
APOE a BDNF jako rizikové genetické markery pro predikci nástupu a rozvoje kognitivního deficitu při Alzheimerově nemoci
APOE and BDNF as genetic risk markers for predicting the onset and development of cognitive deficits due to Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that is typically initialized by neuronal death in the hippocampus and mediotemporal structures with characteristic episodic memory impairment. However, what is different among AD patients is the age of onset and progression of the disease. It has been suggested that the major modulators of these factors appear to be genetic polymorphisms in apolipoprotein E (APOE) and brain-derived neurotrophic factor (BDNF) genes. APOE e4 allele is the primary genetic determinant of risk for late-onset AD. BDNF Val66Met polymorphism has been shown to alter the risk for developing cognitive impairment and disease progression, both directly and indirectly through an interaction with the APOE genotype. The carriage of both risky variants APOE e4/BDNF Met was associated with episodic memory impairment and faster memory decline compared to the presence of only one or none of these high-risk polymorphisms. This information may be useful for improving the early-detection capability of individuals at risk of developing AD, as well as advancing our understanding of polymorphic combinations that predict the rate of disease progression. Some interventional studies also indicate potential for non-pharmacological interventions in disease prevention in high-risk individuals.
Keywords:
mild cognitive impairment – Alzheimer’s disease – apolipoprotein E – brain-derived neurotrophic factor – gene polymorphisms – Cognition
Authors:
K. Čechová 1,2; Z. Chmátalová 2,3; V. Matušková 1,2; V. Maťoška 4; J. Hort 1,2
Authors‘ workplace:
Kognitivní centrum, Neurologická, klinika 2. LF UK a FN Motol, Praha
1; Mezinárodní centrum klinického, výzkumu, FN u sv. Anny v Brně, Brno
2; Ústav lékařské chemie a klinické biochemie, 2. LF UK a FN Motol, Praha
3; Laboratoř molekulární diagnostiky, Nemocnice Na Homolce, Praha
4
Published in:
Cesk Slov Neurol N 2020; 83/116(3): 257-262
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2020257
Overview
Alzheimerova nemoc (AN) je progresivní neurodegenerativní onemocnění, pro které je charakteristické odumírání neuronů v oblasti hipokampu a mediotemporálních struktur s typicky narušenou epizodickou pamětí. U pacientů se však liší věkem rozvoje a rychlostí progrese onemocnění. Zdá se, že hlavními modifikátory těchto dvou faktorů jsou genetické polymorfizmy v genech pro apolipoprotein E (APOE) a brain-derived neurotrophic factor (BDNF). Hlavní rizikovou genetickou determinantou pro rozvoj AN s pozdním počátkem je alela APOE e4. BDNF Val66Met polymorfizmus se ukazuje jako rizikový pro rozvoj kognitivního deficitu a rychlost progrese onemocnění, ať už přímou nebo nepřímou interakcí s APOE genotypem. U nositelů kombinace obou rizikových polymorfizmů APOE e4/BDNF Met byly pozorovány horší výkon v oblasti epizodické paměti a rychlejší progrese kognitivního deficitu v čase při porovnání s pacienty, kteří nejsou nositeli této rizikové kombinace nebo nejsou nositeli žádného z těchto polymorfizmů. Tato informace může být užitečná pro přesnější identifikaci jedinců v riziku rozvoje AN i pro pravděpodobnou prognózu a další vývoj onemocnění. Zároveň některé intervenční studie naznačují potenciál pro nefarmakologické intervence v prevenci onemocnění u rizikových jedinců.
Klíčová slova:
apolipoprotein E – brain-derived neurotrophic factor – Alzheimerova nemoc – mírná kognitivní porucha – genetické polymorfi - zmy – kognice
Kontinuum Alzheimerovy nemoci
Alzheimerova nemoc (AN) je závažné neurodegenerativní onemocnění mozku s celosvětově stoupající prevalencí. Charakteristickým rysem je odumírání (degenerace) neuronů v důsledku patologického extracelulárního hromadění beta amyloidu (Ab) a intracelulární akumulace tau proteinu [1,2]. K patofyziologickým změnám dochází již přibližně 20–30 let před prvními klinickými projevy [3]. Toto období je označováno jako preklinická fáze nemoci. Na ni v kontinuu onemocnění navazuje časná klinická nebo také prodromální fáze nemoci, za kterou je považován syndrom tzv. mírné kognitivní poruchy (mild cognitive impairment; MCI). MCI je stadium, ve kterém je již objektivně narušena kognice měřená standardizovanými neuropsychologickými testy, ale jedinec je ještě zcela soběstačný [4]. MCI je považována za mezistupeň mezi zdravým stárnutím a syndromem demence. Ne vždy ale tomu tak je. Udává se, že 20–40 % pacientů s MCI konvertuje zpět do normy, což však ukazují převážně populační studie [5,6]. Studie vycházející z klinické populace udávají nižší konverzi mezi 5–16 % [7,8]. Stadiu MCI často předchází stadium tzv. subjektivního kognitivního poklesu (subjective cognitive decline; SCD), což je období, kdy pacient vyhledá lékaře kvůli subjektivním stížnostem na kognici, ale objektivně ve standardizovaných neuropsychologických testech jsou jeho výkony ještě v normě a nesplňuje tedy kritéria MCI. Přesto jsou tito pacienti ve zvýšeném riziku rozvoje AN [9,10]. Někdy se tomuto stadiu říká, že „pacient již ví, zatímco lékař ještě ne“ [1,11].
Pro časnou fázi AN je typická neurodegenerace v oblasti hipokampů a mediotemporálních struktur s charakteristicky narušenou funkcí epizodické paměti [12]. To, v čem se ovšem toto onemocnění mezi jedinci liší, je věk nástupu prvních klinických obtíží a rychlost progrese nemoci. Nedávné studie poukazují na důležitost různých faktorů ovlivňujících nástup a progresi AN, vč. socioekonomického statusu nebo životního stylu jedince. Přesto se ale zdá, že nejzásadnějšími z nich jsou genetické predispozice. Již dříve bylo prokázáno, že rizikové genetické polymorfizmy v genech pro apolipoprotein E (APOE) a TOMM40 zhoršují výkonnost v prostorové navigaci u pacientů s MCI [13–15], a tím zvyšují riziko rozvoje AN u svých nositelů. Existují ale i další intenzivně studované polymorfizmy v patofyziologii AN. V tomto přehledovém článku se zaměříme na polymorfizmy v genech pro APOE a brain-derived neurotrophic factor (BDNF), jejich synergetický vliv na kognitivní funkce a s tím spojené riziko rozvoje AN.
