
Steelov syndróm – prvý prípad vzácnej kostnej dysplázie na Slovensku: kazuistika
Steel syndrome – the first case of rare skeletal dysplasia in Slovakia: a case report
Steel syndrome is an ultra-rare autosomal recessive disease affecting primarily the skeletal system. It is characterized by short stature, bilateral hip and radial head dislocations, carpal coalitions, scoliosis and pes cavus. Other features of the disease are facial dysmorphism, long oval face, prominent forehead, hypertelorism, macrocephaly, low-lying ears, brachydactyly or hearing loss. Steel syndrome is caused by mutations in the Col27A1 gene encoding the pro-alpha chain of fibrillar collagen type XXVII. We report an extremely rare case of an 8-year-old girl with mild skeletal dysplasia, constitutively delayed growth, hypertelorism and facial dysmorphism. In the patient, we identified the presence of two unknown heterozygous variants in the Col27A1 gene by whole exome sequencing: c.1741C>T, p.(Gln581Lys) and c.2858C>T, p. (Pro953Leu). At our department, we diagnosed the second case of Steel syndrome in Europe and at the same time it is the first case of this rare disease in the Slavic population.
Keywords:
Col27A1 gene – skeletal dysplasia – Steel syndrome – whole exome sequencing
Authors:
Oravcová Lucia 1; Skalická Katarína 1; Pribilincová Zuzana 1; Tichá Ubica 1; Hamidová Olívia 1; Ilčík Milan 2; Podracká Udmila 1
Authors‘ workplace:
Detská klinika LF UK a NÚDCH, Bratislava
1; Rádiologické oddelenie LF UK a NÚDCH, Bratislava
2
Published in:
Clinical Osteology 2021; 26(1): 29-34
Category:
Overview
Steelov syndróm je mimoriadne zriedkavé autosómovo-recesívne ochorenie postihujúce primárne skeletálny systém. Charakterizované je nízkym vzrastom, bilaterálnymi dislokáciami bedier a hlavy rádia, fúziou karpálnych kostí, skoliózou a pes cavus. Ďalšími znakmi ochorenia sú faciálna dysmorfia, dlhá oválna tvár, prominujúce čelo, hypertelorizmus, makrocefália, nízko položené uši, brachydaktýlia či poruchy sluchu. Príčinou vzniku ochorenia sú mutácie v géne Col27A1 kódujúceho alfa 1 reťazec fibrilárneho kolagénu typu 27. V práci publikujeme extrémne vzácny prípad 8-ročného dievčaťa s miernou kostnou dyspláziou, konštitučne oneskoreným rastom, hypertelorizmom a faciálnou dysmorfiou. U pacientky sme celoexómovým sekvenovaním identifikovali prítomnosť 2 neznámych heterozygotných variantov v géne Col27A1: c.1741C>T, p.(Gln581Lys) a c.2858C>T, p.(Pro953Leu). Na našom pracovisku sme diagnostikovali druhý prípad Steelovho syndrómu v Európe a zároveň prvý prípad tohto raritného ochorenia v slovanskej populácii.
Klíčová slova:
celoexómové sekvenovanie – gén Col27A1 – skeletálna dysplázia – Steelov syndróm
Úvod
Steelov syndróm je zriedkavé autosómovo-recesívne ochorenie, ktoré prvýkrát opísal Steel v roku 1993 na jedinečnom súbore 23 detí z Portorika [1]. Spoločným fenotypovým znakom všetkých detí bol nízky telesný vzrast asociovaný s nápadnými skeletálnymi dysplastickými znakmi, ako je bilaterálna dislokácia bedrového kĺbu a hlavy rádia, fúzia karpálnych kostí, skolióza a pes cavus. K ďalším uniformným syndromologickým charakteristikám patrila mierna faciálna dysmorfia, prominujúce čelo, hypertelorizmus, nízko položené uši a široký koreň nosa [1,2].
