
Septo-optická dysplázia – opomínaná medziodborová klinická jednotka: popis troch prípadov
Septo-optic Dysplasia – Omitted Interdisciplinary Clinic Entity: Report on Three Patients
On example of three patients with septo-optic dysplasia the authors present a rare clinical entity involving combined disturbances of endocrine and neurologic systems and variable expressed clinical triad: 1) pituitary aplasia/dysplasia with pituitary hormones deficiency, 2) developmental disturbance of the middle-brain structures (corpus callosum and septum pellucidum), and 3) dysplastic changes of the optic nerve. The knowledge about components belonging to the competence of other specialists and awareness of consequences of untreated hypopituitarism, are imperatives for interdisciplinary cooperation of ophthalmologist, neurologist, radiologist, and endocrinologist. Moreover, they predict early initiation of the adequate and often vital therapy. Molecular-genetic studies in patients with septo-optic dysplasia represent a way to better knowledge about early stages of the pituitary gland and brain development.
Key words:
septo-optic dysplasia, hypopituitarism, combined pituitary hormone deficiency, optic nerve hypoplasia, gene HESX1
:
K. Podobová 1*; H. Urbánková 2*; S. Kriššáková 3; M. Kantorová 4; M. Šmoldas 5; M. Pura 2
:
títo autori prispeli k práci rovnocenne
*; Očné oddelenie, Ústredná vojenská nemocnica, Ružomberok, Slovensko, vedúci prim. MUDr. Ján Čuvala, Ph. D.
1; Endokrinologické oddelenie, Národný endokrinologický a diabetologický ústav, Ľubochňa, Slovensko, vedúci prim. MUDr. Peter Vaňuga, Ph. D.
2; Pracovisko magnetickej rezonancie, Medicínske centrum NovaMed, Banská Bystrica, Slovensko, vedúci prim. MUDr. Ľubomír Miština, CSc.
3; Laboratórium evokovaných potenciálov, Neurologická klinika MFN a JLF UK, Martin, Slovensko, prednosta doc. MUDr. Egon Kurča, Ph. D.
4; Detské oddelenie, Národný endokrinologický a diabetologický ústav, Ľubochňa, Slovensko, vedúci prim. MUDr. Eva Mendelová
5
:
Čes. a slov. Oftal., 65, 2009, No. 2, p. 59-63
Na príklade kazuistík troch pacientov so septo-optickou dyspláziou autori prezentujú zriedkavú klinickú jednotku zahŕňajúcu kombinované postihnutie endokrinného a neurologického systému v podobe variabilne vyjadrenej triády: 1. aplázie/dysplázie hypofýzy s deficitom hormónov adenohypofýzy, 2. poruchy vývoja stredočiarových štruktúr mozgu (corpus callosum a septum pellucidum) a 3. dysplastických zmien optických nervov. Vedomosť o ďalších súčastiach ochorenia, ktoré nepatria do kompetencie toho ktorého špecialistu a predovšetkým uvedomenie si následkov neliečeného hypopituitarizmu sú imperatívom medziodborovej spolupráce oftalmológa, neurológa, rádiológa a endokrinológa, ako aj predpokladom včasného zahájenia adekvátnej a často vitálnej liečby. Realizácia molekulárno-genetických štúdií u pacientov so septo-optickou dyspláziou je cestou k prehĺbeniu nášho poznania o včasných štádiách vývojových procesov hypofýzy a mozgu.
Kľúčové slová:
septo-optická dysplázia, hypopituitarizmus, kombinovaný deficit hormónov hypofýzy, hypoplázia optického nervu, gén HESX1
Úvod
Septo-optická dysplázia (SOD) (OMIM # 182230), syndróm popísaný v roku 1956 švajčiarskym neurológom Georgesom de Morsierom [7] (syn. de Morsierov syndróm) je klinickou jednotkou, ktorú charakterizuje konštelácia: 1. vývojových porúch stredočiarových štruktúr mozgu, 2. hypoplázie optických nervov a 3. hypoplázie hypofýzy s hypopituitarizmom. Diagnóza SOD je podmienená prítomnosťou dvoch z troch uvedených súčastí, u tretiny pacientov je vyjadrená kompletná triáda [1]. V niektorých prípadoch boli navyše popísané aj abnormality prstov ako syndaktýlia, polydaktýlia, polysyndaktýlia a hypoplázia prstov [4, 9, 10]. U časti pacientov sú podkladom ochorenia mutácie génu HESX1 (Homeobox gene expressed in embryonic stem cells) [5], lokalizovaného na krátkom ramienku 3. chromozómu (3p21.2-21.1) [3]. Zatiaľ bolo identifikovaných päť mutácii spojených s autozomálne recesívnou formou ochorenia a päť mutácií spájajúcich sa s autozomálne dominantnou formou ochorenia [6]. Aj keď je SOD pravidelne uvádzanou „učebnicovou“ etiologickou príčinou vrodených foriem hypopituitarizmu, v klinickej praxi sa s pacientmi s diagnózou SOD stretávame iba zriedka. V zahraničnej, ako aj československej odbornej spisbe je SOD predovšetkým predmetom záujmu endokrinológov [13]. S cieľom upozorniť oftalmologickú odbornú obec na toto „medziodborové“ ochorenie, v nasledujúcom texte autori podávajú popis troch pacientok s variabilne vyjadreným fenotypom SOD.