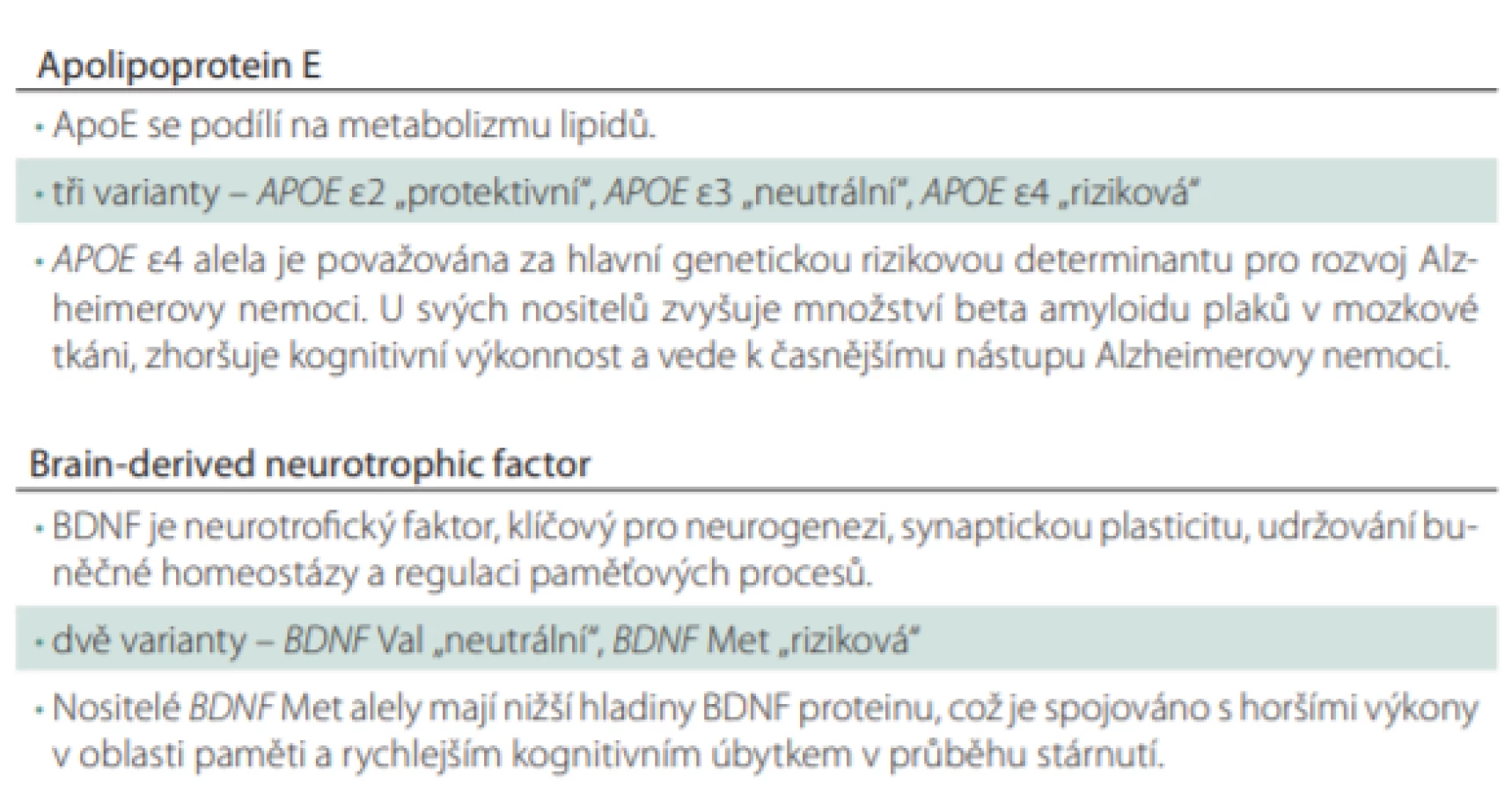
Polymorfizmy v genech pro APOE a BDNF
Jednonukleotidový polymorfizmus (single nucleotide polymorphism; SNP) v genu pro APOE vede k substituci aminokyseliny cysteinu (Cys) za arginin (Arg) na kodónech 112 a 158. Dle daného polymorfizmu jsou alely označovány jako e2 (Cys112, Cys158), e3 (Cys112, Arg158) a e4 (Arg112, Arg158), přičemž riziková pro rozvoj AN je prezentace alely e4. APOE e4 je považována za hlavní genetický rizikový faktor pro nástup AN s pozdním počátkem, tedy po 65. roce života, kdy nositelé jedné rizikové alely (APOE e3/e4) mají 3–4× vyšší pravděpodobnost rozvoje AN. U homozygotů e4 (APOE e4/e4) je toto riziko dokonce až 15× vyšší v porovnání s jedinci bez rizikové alely [16–18]. Zároveň se udává, že pravděpodobnost klinické manifestace AN je u těchto jedinců 80 % do 80 let života [16,19]. Také věk nástupu klinických příznaků se snižuje se zvyšujícím se počtem rizikových APOE e4 [20]. Apolipoprotein E (ApoE) je v mozku produkován převážně astrocyty (a částečně mikrogliemi a buňkami choroidálního plexu [21]) a jeho hlavní funkcí je transport cholesterolu a jiných lipidů k neuronům skrze vazbu na ApoE receptory [22]. Cholesterol je v mozkové tkáni klíčový pro tvorbu a udržování synaptických spojů mezi neurony, ApoE se tedy podílí na synaptické plasticitě. V případě narušení lipidové homeostázy dochází k degeneraci synapsí a dendritických trnů a zhoršení neurotransmise, což významně přispívá k neurodegenerativních změnám [23]. Mechanizmy, kterými se odlišné ApoE izoformy podílí na lipidovém metabolizmu v mozkové tkáni, mohou být skrze jejich rozdíly v konformaci, posttranslačních modifikacích, odlišnou preferencí lipoproteinů nebo afinitou k receptorům. Nicméně jejich přesná role nebyla doposud objasněna [24]. Je ale známo, že izoformy ApoE se liší ve své afinitě k Ab v mozkové tkáni. Akumulace Ab je považována za počátek toxické kaskády, která vede k synaptické dysfunkci a následnému odumírání neuronů a neurodegeneraci [25], přestože tato teorie byla některými autory zpochybňována [26]. Nejvyšší afinitu k Ab má ApoE2, poté ApoE3 a nejnižší ApoE4 protein. Předpokládá se, že díky silnější vazbě se ApoE2 a ApoE3 podílí na efektivnějším odstraňování extracelulárního Ab ve srovnání s ApoE4, u kterého je akumulace nejvýraznější [27]. U nositelů rizikové APOE e4 alely bylo opakovaně pozorováno vyšší hromadění patologického Ab a zároveň bylo v mozku narušeno jeho odbourávání [28,29]. Mezi pacienty s pozitivním nálezem Ab v mozku je prevalence této rizikové alely vysoká. Konkrétně 66% u pacientů s demencí při AN a dokonce 64 % u jedinců ve stádiu MCI [30]. Kromě toho byl u jedinců s APOE e4 pozorován vztah mezi prezentací Ab plaků a kognitivní výkonností. Ukázalo se, větší množství Ab plaků v mozkové tkáni u nositelů APOE e4 je spojeno s výraznějším poklesem v kognitivní výkonnosti, zejména v oblasti globálního kognitivního výkonu, epizodické paměti a vizuospaciálních funkcích [31]. V longitudinálním sledování kognitivně zdravých jedinců byl zaznamenán počátek poklesu epizodické paměti kolem 60. roku věku, který byl u nositelů APOE e4 následován výrazně rychlejší progresí paměťového deficitu v porovnání s nositeli e3 a e2 alel. Podobný trend, ale méně významný, byl pozorován i v oblasti globálního hodnocení kognice a vizuospaciálních funkcí [32].