Aj napriek tomu, že Steelov syndróm je známy už 28 rokov, gén zodpovedný za toto zriedkavé ochorenie bol objavený až v roku 2015. Gén Col27A1 kóduje alfa 1 reťazec kolagénu typu 27. Spolu s ďalšími génmi ako Col5A1, Col5A3, Col11A1, Col11A2, Col24A1 patrí do skupiny fibrilárnych kolagénov. Expresiu kauzálneho génu reguluje transkripčný faktor SOX9, hlavný regulátor chondrogenézy u stavovcov. Vysoká expresivita génu vo vyvíjajúcej sa chrupavke naznačuje jeho významnú úlohu v skeletogenéze a mineralizácii chrupavky. V menšej miere je exprimovaný v iných tkanivách, ako oči, uši, pľúca, hrubé črevo a žalúdok [3,4].
Celosvetovo je v literatúre opísaných len 20 pacientov s molekulovo-geneticky potvrdeným Steelovým syndrómom, z toho 9 mužov a 11 žien (tab. 1). 11 pacienti z Portorika z pôvodného súboru mali identifikovanú rovnakú homozygotnú mutáciu (Gly697Arg), čo naznačuje jej zakladateľský efekt [4]. Ďalšie homozygotné mutácie sa našli u 2 pacientov; zo Spojených arabských emirátov a Jemenu (c.3556–2A>G a c.4261 - 1G>A) [5,6]. Dvaja pacienti, indického a sýrskeho pôvodu, boli identifikovaní ako zlúčení heterozygoti (p.(Cys174Serfs*34) a p.(Arg707*) a p.(Phe32Leufs*71) a p.(Lys1026Argfs*33) [7,2]. Homozygotná mutácia p.(Gly904Arg) sa potvrdila u pacienta z Iraku [8]. Nedávno boli publikované 3 nové homozygotné mutácie u tureckých pacientov: c.2683G>A p.(Gly895Arg); c.4976_4980del; p.(Gly1660Aspfs*3) a delécia c.63–4_69del; p.(Gly22Serfs*6) [9]. Prvý prípad s geneticky potvrdeným Steelovým syndrómom v Európe dokumentovali u 4-ročného chlapca z Cypru, ktorý je nositeľom homozygotnej mutácie p.(Gly802Glu) [10].
![Fenotypové porovnanie molekulovo-geneticky charakterizovaných pacientov so Steelovým syndrómom.
Upravené podľa [9–10]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image_pdf/12e0ad6f692c62d4547b4715dc2a7da2.png)
Vzhľadom k vysokej variabilite klinických znakov je diagnostika skeletálnych dysplázií veľmi náročná. Celoexómové sekvenovanie sa v čoraz väčšej miere stáva zlatým štandardom pre stanovenie diagnózy a prognózy ochorenia. Retrospektívna štúdia klinických prejavov u pacientov so Steelovym syndrómom preukázala, že ortopedické intervencie kostných deformít u postihnutých jedincov nie sú efektívne a nepriniesli očakávaný benefit [11]. Molekulovo-genetická analýza kauzálnych génov tak prispieva k voľbe najvhodnejšej liečby a môže zabrániť zbytočným invazívnym zásahom.
V článku demonštrujeme prvé dieťa so Steelovým syndrómom v slovanskej populácii a zároveň druhý prípad tohto vzácneho ochorenia v Európe.
