of undetermined significance: a study in highly purified aberrant plasma cells. Haematologica 2013; 98(2): 279–287. doi: 10.3324/ haematol.2011.060632.
20. Yan Y, Qin X, Liu J et al. Clonal phylogeny and evolution of critical cytogenetic aberrations in multiple myeloma at single-cell level by QM-FISH. Blood Adv 2022; 6(2): 441–451. doi: 10.1182/ bloodadvances.2021004992.
21. Chretien ML, Corre J, Lauwers-Cances V et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood 2015; 126(25):
2713–2719. doi: 10.1182/ blood-2015-06-650242.
22. Walker BA, Wardell CP, Murison A et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun 2015; 6(1): 6997. doi: 10.1038/ ncomms7997.
23. Kuglík P, Filková H, Oltová A et al. Význam a současné možnosti diagnostiky cytogenetických změn u mnohočetného myelomu. Vnitr Lek 2006; 52 (Suppl 2): 76–78.
24. Moreau P, Facon T, Leleu X et al. Recurrent 14q32 translocations determine the prognosis of multiple myeloma, especially in patients receiving intensive chemotherapy. Blood 2002; 100(5): 1579–1583. doi: 10.1182/ blood-2002-03-0749.
25. Walker BA, Wardell CP, Johnson DC et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood 2013; 121(17):
3413–3419. doi: 10.1182/ blood-2012-12-471888.
26. Chesi M, Bergsagel PL, Brents LA et al. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood 1996; 88(2): 674–681.
27. Leiba M, Duek A, Amariglio N et al. Translocation t(11;14) in newly diagnosed patients with multiple myeloma: is it always favorable? Genes Chromosomes Cancer 2016; 55(9): 710–718. doi: 10.1002/ gcc.22372.
28. An G, Xu Y, Shi L et al. T(11;14) multiple myeloma: a subtype associated with distinct immunological features, immunophenotypic characteristics but divergent outcome. Leuk Res 2013; 37(10): 1251–1257. doi: 10.1016/ j.leukres.2013.06.020.
29. Lauring J, Abukhdeir AM, Konishi H et al. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 2008; 111(2): 856–864. doi: 10.1182/ blood-2007-05-088674.
30. Nemec P, Zemanova Z, Kuglik P et al. Complex karyotype and translocation t(4;14) define patients with high-risk newly diagnosed multiple myeloma: results of CMG2002 trial. Leuk Lymphoma 2012; 53(5): 920–927. doi: 10.3109/ 10428194.2011.634042.
31. Stong N, Ortiz-Estévez M, Towfic F et al. The location of the t(4;14) translocation breakpoint within the NSD2 gene identifies a subset of patients with high--risk NDMM. Blood 2023; 141(13): 1574–1583. doi: 10.1182/ blood.2022016212.
32. Hoang PH, Cornish AJ, Dobbins SE et al. Mutational processes contributing to the development of multiple myeloma. Blood Cancer J 2019; 9(8): 60. doi: 10.1038/ s41408-019-0221-9.
33. Shaughnessy J, Gabrea A, Qi Y et al. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood 2001; 98(1): 217–223. doi: 10.1182/ blood.V98.1.217.
34. Campo E, Jaffe ES, Cook JR et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 2022; 140(11): 1229–1253. doi: 10.1182/ blood.2022015851.
35. Oben B, Froyen G, Maclachlan KH et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun 2021; 12(1): 1861. doi: 10.1038/ s41467-021-22140-0.
36. Barwick BG, Neri P, Bahlis NJ et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun 2019; 10(1): 1911. doi: 10.1038/ s41467-019-09555-6.
37. Walker BA, Leone PE, Chiecchio L et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010; 116(15): e56–e65. doi: 10.1182/ blood-2010-04-279
596.
38. Shi L, Wang S, Zangari M et al. Over-expression of CKS1B activates both MEK/ ERK and JAK/ STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010; 1(1): 22–33. doi: 10.18632/ oncotarget.105.
39. Smetana J, Berankova K, Zaoralova R et al. Gain(1)(q21) is an unfavorable genetic prognostic factor for patients with relapsed multiple myeloma treated with thalidomide but not for those treated with bortezomib. Clin Lymphoma Myeloma Leuk 2013; 13(2): 123–130. doi: 10.1016/ j.clml.2012.11.012.
40. Schavgoulidze A, Talbot A, Perrot A et al. Biallelic deletion of 1p32 defines ultra-high-risk myeloma, but monoallelic del(1p32) remains a strong prognostic factor. Blood 2023; 141(11): 1308–1315. doi: 10.1182/ blood.2022017863.
41. Weinhold N, Ashby C, Rasche L et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016; 128(13):
1735–1744. doi: 10.1182/ blood-2016-06-723007.
42. Ansari-Pour N, Samur M, Flynt E et al. Whole-genome analysis identifies novel drivers and high-risk double--hit events in relapsed/ refractory myeloma. Blood 2023; 141(6): 620–633. doi: 10.1182/ blood.2022017010.
43. Shirazi F, Jones RJ, Singh RK et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplasmic reticulum stress in multiple myeloma. Proc Natl Acad Sci U S A 2020; 117(33):
20004–20014. doi: 10.1073/ pnas.2005052117.
44. Rustad EH, Yellapantula VD, Glodzik D et al. Revealing the impact of structural variants in multiple myeloma. Blood Cancer Discov 2020; 1(3): 258–273. doi: 10.1158/ 2643-3230.BCD-20-0132.