Nositelé APOE e4 tedy rozvinou nemoc častěji, v nižším věku a může mít u nich rychlejší průběh.
Nicméně rychlost progrese onemocnění se v kombinaci s dalšími genetickými polymorfizmy liší. V této souvislosti je studován zejména polymorfizmus v genu pro BDNF. SNP vede k nahrazení aminokyseliny valinu (Val) methioninem (Met) na 66. kodonu, což negativně ovlivňuje produkci a sekreci BDNF proteinu [33]. BDNF patří do rodiny proteinů označovaných jako neurotrofiny, což jsou látky, které v rámci nervového systému ovlivňují růst, diferenciaci a buněčný cyklus neuronů i glií [34]. Během ontogeneze centrální nervové soustavy je BDNF klíčový pro růst a směrování axonů [35], podporuje růst dendritických trnů a moduluje tvorbu synapsí [36]. Nicméně se ukazuje, že i během dospělého života se tento neurotrofin podílí na vzniku nových neuronů, tzv. neurogenezi, a to zejména v oblasti hipokampu [37], čímž se významně účastní na tvorbě paměťové stopy [38,39]. BDNF v organizmu vzniká enzymatickým štěpením ze svého prekurzoru zvaného proBDNF pomocí plazminu. Uvedená přeměna z proBDNF na BDNF je důležitým procesem regulujícím neuronální aktivitu a paměťové procesy z důvodu zcela opačného efektu na funkce neuronu. ProBDNF se na buněčné membráně váže na receptor p75NTR (neurotrophin receptor p75), čímž v oblasti hipokampu spouští kaskádu vedoucí ke snižování synaptické aktivity, zvyšování dlouhodobé deprese mezi neurony a apoptotickému zániku buňky. Oproti tomu BDNF se na membráně váže na tyrozinkinázový receptor B (tropomyosin receptor kinase B; TrkB), kdy jeho navázáním dochází k dimerizaci a následné autofosforylaci, která posléze spustí zcela odlišnou kaskádu dějů. Výsledkem je posílení synaptické aktivity mezi neurony, zvýšení dlouhodobé potenciace, a tím posílení tvorby paměťové stopy [40–42]. Právě kvůli své roli v dlouhodobé potenciaci je BDNF považován za klíčovou molekulu podporující tvorbu a konsolidaci paměťové stopy.
Nižší hladiny sérového BDNF jsou spojovány s horšími výsledky v některých paměťových testech u zdravých jedinců [43]. Z těchto důvodů se začalo o sérovém BDNF uvažovat jako o možném rizikovém faktoru AN. A skutečně se ukazuje, že u pacientů ve stadiu MCI i ve stadiu syndromu demence při AN jsou ve srovnání se zdravými kontrolami patrny výrazně snížené sérové hladiny BDNF [44]. Tyto snížené hladiny se u pacientů ve stadiu AN ukázaly jako prediktory pro progresi kognitivního deficitu. Pacienti s nižší hladinou BDNF měli výrazně rychlejší pokles kognice během jednoho roku než pacienti s vyšší hladinou BDNF [45]. Podobně nižší hladiny BDNF v mozkomíšním moku měly za následek rychlejší progresi z MCI do AN [46].
Předpokládá se, že hladiny BDNF jsou ovlivněny genetickým polymorfizmem v genu pro BDNF [33,47]. Samotné nositelství rizikové alely BDNF Met je spojováno s narušením výkonu v oblasti paměti u kognitivně zdravých, které je akcentováno v průběhu stárnutí. Paměťový deficit je přítomen v oblasti deklarativní epizodické paměti [48], která je závislá zejména na funkci hipokampu [49], pro jehož správnou funkci je neurotrofin BDNF klíčový [50]. Současně s horšími výsledky v testech epizodické paměti byla u kognitivně zdravých nositelů patologické BDNF Met alely pozorována abnormální aktivace hipokampu při snímání mozku pomocí funkční MR ve srovnání s nositeli fyziologické BDNF Val alely [33,51]. Výsledky v testech závislých na funkcích prefrontálních a frontálních oblastí se mezi oběma skupinami nelišily [33]. V 3letém pozorování kognitivně zdravých nositelů BDNF Met alely se zvýšeným množstvím Ab plaků v mozku byl přítomen výraznější pokles v oblasti epizodické paměti, exekutivních funkcí, řeči a také výraznější atrofie v oblasti hipokampu v porovnání s BDNF Val nositeli, kteří měli rovněž zvýšené množství Ab plaků v mozku [52]. Ačkoliv BDNF se nezdá být specifickým biomarkerem pro neurodegenerativní onemocnění kvůli jeho zapojení v mnoha patologických procesech, lze ho považovat za marker progrese paměťového deficitu [53,54].