Kazuistika
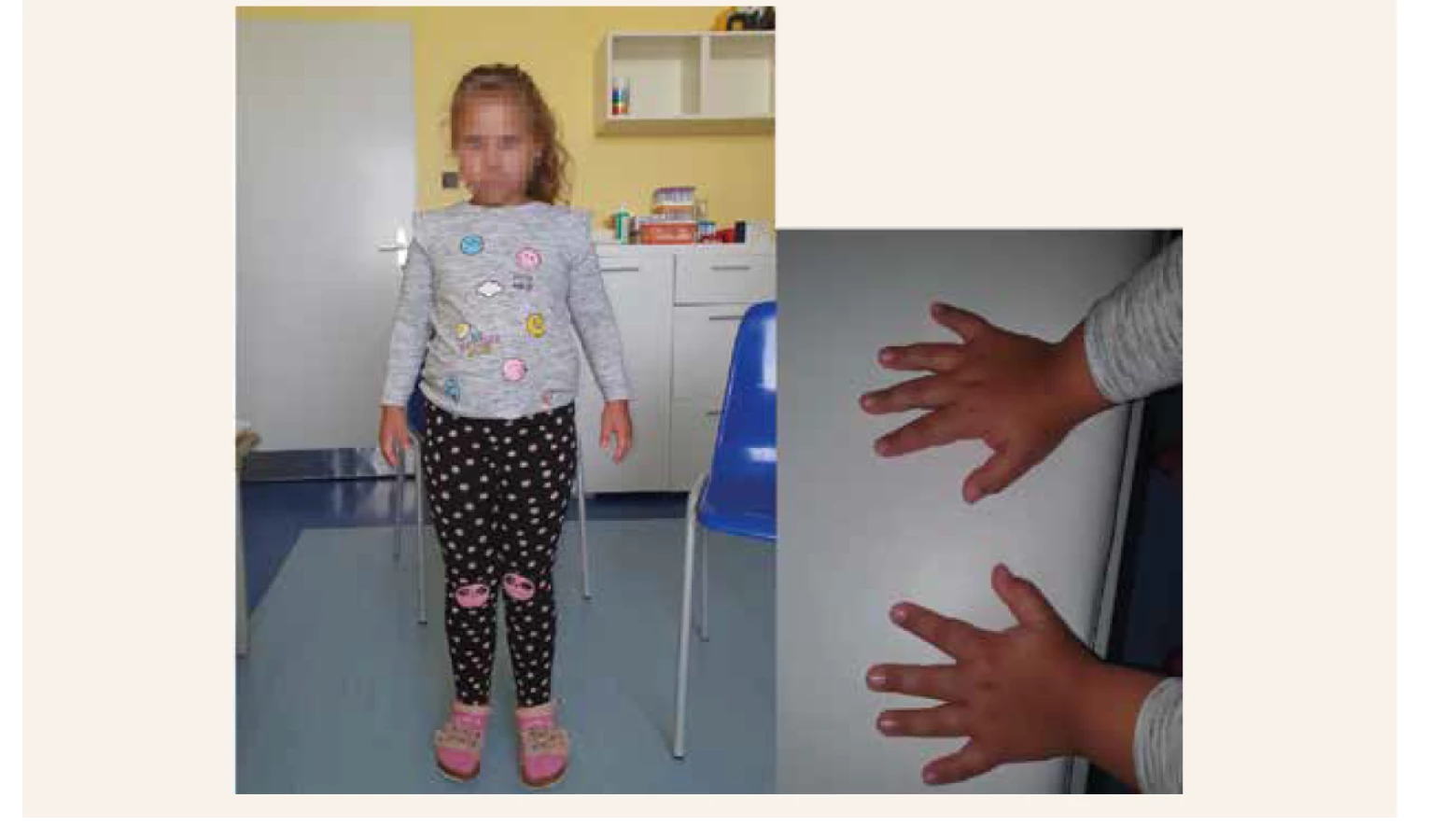
8-ročné dievčatko (obr. 1) je sledované od 16. mesiaca života v endokrinologickej ambulancii Detskej kliniky LF UK a NÚDCH pre intrauterinnú rastovú retardáciu. Narodila sa v 37. týždni tehotenstva nekonsangvinným rodičom. Tehotenstvo bolo predčasne ukončené kvôli nízkemu prietoku krvi placentou, jej pôrodná hmotnosť bola 2 130 g, pôrodná dĺžka 46 cm (≥ -2SD) zodpovedajúca gestačnému veku [14]. Peri - a postanatálny priebeh bol bez komplikácií, psychomotorický vývoj primeraný. Rastová krivka sa udržiavala na 3. percentile s miernym zlepšením po dosiahnutí 5. roku života, v rozmedzí prediktívnej výšky rodičov. Vo veku 8 rokov sa rýchlosť rastu spomalila a manifestovali sa známky disproporcionality. Antropometrické parametre potvrdili relatívne krátke ramená, nízky rast pri výške -2 SD a zvýšenom BMI (+3,4 SD) s brušnou adipozitou, široký hrudník (obvod hrudníka +4,2 SD) a panvu (+2,8 SD). Kraniálny index zodpovedal brachycefálii. U pacientky sme pre rozvoj dysmorfických znakov (lordotická chôdza, genua valga, disproporčný rast, široký hrudník, krátke akrálne časti a klinodaktýlia, hypertelorizmus, prominujúce čelo, nízko posadené uši) a RTG-nález na epifýzach rúk (zobákovité epifýzy rádia a širšie kostičky zápästných kostí) indikovali genetické vyšetrenie. Endokrinné alebo iné organické príčiny nízkeho vzrastu sa vylúčili. Audiometrické vyšetrenie bolo v norme. Rádiologické znaky zodpovedajúce miernej forme Steelovho syndrómu sú znázornené na obr. 2.