Popis prípadu
Prípad č. 1
Pacientka dnes 25-ročná (165 cm, 68 kg), dieťa z druhej tehotnosti fyziologického priebehu, narodená v termíne, spontánne, záhlavím. Pôrodné parametre 3150 g/50 cm, popôrodná adaptácia alterovaná, diagnostikovaná bola hemodynamicky nezávažná istmická stenóza aorty, pre ktorú bola prechodne dispenzarizovaná v kardiologickej ambulancii. Vo veku 11 rokov a 11/12 mesiacov diagnostikovaná centrálna hypotyreóza (TSH 1,060 mU/l, fT4 8,7 pmol/l), vo veku 14 rokov a 3/12 mesiacov ozrejmený centrálny hypokorticizmus (nízko polohovaná kortizoliémia 42 nmol/l, s neadekvátne nízkou hodnotou ACTH 22,7 pg/ml) a hypogonadizmus, keď klinicky bola konštatovaná absencia axilárneho a pubického ochlpenia, ultrasonograficky hypoplázia uteru infantilného typu bez diferencovateľných ovárií a laboratórne konštelácia centrálneho hypogonadizmu (LH 2,0 U/l, FSH 2,5 U/L, estradiol 0,00 nmol/l). Vzhľadom na telesné parametre – 59 kg/162 cm (+ 0,7 SDS) nebolo vyslovené podozrenie na deficit rastového hormónu, tento bol potvrdený až vo veku 22 rokov a 2/12 mesiace (tab. 1), kedy bola konštatovaná aj funkčná hyperprolaktinémia mierneho stupňa (PRL 1103 mU/l). Pri vyšetrení magnetickou rezonanciou (MR) popísaná hypoplastická hypofýza „piškótovitého“ tvaru, bez zobrazenia hypofyzárnej stopky, slepo sa končiace infundibulum; stredočiarové štruktúry bez presunu a hypoplázia optických nervov. Po prechode do dospelej endokrinologickej starostlivosti doplnené kompletné oftalmologické vyšetrenie. Pri vyšetrení očného pozadia konštatované nablednutie terčov zrakových nervov (obr. 1), počítačovým perimetrom ozrejmená temporálna hemianopsia vpravo, vľavo depresia zorného poľa v hornom kvadrante so skotomizáciou v dolnom temporálnom, ako aj nazálnom kvadrante (obr. 2). Pri vyšetrení vizuálnych evokovaných potenciálov potvrdená demyelinizačná lézia zrakovej dráhy vpravo ľahkého stupňa, s axonálnou komponentou lézie (stimulácia zrakovej dráhy vľavo: P100 100,3 ms, amplitúda 3,7 μV, stimulácia zrakovej dráhy vpravo: P100 116,5 ms, amplitúda 1,5 μV) (obr. 3). Nálezy svedčia pre dyspláziu optických nervov, výraznejšie vyjadrenú vpravo.




Prípad č. 2
Pacientka dnes 26-ročná (150 cm, 84 kg), dieťa z prvej tehotnosti zdravých rodičov, narodená v 43. týždni indukovaným pôrodom, záhlavím, s pôrodnými parametrami 3280 g/50 cm, s dobrou včasnou popôrodnou adaptáciou. Vo veku 7 mesiacov diagnostikovaná amauróza na podklade hypoplázie optických nervov, konvergentný strabizmus s nystagmom. Vo veku 14 rokov a 9/12 mesiacov odoslaná na naše pracovisko pre zaostávanie v raste (140 cm; -3 SDS). V stimulačnom teste s postinzulínovou hypoglykémiou potvrdený hyposomatotropizmus a hypokorticizmus (tab. 1). Zároveň konštatovaná primárna amenorea, chýbajúce pubické a axilárne ochorenie, pri USG vyšetrení výrazná hypoplázia vnútorného genitálu, hodnoty gonadotropínov a estradiolu v konštelácii centrálneho hypogonadizmu (LH 1,15 U/l, FSH 3,83 U/l, estradiol 0,01 nmol/l). Pri USG vyšetrení hypoplastická štítna žľaza (ľavý lalok s rozmermi 8x8x23 mm, objem 0,7 ml, pravý lalok s rozmermi 9 x 8 x 30 mm, objem 1,03 ml, spolu 1,73 ml), laboratórne centrálna hypotyreóza (TSH 0,044 mU/l, fT4 12,7 pmol/l), u pacientky bola vzápätí zahájená substitučná panhypopituitarizmu. Pri následne prevedenom vyšetrení MR ozrejmená anomália hypotalamo-hypofyzárnej osi – absencia infundibula hypofýzy, ektopia neurohypofýzy, zároveň verifikovaná ľahká hypoplázia optických nervov intraorbitálne a hypoplázia optických nervov ťažkého stupňa v prechiazmatickej časti a v oblasti chiazmy.