Vliv kombinace APOE ε4/ BDNF Met na kognitivní výkonnost
APOE e4 i BDNF Met jsou dva genetické polymorfizmy ovlivňující riziko nástupu a rozvoje AN. APOE jako nejrizikovější genetický prediktor pro rozvoj AN s pozdním nástupem a BDNF pro jeho roli v regulaci paměťových procesů (Tab. 1). V současnosti jsou intenzivně studovány interakce mezi těmito geny a vliv možného aditivního efektu BDNF na APOE [55]. Ukazuje se, že kombinace těchto rizikových polymorfizmů se na rozvoji kognitivního deficitu podílí významněji než polymorfizmy samotné. U kognitivně zdravých stárnoucích osob nositelství APOE e4/ BDNF Met negativně ovlivňuje paměťové funkce, a to selektivně v oblasti epizodické paměti [56]. U jedinců v preklinickém stadiu AN, tedy u jedinců bez objektivizovatelného kognitivního deficitu, kteří byli kognitivně zdraví, ale s patologickou akumulací Ab v mozku, byl pozorován výraznější pokles epizodické paměti v čase: 1) nositelé kombinace APOE e4/ BDNF Met dosáhli klinicky signifikantního poškození epizodické paměti během 3 let; 2) u nositelů kombinace APOE e4/ BDNF Val se stejný deficit objevil až v horizontu 10 let a 3) nositelé APOE e3 by srovnatelného kognitivního deficitu dosáhli za 27 let [57]. Otázkou však zůstává, jaký vliv má přítomnost těchto rizikových polymorfizmů u jedinců v prodromálním stadiu onemocnění, tedy u těch, kteří již klinické symptomy rozvinuli. Tuto otázku se výzkumníci pokusili odpovědět v nedávné průřezové studii, která testovala vliv kombinace rizikových genetických polymorfizmů na kognitivní výkonnost a mozkové struktury klíčové pro paměť. Specificky byly měřeny objem hipokampů a tloušťka parahipokampální a entorhinální kůry u pacientů ve stadiu amnestické MCI (aMCI). Bylo zjištěno, že přestože se jedinci nelišili v demografických charakteristikách, skóru Mini-Mental State Examination (MMSE) ani míře depresivity, nositelé APOE e4/ BDNF Met měli selektivně narušenu oddálenou výbavnost paměti. Ta bývá považována za reprezentaci epizodické paměti, která je typicky narušena u AN. U rizikových jedinců navíc nebyla pozorována výraznější atrofie v žádné z klíčových oblastí pro paměť. Je tedy pravděpodobné, že právě tito jedinci rozvinou syndrom demence v čase dříve, protože funkční změny v kognici jsou nejsilnějším prediktorem rozvoje syndromu demence [58].
Zdá se, že APOE je hlavní determinantou nástupu onemocnění a BDNF je regulátorem jeho progrese. Nicméně jakým způsobem tyto geny interagují, není v současné době zcela objasněno. Nedávná studie poprvé popsala epigenetickou interakci produktů těchto genů. Bylo demonstrováno, že odlišné ApoE izoformy regulují sekreci a štěpení BDNF proteinu v hipokampálních astrocytech. Sen et al [59] prokázali u těchto buněk 38,4× zvýšenou produkci BDNF proteinu, pokud byly v médiu s ApoE3 ve srovnání s kontrolní skupinou buněk. Buňky v médiu s ApoE4 proteinem produkovaly zanedbatelné množství BDNF proteinu. Výsledky dále prokázaly, že ApoE4 zvyšuje translokaci histon deacetylázy do jádra, což vede k deacetylaci histonů a negativně to ovlivňuje transkripci BDNF genu. V důsledku výše popsaných procesů dochází k nižší produkci BDNF proteinu. Jedním z možných vysvětlení negativního účinku pozorovaného u APOE e4/ BDNF Met nositelů tedy je, že ApoE4 ještě více redukuje sekreci BDNF proteinu u Met nositelů. Pro ověření této hypotézy u klinické populace byla provedena pilotní studie a u podskupiny pacientů s aMCI, kde byly testovány hladiny BDNF v krevním séru. Výsledky naznačily, že společně se selektivně narušenou epizodickou pamětí je u pacientů s aMCI pozorováno signifikantně snížené množství BDNF proteinu v krevním séru u nositelů rizikové kombinace polymorfizmů APOE e4/ BDNF Met [60,61]. Pokud vezmeme v úvahu roli BDNF proteinu v neuroprotekci a synaptické plasticitě, zdá se tedy, že společné působení těchto polymorfizmů vede k vyššímu riziku rozvoje AN a její horší klinické manifestaci skrze snižování efektivního množství BDNF proteinu. Nicméně pro ověření této teorie bude zapotřebí více prací studujících interakci mezi proteiny ApoE a BDNF. Dále je pravděpodobné, že právě nositelé genu APOE e4 jsou ti, u kterých v mozku dochází k dřívější akumulaci patologického Ab. S ohledem na tuto skutečnost je pravděpodobné, že se u těchto pacientů nejvíce projeví negativní efekt mutace genu pro BDNF protein. Zdá se totiž, že nositelství BDNF Met zvyšuje náchylnost mozku k toxickému efektu Ab [62]. Negativní efekt pozorovaný u nositelů APOE e4/ BDNF Met je tedy zřejmě zprostředkován skrze patologický Ab v mozku.
Epigenetické ovlivnění kognitivní výkonnosti
Přestože genetické polymorfizmy jsou hlavní determinanty nástupu a progrese onemocnění, zdá se, že faktory životního stylu s nimi mohou interagovat. Pravidelná fyzická aktivita zvyšuje objem šedé hmoty v oblasti hipokampů, a tím i zlepšuje kognitivní výkonnost v oblasti paměti [63]. Studie zdravých stárnoucích jedinců ukázaly, že vyšší intenzita fyzické aktivity vede k lepší kognitivní výkonnosti, snižuje riziko kognitivního poklesu a atrofie hipokampů, a to výrazněji právě u nositelů rizikové alely APOE e4 oproti neutrální e3 [64,65]. Podobné výsledky jsou pozorovány u pacientů ve stadiu MCI. Fyzicky aktivní e4 nositelé mají stabilní výkon v kognici, zatímco u neaktivních e4 nositelů dochází ke kognitivnímu poklesu [66]. Zároveň u fyzicky aktivních e4 nositelů bylo pozorováno nižší ukládání Ab v mozku ve srovnání s méně aktivními nositeli rizikové alely, ale u nositelů e3 tento efekt pozorován nebyl [66].