Diagnostiku skeletálnych dysplázií sme realizovali celoexómovým sekvenovaním. Na prípravu knižníc bol použitý kit Twist Human Core Exome Kit (Twist Bioscience). Sekvenačná analýza bola realizovaná na prístroji NovaSeq6000. Sekvenačné a CNV-dáta sme hodnotili pomocou softvérov NextGENe a GeneticistAssistant (Softgenetics), bioinformatickou analýzou 452 génov asociovaných so vznikom skeletálnych dysplázií (tab. 2).

U pacientky sme identifikovali prítomnosť 2 neznámych heterozygotných variantov v géne Col27A1: c.1741C>T, p.(Gln581Lys) a c.2858C>T, p.(Pro953Leu). Kauzalitu identifikovaných genetických variantov sme overili segregačnou analýzou vyšetrením oboch rodičov pacientky. Zistili sme, že jeden variant zdedila pacientka od matky (c.2858C> T) a druhý od otca (c.1741C> T) (obr. 3). Vzhľadom na autosómovo-recesívny typ dedičnosti Steelovho syndrómu sme potvrdili kauzalitu identifikovaných variantov a ich asociáciu s ochorením u probandky.

Diskusia
Skeletálne dysplázie predstavujú rozsiahlu skupinu vývojových a štrukturálnych porúch, ktoré môžu byť spôsobené mutáciami v rôznych génoch zodpovedných za vývoj chondrocytov, diferenciáciu, mineralizáciu a skorú osifikáciu [4]. Na základe dokumentovaných klinických a genetických analýz sa Steelov syndrómom dá odlíšiť od iných dobre charakterizovaných skeletálnych dysplázií a porúch spojivového tkaniva [9].
Donedávna sa predpokladalo, že Steelov syndróm je dôsledkom zakladateľskej mutácie Gly697Arg centralizovaný v hispánskej oblasti, v ktorej sa diagnostikovali prví 11 pacienti s touto mutáciou. Postupne však pribúdajú geneticky potvrdené prípady Steelovho syndrómu aj z iných oblastí sveta. Predpokladá sa, že skutočná prevalencia tohto vzácneho syndrómu je značne poddimenzovaná a celosvetovo je počet pacientov s týmto ochorením vyšší.