45. Holland AJ, Cleveland DW. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med 2012; 18(11): 1630–1638. doi: 10.1038/ nm.2988.
46. Guo W, Comai L, Henry IM. Chromoanagenesis in plants: triggers, mechanisms, and potential impact. Trends Genet 2023; 39(1): 34–45. doi: 10.1016/ j.tig.2022.08.003.
47. Liu P, Erez A, Nagamani SCS et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2011; 146(6): 889–903. doi: 10.1016/ j.cell.2011.07.042.
48. Berry NK, Dixon-McIver A, Scott RJ et al. Detection of complex genomic signatures associated with risk in plasma cell disorders. Cancer Genet 2017; 218–219 : 1–9. doi: 10.1016/ j.cancergen.2017.08.004.
49. Maura F, Bolli N, Angelopoulos N et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun 2019; 10(1): 3835. doi: 10.1038/ s41467-019-11680-1.
50. Baca SC, Prandi D, Lawrence MS et al. Punctuated evolution of prostate cancer genomes. Cell 2013; 153(3):
666–677. doi: 10.1016/ j.cell.2013.03.021.
51. Stephens PJ, Greenman CD, Fu B et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144(1):
27–40. doi: 10.1016/ j.cell.2010.11.055.
52. Ly P, Teitz LS, Kim DH et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 2017; 19(1): 68–75. doi: 10.1038/ ncb3450.
53. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell 2013; 152(6):
1226–1236. doi: 10.1016/ j.cell.2013.02.023.
54. Hadi K, Yao X, Behr JM et al. Distinct classes of complex structural variation uncovered across thousands of cancer genome graphs. Cell 2020; 183(1): 197–210.e32. doi: 10.1016/ j.cell.2020.08.006.
55. Stevens JB, Abdallah BY, Liu G et al. Diverse system stresses: common mechanisms of chromosome fragmentation. Cell Death Dis 2011; 2(6): e178. doi: 10.1038/ cddis.2011.60.
56. Zhang CZ, Spektor A, Cornils H et al. Chromothripsis from DNA damage in micronuclei. Nature 2015; 522(7555): 179–184. doi: 10.1038/ nature14493.
57. Liu S, Kwon M, Mannino M et al. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018; 561(7724): 551–555. doi: 10.1038/ s41586-018-0534-z.
58. Umbreit NT, Zhang CZ, Lynch LD et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020; 368(6488): eaba0712. doi: 10.1126/ science.aba0712.
59. Shimizu N, Shingaki K, Kaneko-Sasaguri Y et al. When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp Cell Res 2005; 302(2): 233–243. doi: 10.1016/ j.yexcr.2004.09.001.
60. Maciejowski J, Li Y, Bosco N et al. Chromothripsis and kataegis induced by telomere crisis. Cell 2015; 163(7): 1641–1654. doi: 10.1016/ j.cell.2015.11.054.
61. Aaltonen LA, Abascal F, Abeshouse A et al. Pan-cancer analysis of whole genomes. Nature 2020; 578(7793):
82–93. doi: 10.1038/ s41586-020-1969-6.
62. Ashby C, Boyle EM, Bauer MA et al. Structural variants shape the genomic landscape and clinical outcome of multiple myeloma. Blood Cancer J 2022; 12(5): 1–9. doi: 10.1038/ s41408-022-00673-x.
63. Shorokhova M, Nikolsky N, Grinchuk T. Chromothripsis-explosion in genetic science. Cells 2021; 10(5): 1102. doi: 10.3390/ cells10051102.
64. Cortés-Ciriano I, Lee JJK, Xi R et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 2020; 52(3):
331–341. doi: 10.1038/ s41588-019-0576-7.
65. Voronina N, Wong JKL, Hübschmann D et al. The landscape of chromothripsis across adult cancer types. Nat Commun 2020; 11(1): 2320. doi: 10.1038/ s41467-020-16134-7.
66. Holstein SA, Asimakopoulos F, Azab AK et al. Proceedings from the blood and marrow transplant clinical trials network myeloma intergroup workshop on immune and cellular therapy in multiple myeloma. Transplant Cell Ther 2022; 28(8): 446–454. doi: 10.1016/ j.jtct.2022.05.
019.
67. Maura F, Boyle EM, Rustad EH et al. Chromothripsis as a pathogenic driver of multiple myeloma. Semin Cell Dev Biol 2022; 123 : 115–123. doi: 10.1016/ j.semcdb.2021.04.014.
68. Maclachlan KH, Rustad EH, Derkach A et al. Copy number signatures predict chromothripsis and clinical outcomes in newly diagnosed multiple myeloma. Nat Commun 2021; 12(1): 5172. doi: 10.1038/ s41467-021-254
69-8.
69. Smetana J, Oppelt J, Štork M et al. Chromothripsis 18 in multiple myeloma patient with rapid extramedullary relapse. Mol Cytogenet 2018; 11 : 7. doi: 10.1186/ s13039-018-0357-5.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2024 Issue 1
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Molekulární podstata mnohočetného myelomu
- Střevní mikrobiota a karcinom pankreatu
- Analýza efektu detekce časné clearance ct-DNA při baseline na přežití u pacientů s pokročilým EGFR mutovaným nemalobuněčným karcinomem plic
- Převodní systém srdeční jako nový rizikový orgán v radioterapii