Protein ApoE hraje důležitou roli také v metabolizmu cholesterolu a lipidů. Přítomnost APOE e4 zvyšuje hladiny celkového cholesterolu a LDL (low-density lipoproteins) cholesterolu, a tím u svých nositelů negativně ovlivňuje efekt na nervové funkce [67]. Vysokokalorická dieta a zvýšený příjem tuků byl ve 4letém pozorování kognitivně zdravých stárnoucích spojen s častějším výskytem AN u nositelů e4 a nikoliv e3 alely [68]. Na druhé straně změna vysokokalorické diety za stravu obsahující sacharidy s nízkým glykemickým indexem (např. čerstvá zelenina, luštěniny, ořechy) vede u nositelů e4 ke snižování LDL cholesterolu ve srovnání s e3 nositeli [67].
Ve 2leté intervenční studii u zdravých stárnoucích nositelů APOE e4 vedla kombinace zvýšené fyzické aktivity, diety s nízkokalorickým příjmem a kognitivního tréninku ke zlepšení kognitivních funkcí oproti kontrolní skupině [69]. Z těchto výsledků je patrno, že jedinci ve zvýšeném riziku rozvoje AN mohou svým životním stylem toto riziko snižovat.
Ovšem jinak je tomu u polymorfizmu v genu pro BDNF. Uvažuje se, že příznivý efekt fyzické aktivity na mozek a kognici je mediován skrze zvýšenou expresi BDNF proteinu během cvičení, a to zejména v oblasti hipokampů, kde přispívá k neurogenezi a synaptické plasticitě [70]. Benefit pro mozek a kognici však přináší fyzická aktivita zřejmě pouze nositelům nerizikové BDNF Val alely. Pouze u fyzicky aktivních BDNF Val nositelů byly pozorovány zvýšené hladiny BDNF proteinu a zároveň výrazně lepší výsledky v testech epizodické paměti v porovnání se stejně aktivními nositeli rizikové Met alely [71]. Podobné výsledky zaznamenala intervenční studie s pacienty ve stadiu MCI. Po absolvování 16týdenního tréninku bylo pozorováno signifikantní zvýšení hladin tohoto proteinu pouze u nositelů BDNF Val, a nikoliv u nositelů rizikové Met alely. Nebyl sice pozorován žádný efekt na kognici [72], přesto se na základě znalostí efektu BDNF na mozkové funkce dá předpokládat, že u pacientů s vyšší hladinou BDNF proteinu bude konverze do syndromu demence pomalejší. Dosud ale není známa studie, která by tuto hypotézu ověřila v longitudinálním sledování. Zdá se tedy, že zvýšení exprese BDNF při pohybové aktivitě naráží na určité limity v závislosti na daném polymorfizmu, a tak jedinci s rizikovou alelou nemohou fyzickou aktivitou kompenzovat sníženou produkci BDNF. Zvyšování hladin BDNF je pozorováno také při praktikování jógy nebo meditace [73], které jsou známy pro své neuroprotektivní účinky a prevenci kognitivního deficitu [74]. Není ale známa studie, která by tyto aktivity měřila ve vztahu k polymorfizmům.
Sdělení výsledků genetického vyšetření
Znalost polymorfizmu v genu pro APOE poskytuje lékaři důležitou informaci o pravděpodobnosti rozvoje AN. Stanovení APOE polymorfizmu se ale v běžné klinické praxi nedoporučuje a je určeno spíše pro vědecké účely vzhledem k tomu, že se jedná o rizikový a nikoliv determinační faktor onemocnění [75]. Informování pacientů také může vést k následnému ovlivnění jejich stížností, kognitivní výkonnosti i změnám životního stylu. Nedávná studie naznačila, že kognitivně zdraví stárnoucí nositelé APOE e4, kteří znali svůj genotyp, subjektivně hodnotili svou paměť jako horší a zároveň skórovali v testech paměti výrazně hůře než nositelé APOE e4, kteří tuto informaci nevěděli [76]. To může částečně souviset s faktem, že znalost nositelství e4 vede u jedinců k většímu stresu během kognitivního testování [77]. Přesto se neprokázalo, že by tato informace vedla k vyšší depresivní nebo úzkostné symptomatice [77], naopak vedla ke zlepšení životního stylu. U nositelů APOE e4 se téměř 3× zvýšila pravděpodobnost zvýšení fyzické aktivity, zdravějšího stravování a užívání vitaminů [78]. Proto je na lékařském úsudku, zda informaci o genotypu pacientovi sdělit.
Závěr
Genetika v mnoha oblastech medicíny determinuje vznik onemocnění. Ačkoliv u AN s pozdním počátkem takový gen nemáme, může nám genetický skríning poskytnout důležité informace o nástupu a prognóze onemocnění. Rutinní stanovení APOE ani BDNF polymorfizmu není zatím součástí českých doporučených postupů pro diagnostiku AN z roku 2007 [79], ale my jej považujeme za významný faktor pro predikci kognitivního deficitu u pacientů ve zvýšeném riziku rozvoje AN, zejména v kombinaci s dalšími rizikovými genetickými [58] nebo metabolickými biomarkery AN [80]. V souvislosti se současným vývojem nových farmakologických terapeutických intervencí také znalost genetického polymorfizmu výrazně přispěje k přesnější identifikaci vhodných jedinců pro včasnou léčbu. Specificky stanovení APOE polymorfizmu poskytne lékařům informaci o pravděpodobnosti rozvoje AN a stanovení polymorfizmu BDNF o rychlosti její progrese. Znalost obojího zároveň pomůže doporučit účinnou intervenci. Přestože znalost nositelství rizikových genetických polymorfizmů u jedinců ovlivňuje míru stížností i paměťové výkony, zároveň tato informace může vést ke zlepšení životního stylu jedince, a tím snížit riziko rozvoje AN nebo oddálit nástup prvních obtíží.
Konflikt zájmů
Autoři deklarují, že v souvislosti s předmětem studie nemají žádný konflikt zájmů.