Vzhľadom na raritný výskyt sú publikované údaje o Steelovom syndróme iba ojedinelé. Doteraz najrozsiahlejšou klinickou štúdiou v literatúre je retrospektívna analýza jedinečnej kohorty 32 probandov (18 pacientov z originálnej Steelovej štúdie a 14 noví pacienti), v ktorej autori sledovali vplyv chirurgických intervencií osteoskletálnych deformácií na dlhodobú morbiditu a kvalitu života chorých [11]. Celkové klinické charakteristiky a rádiologické nálezy sa porovnávali v dvoch skupinách chorých (pacienti bez liečby vs pacienti, ktorí podstúpili ortopedické intervencie). Priemerný vek pacientov v čase zaradenia do sledovania bol 17,8 roka, 7 probandov v súbore bolo dospelých. Dĺžka sledovaného obdobia bola 13,7 rokov. Ani jeden pacient nemal kognitívny deficit a všetky deti mali primeraný neurologický vývoj. Zaradení probandi mali nízku postavu, vrodenú bilaterálnu dislokáciu bedier, oválnu tvár, prominujúce čelo a hypertelorizmus. Ortopedické intervencie bedrových kĺbov sprevádzali u 61 % operovaných (39/64) rôzne komplikácie. Až 85 % (33/39) bedrových kĺbov ostalo aj po operácii dislokovaných, v ostatných prípadoch sa pozorovala subluxácia, dysplázia acetábula a chronická bolesť vyžadujúca liečbu. Skupina chorých s neliečenými dislokáciami udávala prekvapivo menej celkových komplikácií a limitácií v denných aktivitách. Resekcia hlavy rádia obmedzovala rozsah pohybu či funkcie kĺbu, čo poukazuje na to, že sa jedná o vrodenú deformitu, ktorá nevyžaduje liečbu, pokiaľ nie je výrazne symptomatická. Pri hodnotení lakťových kĺbov sa v 87 % (56/64 lakťov) pozorovala dislokácia hlavy rádia, v 73 % prípadoch (47/64 zápästí) došlo k fúzií karpálnych kostí. Polovica pacientov (17/32) mala skoliózu, tretina (22/64 nôh) pes cavus a 10 % (3/32) pacientov vykazovalo anomálie krčnej chrbtice [11]. Výsledky klinicko-rádiologickej korelačnej analýzy podporujú závery originálneho sledovania Steela et al [1] o relatívnej kontraindikácii chirurgických korekcií skeletu u týchto pacientov.
Klinický obraz našej pacientky zodpovedá miernej forme Steelovho syndrómu. Probandka má prejavy kostnej dysplázie s jemnými skeletálnymi znakmi a faciálnou dysmorfiou. Treba však zohľadniť nízky biologický vek dievčatka (8 rokov), neúplnú osifikáciu kostí a rozvoj ďalších symptómov ochorenia v staršom veku.
K častým prejavom Steelovho syndrómu patrí strata sluchu. Jej kauzálny súvis s entitou Steelovho syndrómu potvrdzujú funkčné štúdie na myšacích modeloch, ktoré demonštrovali expresiu génu Col27A1 v chrupavkových štruktúrach vnútorného ucha a v kochleárnom epiteli [15]. Experimentálne dôkazy korešpondujú so stratou sluchu u pacientov so Steelovým syndrómom so závažnými genetickými variantami v géne Col27A1, ktoré majú za následok stratu funkcie proteínu. Donedávna sa predpokladalo, že s týmto prejavom sú asociované závažnejšie typy mutácií, napríklad zostrihové mutácie. Túto hypotézu však vyvracajú prípady zlúčených heterozygotných ako aj homozygotných missense variantov identifikovaných v géne Col27A1. Zaujímavé je, že v rodine z Portorika sa u 3 dospelých príbuzných so zakladateľskou mutáciou Gly697Arg vyvinula obojstranná strata sluchu až po 30. roku života [9]. U našej pacientky sme poruchu sluchu zatiaľ nediagnostikovali, hoci je nositeľkou miernejších mutácií (c.1741C>T, p.(Glu581Lys) a c.2858C>T, p.(Pro953Leu)). Pre nástup hluchoty u probandov vo vyššom veku sa odporúča pravidelné monitorovanie sluchu u každého jedinca so Steelovým syndrómom.