Poděkování
Finančně podpořeno projektem č. LQ1605 z Národního programu udržitelnosti II (MŠMT), MZ ČR – RVO, FN v Motole 00064203, projektem GA UK č. 176317, projektem AZV 19-04-00560, programem IPE 2. LF UK 699012 a Alzheimer nadačním fondem.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Mgr. Kateřina Čechová
Kognitivní centrum Neurologická klinika
2. LF UK a FN Motol V Úvalu 84
150 06 Praha 5
e-mail: katerina.cechova@lfmotol.cuni.cz
Přijato k recenzi: 4. 2. 2020
Přijato do tisku: 28. 4. 2020
Sources
1. Čechová K, Marková H, Mazancova AF et al. V bludišti jménem Alzheimer. Praha: Albatros media a. s. 2019.
2. Hort J, Glosová L, Vyhnálek M et al. Tau protein a beta amyloid v likvoru u Alzheimerovy choroby. Cesk Slov Neurol N 2007; 70/103 (1): 30–36.
3. Braak H, Thal DR, Ghebremedhin E et al. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011; 70 (11): 960–969. doi: 10.1097/NEN.0b013e318232a379.
4. Petersen RC. Mild cognitive impairment. N Engl J Med 2011; 364 (23): 2227–2234. doi: 10.1056/NEJMcp0910237.
5. Busse A, Hensel A, Guhne U et al. Mild cognitive impairment: long-term course of four clinical subtypes. Neurology 2006; 67 (12): 2176–2185. doi: 10.1212/01.wnl.0000249117.23318.e1.
6. Gao Q, Gwee X, Feng L et al. Mild Cognitive impairment reversion and progression: rates and predictors in community-living older persons in the Singapore longitudinal ageing studies cohort. Dement Geriatr Cogn Dis Extra 2018; 8 (2): 226–237. doi: 10.1159/000488936.
7. Grande G, Cucumo V, Cova I et al. Reversible mild cognitive impairment: the role of comorbidities at baseline evaluation. J Alzheimers Dis 2016; 51 (1): 57–67. doi: 10.3233/JAD-150786.
8. Koepsell TD, Monsell SE. Reversion from mild cognitive impairment to normal or near-normal cognition: Risk factors and prognosis. Neurology 2012; 79 (15): 1591–1598. doi: 10.1212/WNL.0b013e31826e26b7.
9. Jessen F, Amariglio RE, van Boxtel M et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 2014; 10 (6): 844–852. doi: 10.1016/j.jalz.2014.01.001.
10. Markova H, Nikolai T, Mazancova AF et al. Differences in subjective cognitive complaints between non-demented older adults from a memory clinic and the community. J Alzheimers Dis 2019; 70 (1): 61–73. doi: 10.3233/JAD-180630.
11. Reisberg B, Prichep L, Mosconi L et al. The pre-mild cognitive impairment, subjective cognitive impairment stage of Alzheimer’s disease. Alzheimers Dement 2008; 4 (1 Suppl 1): S98–S108. doi: 10.1016/j.jalz.2007.11.017.
12. Hort J, Laczó J, Vyhnálek M. Alzheimerova nemoc. In: Rusina R, Matěj R (eds). Neurodegenerativní onemocnění. Praha: Mladá fronta 2014 : 102–112.
13. Laczó J, Andel R, Vyhnalek M et al. APOE and spatial navigation in amnestic MCI: results from a computer-based test. Neuropsychology 2014; 28 (5): 676–684. doi: 10.1037/neu0000072.
14. Laczó J, Andel R, Vyhnalek M et al. The effect of TOMM40 on spatial navigation in amnestic mild cognitive impairment. Neurobiol Aging 2015; 36 (6): 2024–2033. doi: 10.1016/j.neurobiolaging.2015.03.004.
15. Hort J, Laczo J, Vyhnalek M et al. Spatial navigation deficit in amnestic mild cognitive impairment. Proc Natl Acad Sci 2007; 104 (10): 4042–4047. doi: 10.1073/pnas.0611314104.
16. Corder EH, Saunders AM, Strittmatter WJ et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261 (5123): 921–923. doi: 10.1126/science.8346443.
17. Neu SC, Pa J, Kukull W et al. Apolipoprotein E genotype and sex risk factors for Alzheimer’s disease. JAMA Neurol 2017; 74 (10): 1178–1189. doi: 10.1001/jamaneurol.2017.2188.
18. Farrer LA, Cupples LA, Haines JL et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997; 278 (16): 1349–1356.
19. Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 2016; 18 (5): 421–430. doi: 10.1038/gim.2015.117.
20. Kwon OD, Khaleeq A, Chan W et al. Apolipoprotein E polymorphism and age at onset of Alzheimer’s disease in a quadriethnic sample. Dement Geriatr Cogn Disord 2010; 30 (6): 486–491. doi: 10.1159/000322368.
21. Xu Q, Bernardo A, Walker D et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 2006; 26 (19): 4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006.
22. Grehan S, Tse E, Taylor JM. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci 2001; 21 (3): 812–822.
23. Koudinov AR, Koudinova N V. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J 2001; 15 (10): 1858–1860. doi: 10.1096/fj.00-0815fje.
24. Kanekiyo T, Xu H, Bu G. ApoE and Ab in Alzheimer’s disease: accidental encounters or partners? Neuron 2014; 81 (4): 740–754. doi: 10.1016/j.neuron.2014.01.045.
25. Castellano JM, Kim J, Stewart FR et al. Human apoE isoforms differentially regulate brain amyloid-peptide clearance. Sci Transl Med 2011; 3 (89): 89ra57-89ra57. doi: 10.1126/scitranslmed.3002156.
26. Kalvach P, Kupka K, Vogner M. Je amyloid podstatný pro senilní demenci? Cesk Slov Neurol N 2018; 81/114 (2): 164–170. doi: 10.14735/amcsnn2018csnn.eu1
27. Tokuda T, Calero M, Matsubara E et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem J 2000; 348 (Pt 2): 359–365.
28. Verghese PB, Castellano JM, Garai K et al. ApoE influences amyloid-b (Ab) clearance despite minimal apoE/Ab association in physiological conditions. Proc Natl Acad Sci U S A 2013; 110 (19): E1807–E1816. doi: 10.1073/pnas.1220484110.
29. Lim YY, Mormino EC, Alzheimer’s disease neuroimaging initiative. APOE genotype and early b-amyloid accumulation in older adults without dementia. Neurology 2017; 89 (10): 1028–1034. doi: 10.1212/WNL.0000000000004336.
30. Mattsson N, Groot C, Jansen WJ et al. Prevalence of the apolipoprotein E e4 allele in amyloid b positive subjects across the spectrum of Alzheimer’s disease. Alzheimers Dement 2018; 14 (7): 913–924. doi: 10.1016/j.jalz.2018.02.009.
31. Kantarci K, Lowe V, Przybelski SA et al. APOE modifies the association between A load and cognition in cognitively normal older adults. Neurology 2012; 78 (4): 232–240. doi: 10.1212/WNL.0b013e31824365ab.
32. Caselli RJ, Dueck AC, Osborne D et al. Longitudinal modeling of age-related memory decline and the APOE e4 Effect. N Engl J Med 2009; 361 (3): 255–263. doi: 10.1056/NEJMoa0809437.
33. Egan MF, Kojima M, Callicott JH et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003; 112 (2): 257–269. doi: 10.1016/s0092-8674 (03) 00035-7.
34. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001; 24 : 677–736. doi: 10.1146/annurev.neuro.24.1.677.
35. Liao GY, Bouyer K, Kamitakahara A et al. Brain-derived neurotrophic factor is required for axonal growth of selective groups of neurons in the arcuate nucleus. Mol Metab 2015; 4 (6): 471–482. doi: 10.1016/j.molmet.2015.03.003.
36. Gao X, Smith GM, Chen J. Impaired dendritic development and synaptic formation of postnatal-born dentate gyrus granular neurons in the absence of brain-derived neurotrophic factor signaling. Exp Neurol 2009; 215 (1): 178–190. doi: 10.1016/j.expneurol.2008.10.009.
37. Li T, Jiang L, Zhang X et al. In-vitro effects of brain-derived neurotrophic factor on neural progenitor/stem cells from rat hippocampus. Neuroreport 2009; 20 (3): 295–300. doi: 10.1097/WNR.0b013e32832000c8.
38. Deinhardt K, Chao M V. Shaping neurons: long and short range effects of mature and proBDNF signalling upon neuronal structure. Neuropharmacology 2014; 76 (Pt C): 603–609. doi: 10.1016/j.neuropharm.2013.04.054.
39. Panja D, Bramham CR. BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology 2014; 76 (Pt C): 664–676. doi: 10.1016/j.neuropharm.2013.06.024.
40. Angelucci F, Čechová K, Průša R et al. Amyloid beta soluble forms and plasminogen activation system in Alzheimer’s disease: consequences on extracellular maturation of brain-derived neurotrophic factor and therapeutic implications. CNS Neurosci Ther 2019; 25 (3): 303–313. doi: 10.1111/cns.13082.
41. Hashimoto K. Regulation of brain-derived neurotrophic factor (BDNF) and its precursor proBDNF in the brain by serotonin. Eur Arch Psychiatry Clin Neurosci 2016; 266 (3): 195–197. doi: 10.1007/s00406-016-0682-9.
42. Yang J, Harte-Hargrove LC, Siao CJ et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep 2014; 7 (3): 796–806. doi: 10.1016/j.celrep.2014.03.040.
43. Yu H, Zhang Z, Shi Y et al. Cognitive function, serum BDNF levels and BDNF gene Val66Met polymorphism in amnestic mild cognitive impairment. J Cent South Univ Med Sci 2008; 33 (4): 321–325.
44. Forlenza OV, Miranda AS, Guimar I et al. Decreased neurotrophic support is associated with cognitive decline in non-demented subjects. J Alzheimers Dis 2015; 46 (2): 423–429. doi: 10.3233/JAD-150172.
45. Laske C, Stellos K, Hoffmann N et al. Higher BDNF serum levels predict slower cognitive decline in Alzheimer’s disease patients. Int J Neuropsychopharmacol 2011; 14 (3): 399–404. doi: 10.1017/S1461145710001008.
46. Forlenza OV, Diniz BS, Teixeira AL et al. Lower cerebrospinal fluid concentration of brain-derived neurotrophic factor predicts progression from mild cognitive impairment to Alzheimer’s disease. NeuroMolecular Med 2015; 17 (3): 326–332. doi: 10.1007/s12017-015-8361-y.
47. Ozan E, Okur H, Eker Ç et al. The effect of depression, BDNF gene val66met polymorphism and gender on serum BDNF levels. Brain Res Bull 2010; 81 (1): 61–65. doi: 10.1016/j.brainresbull.2009.06.022.
48. Kennedy KM, Reese ED, Horn MM et al. BDNF val66met polymorphism affects aging of multiple types of memory. Brain Res 2015; 1612 : 104–117. doi: 10.1016/j.brainres.2014.09.044.
49. Squire LR, Ojemann JG, Miezin FM et al. Activation of the hippocampus in normal humans: a functional anatomical study of memory. Proc Natl Acad Sci U S A 1992; 89 (5): 1837–1841. doi: 10.1073/pnas.89.5.1837.
50. Brown DT, Vickers JC, Stuart KE et al. The BDNF Val66Met polymorphism modulates resilience of neurological functioning to brain ageing and dementia: a narrative review. Brain Sci 2020; 10 (4). pii: E195. doi: 10.3390/brainsci10040195.
51. Hariri AR, Goldberg TE, Mattay VS et al. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci 2003; 23 (17): 6690–6694.
52. Lim YY, Villemagne VL, Laws SM et al. BDNF Val66Met, Ab amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol Aging 2013; 34 (11): 2457–2464. doi: 10.1016/j.neurobiolaging.2013.05.006.
53. Combarros O, Infante J, Llorca J et al. Polymorphism at Codon 66 of the Brain-derived neurotrophic factor gene is not associated with sporadic Alzheimer’s disease. Dement Geriatr Cogn Disord 2004; 18 (1): 55–58. doi: 10.1159/000077736.
54. Lambert JC, Ibrahim-Verbaas CA, Harold D et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 2013; 45 (12): 1452–1458. doi: 10.1038/ng.2802.
55. Tsai SJ, Gau YT, Liu ME et al. Association study of brain-derived neurotrophic factor and apolipoprotein E polymorphisms and cognitive function in aged males without dementia. Neurosci Lett 2008; 433 (2): 158–162. doi: 10.1016/j.neulet.2007.12.057.