Súčasné metódy sekvenovania novej generácie umožňujú diagnostiku ochorení prostredníctvom celoexómového sekvenovania. Identifikácia kauzálnych variantov v géne Col27A1 asociovaných so Steelovým syndrómom je nevyhnutná nielen pre diagnostiku ochorenia, ale aj pre genetické poradenstvo nositeľov mutácie, čiže potenciálnych prenášačov ochorenia. Pacienti s poruchou rastu, ktorá je asociovaná s dysmorfnými črtami, majú vďaka modernej molekulovo-genetickej diagnostike šancu na identifikáciu základného ochorenia a určenie optimálnej liečby. U jedincov s charakteristickou klinickou manifestáciou sa odporúča cielená molekulová analýza na detekciu možných genetických variantov v géne Col27A1, ktorá určí asociáciu so suponovaným ochorením a prispeje k mapovaniu frekvencie aliel v ostatných populáciách vo svete. Na dôkaz vplyvu týchto variantov a stanovenie genotypovo-fenotypovej korelácie sú potrebné viaceré funkčné štúdie a analýzy na väčšom počte pacientov.
Záver
Variabilita klinických prejavov ako aj prekrývajúce sa fenotypové znaky skeletálnych dysplázií sťažujú ich exaktnú diagnostiku. Nárast identifikovaných génov asociovaných so skeletálnymi dyspláziami poukazuje na ich poddimenzovanú diagnostiku a potrebu multidisciplinárneho prístupu v personalizovanom manažmente pacientov. Molekulovo-genetické testovanie prispieva k stanoveniu definitívnej diagnózy, poskytuje cielené genetické poradenstvo a komplexné informácie o možnom riziku pre ďalších potomkov. Zároveň pomáha monitorovať možné komplikácie v budúcnosti, zvoliť optimálnu liečbu a predísť rizikám nevhodne indikovanej terapie s nepriaznivým dopadom na pacientov. Z klinického hľadiska je sledovanie pacientov kľúčom k indikácii na genetické vyšetrenie vzhľadom na možné zmeny počas ich vývoja. Rovnako potrebná je aj revízia doterajších, doposiaľ geneticky nediagnostikovaných pacientov.
Práca bola finančne podporená grantom Ministerstva zdravotníctva Slovenskej republiky (MZSR 2018/36-LFUK-10)
RNDr. Lucia Oravcová, PhD.
www.nudch.eu
Received | Doručené do redakcie | Doručeno do redakce 28. 2. 2021
Accepted | Prijaté po recenzii | Přijato po recenzi 11. 3. 2021
Received | Doručeno do redakce | Doručené do redakcie 28. 2. 2021
Accepted | Přijato po recenzi | Prijaté po recenzii 11. 3. 2021
Sources
- Steel HH, Piston RW, Clancy M et al. 1993. A syndrome of dislocated hips and radial heads, carpal coalition, and short stature in Puerto Rican children. J Bone Joint Surg Am 1993; 75(2): 259–264. Dostupné z DOI: <https://doi.org/10.2106/00004623–199302000–00013>.
- Pölsler L, Schatz UA, Simma B et al. A Syrian patient with Steel syndrome due to compound heterozygous COL27A1 mutations with colobomata of the eye. Am J Med Genet A 2020; 182(4): 730–734. Dostupné z DOI: <https://doi.org/10.1002/ajmg.a.61478>.
- Boot-Handford RP, Tuckwell DS, Plumb DA et al. A novel and highly conserved collagen (pro(alpha)1(XXVII)) with a unique expression pattern and unusual molecular characteristics establishes a new clade within the vertebrate fibrillar collagen family. J Biol Chem. 2003; 278(33): 31067–31077. Dostupné z DOI: <http://dx.doi.org/10.1074/jbc.M212889200>.
- Gonzaga-Jauregui C, Gamble CN, Yuan B et al. Mutations in COL27A1 cause Steel syndrome and suggest a founder mutation effect in the Puerto Rican population. Eur J Hum Genet 2015; 23(3): 342–346. Dostupné z DOI: <https://doi.org/10.1038/ejhg.2014.107>.
- Gariballa N, Ben-Mahmoud A, Komara M et al. 2017. A novel aberrant splice site mutation in COL27A1 is responsible forSteel syndrome and extension of the phenotype to include hearing loss. Am J Med Genet 2017; 173(5): 1257–1263. Dostupné z DOI: <https://doi.org/10.1002/ajmg.a.38153>.
- Maddirevula S, Alzahrani F, Al-Owain M et al. Autozygome and high throughput confirmation of diseasegenes candidacy. Genet Med 2019; 21(3): 736–742. Dostupné z DOI: <http://dx.doi.org/10.1038/s41436–018–0138-x>.
- Kotabagi S, Shah H, Shukla A et al. Second family provides further evidence for causation of Steel syndrome by biallelic mutations in COL27A1. Clin Genet 2017; 92, 323–326. Dostupné z DOI: <https://doi.org/10.1111/cge.13006>.
- Thuresson A, Soussi Zander C, Zhao JJ et al. Whole genome sequencing of consanguineous families reveals novel pathogenic variants in intellectual disability. Clin Genet 2019; 95(3): 436–439. Dostupné z DOI: <https://doi.org/10.1111/cge.13470>,
- Gonzaga-Jauregui C, Yesil G, Nistala H et al. Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. Eur J Hum Genet 2020; 28(9): 1243–1264. <http://dx.doi.org/10.1038/s41431–020–0632-x>.
- Kritioti E., Theodosiou A., Nicolaou N et al. First reported case of Steel syndrome in the European population: A novel homozygous mutation in COL27A1 and review of the literature. Eur J Med Genet 2020; 63(7):103939. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejmg.2020.103939>.
- Flynn JM, Ramirez N, Betz R et al. Steel syndrome: dislocated hips and radial heads, carpal coalition, scoliosis, short stature, and characteristic facial features. J Pediatr Orthop 2010; 30(3): 282–288. <http://dx.doi.org/10.1097/BPO.0b013e3181d3e464>.
- Belbin GM, Odgis J, Sorokin EP et al. Genetic identification of a common collagen disease in Puerto Ricans via identity-bydescent mapping in a health system. Elife 2017; 6: e25060. Dostupné z DOI: <https://doi.org/10.7554/eLife.25060>.
- Amlie‐Wolf L, Moyer‐Harasink S, Carr A et al. Three new patients with Steel syndrome and a Puerto Rican specific COL27A1 mutation. Am J Med Genet A 2020; 182(4): 798–803. Dostupné z DOI: <https://doi.org/10.1002/ajmg.a.61465>.
- Lawrence C, Fryer JG, Karlberg P et al. Modelling of reference values for size at birth. Acta Paediatr Scand Suppl 1989; 350 : 55–69. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1651–2227.1989.tb11198.x>.
- Plumb DA, Ferrara L, Torbica T et al. Collagen XXVII organises the pericellular matrix in the growth plate. PLoS One 2011; 6(12): e29422. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.0029422>.
Labels
Clinical biochemistry Paediatric gynaecology Paediatric radiology Paediatric rheumatology Endocrinology Gynaecology and obstetrics Internal medicine Orthopaedics General practitioner for adults Radiodiagnostics Rehabilitation Rheumatology Traumatology OsteologyArticle was published in
Clinical Osteology

2021 Issue 1
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
Most read in this issue
- Recidivujúca hyperkalciémia – ťažký diagnostický oriešok: kazuistika
- Osteoporóza asociovaná s těhotenstvím: kazuistika
- Pseudohypoparatyreóza typu Ib: kazuistika a literárny prehľad
- Dodržiavanie nefarmakologických postupov v liečbe postmenopauzálnej osteoporózy: pilotná štúdia