56. Ward D, Summers MJ, Saunders NL et al. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav Brain Res 2014; 271 : 309–315. doi: 10.1016/j.bbr.2014.06.022.
57. Lim YY, Villemagne VL, Laws SM et al. APOE and BDNF polymorphisms moderate amyloid b-related cognitive decline in preclinical Alzheimer’s disease. Mol Psychiatry 2015; 20 (11): 1322–1328. doi: 10.1038/mp.2014.123.
58. Cechova K, Andel R, Angelucci F et al. Impact of APOE and BDNF Val66Met gene polymorphisms on cognitive functions in patients with amnestic mild cognitive impairment. J Alzheimers Dis 2020; 73 (1): 247–257. doi: 10.3233/JAD-190464.
59. Sen A, Nelson TJ, Alkon DL. ApoE4 and a oligomers reduce BDNF expression via HDAC nuclear translocation. J Neurosci 2015; 35 (19): 7538–7551. doi: 10.1523/JNEUROSCI.0260-15.2015.
60. Cechova K, Chmatalova Z, Markova H et al. Impact of genetic variant of APOE E4 and BDNF Met on BDNF levels, cognition and brain morphometry in mild cognitive impairment. J Neurol Sci 2019; 405 : 19–20. doi: 10.1016/j.jns.2019.10.247.
61. Cechova K, Chmatalova Z, Markova H et al. Effect of APOE E4 and BDNF Val66Met polymorphism on BDNF levels and cognitive performance in mild cognitive impairment patients. Alzheimers Dement 2019; 15: P629.
62. Franzmeier N, Ren J, Damm A et al. The BDNFVal66Met SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol Psychiatry 2019 Mar 21 [Online ahead of print]. doi: 10.1038/s41380-019-0404-6.
63. Erickson KI, Voss MW, Prakash RS et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci 2011; 108 (7): 3017–3022. doi: 10.1073/pnas.1015950108.
64. Smith JC, Nielson KA, Woodard JL et al. Physical activity reduces hippocampal atrophy in elders at genetic risk for Alzheimers disease. Front Aging Neurosci 2014; 6 : 61. doi: 10.3389/fnagi.2014.00061.
65. Schuit A, Feskens E, Launer L et al. Physical activity and cognitive decline, the role of the apolipoprotein e4 allele. Med Sci Sports Exerc 2001; 33 (5): 772–777. doi: 10.1097/00005768-200105000-00015.
66. Jensen CS, Simonsen AH, Siersma V et al. Patients with Alzheimer’s disease who carry the APOE e4 allele benefit more from physical exercise. Alzheimers Dement (N Y) 2019; 5 : 99–106. doi: 10.1016/j.trci.2019.02.007.
67. de Chaves EP, Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol 2008; 3 (5): 505–530. doi: 10.2217/17460875.3.5.505.
68. Luchsinger JA, Tang MX, Shea S et al. Caloric intake and the risk of Alzheimer disease. Arch Neurol 2002; 59 (8): 1258–1263. doi: 10.1001/archneur.59.8.1258.
69. Solomon A, Turunen H, Ngandu T et al. Effect of the apolipoprotein E genotype on cognitive change during a multidomain lifestyle intervention: a subgroup analysis of a randomized clinical Trial. JAMA Neurol 2018; 75 (4): 462–470. doi: 10.1001/jamaneurol.2017.4365.
70. Coelho FG de M, Gobbi S, Andreatto CA et al. Physical exercise modulates peripheral levels of brain-derived neurotrophic factor (BDNF): a systematic review of experimental studies in the elderly. Arch Gerontol Geriatr 2013; 56 (1): 10–15. doi: 10.1016/j.archger.2012.06.003.
71. Canivet A, Albinet CT, André N et al. Effects of BDNF polymorphism and physical activity on episodic memory in the elderly: a cross sectional study. Eur Rev Aging Phys Act 2015; 12 : 15. doi: 10.1186/s11556-015-0159-2.
72. Nascimento CM, Pereira JR, Pires de Andrade L et al. Physical exercise improves peripheral BDNF levels and cognitive functions in mild cognitive impairment elderly with different BDNF Val66Met genotypes. J Alzheimers Dis 2014; 43 (1): 81–91. doi: 10.3233/JAD-140576.
73. Cahn BR, Goodman MS, Peterson CT et al. Yoga, meditation and mind-body health: increased BDNF, cortisol awakening response, and altered inflammatory marker expression after a 3-month yoga and meditation retreat. Front Hum Neurosci 2017; 11 : 315. doi: 10.3389/fnhum.2017.00315.
74. Marciniak R, Sheardova K, Cermáková P et al. Effect of meditation on cognitive functions in context of aging and neurodegenerative diseases. Front Behav Neurosci 2014; 8 : 17. doi: 10.3389/fnbeh.2014.00017.
75. Vyhnálek M, Vyhnálková E, Laczó J. Genetika Alzheimerovy nemoci pro klinickou praxi. Neurol praxi 2019; 20 : 338–341.
76. Lineweaver TT, Bondi MW, Galasko D et al. Effect of knowledge of APOE genotype on subjective and objective memory performance in healthy older adults. Am J Psychiatry 2014; 171 (2): 201–208. doi: 10.1176/appi.ajp.2013.12121590.
77. Green RC, Roberts JS, Cupples LA et al. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med 2009; 361 (3): 245–254. doi: 10.1056/NEJMoa0809578.
78. Chao S, Roberts JS, Marteau TM et al. Health behavior changes after genetic risk assessment for Alzheimer disease: the REVEAL study. Alzheimer Dis Assoc Disord 2008; 22 (1): 94–97. doi: 10.1097/WAD.0b013e31815a9dcc.
79. Ressner P, Hort J, Rektorová I et al. Doporučené postupy pro diagnostiku Alzheimerovy nemoci a dalších onemocnění spojených s demencí. Cesk Slov Neurol N 2008; 71/104 (4): 494–501.
80. Ferda J, Ferdová E, Baxa J et al. PET/MR u neurodegenerativních onemocnění s kognitivním deficitem. Ces Radiol 2015; 69 (4): 229–237.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2020 Issue 3
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine Eases Daily Life for Patients and Caregivers
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Glioblastom grade IV – dlouhodobé přežití
- Primární progresivní afázie
- Bolesti hlavy v graviditě
- Kognitivní poruchy u dětí s epilepsií