Prípad č. 3
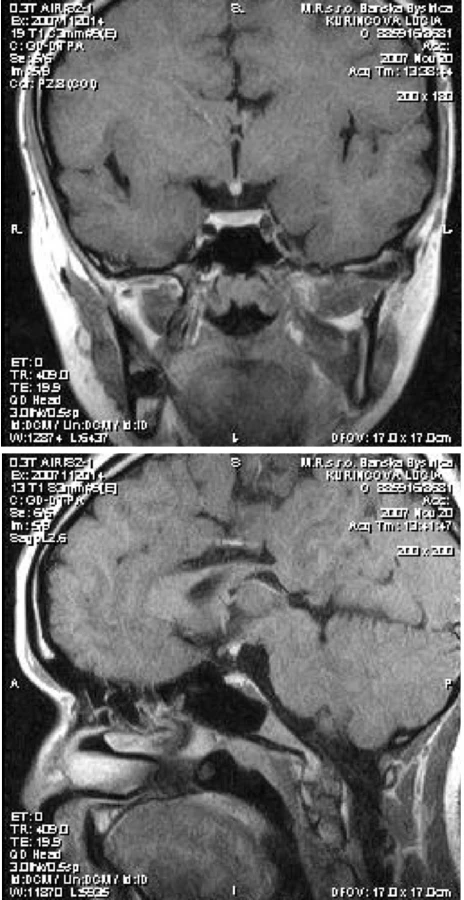
Pacientka t.č. 19-ročná, s aktuálnymi telesnými parametrami 155 cm, 45 kg, z rizikovej gravidity, pôrodu sekciou, s pôrodnými parametrami 3580 g/50 cm, perinatálne konštatovaná fraktúra klavikuly, paréza brachiálneho plexu a pes equinovarus vľavo. Vo veku 6 rokov diagnostikovaný izolovaný deficit rastového hormónu, postupne pridružená centrálna hypotyreóza a centrálny hypokorticizmus. Liečba rastovým hormónom ukončená vo veku 15 rokov pre uzavretie rastových štrbín. Po dosiahnutí dospelosti pacientka detským endokrinológom odoslaná na pracovisko autorov, kde pri prevzatí do starostlivosti indikované morfologické zobrazovacie vyšetrenie hypofýzy MR. Pri vyšetrení (toto prevedené technikou T2, T1 natív COR, T1 Omniscan i. v. dynamické sekvencie COR, oneskorené sekvencie COR, SAG) zobrazená adenohypofýza primeraného uloženia, konfigurácie a veľkosti, diafragma sellae bez elevácie. Po aplikácii kontrastnej látky homogénny enhancement intrasellárnych štruktúr, na oneskorených sekvenciách homogénna intenzita adenohypofýzy. Neurohypofýza ektopicky uložená pri dolnom okraji hypotalamu, tesne za optickou chiazmou. Samotná optická chiazma bez patologického nálezu. Ako vedľajší nález ozrejmená parciálna agenéza tela svorového telesa (truncus corporis callosum) s asymetriou tiel postranných komôr, susp. komunikácia ľavej postrannej komory s interhemisferálnou fisúrou (obr. 4). Vzhľadom na koincidenciu hypopituitarizmu a vývojovej anomálie stredočiarových štruktúr mozgu vyslovujeme podozrenie na SOD a dopĺňame detailné oftalmologické vyšetrenie. Pri tomto bol konštatovaný fyziologický nález na očnom pozadí – papily zrakového disku v niveau sietnice, ružovej farby, bez známok mestnania alebo atrofie, sietnica priložená, makula neporušená, foveolárny reflex živý.
Koincidencia hypopituitarizmu a/alebo vývojových anomálií stredočiarových štruktúr mozgu a hypofýzy a/alebo dysplázie optických nervov nás viedli v prípadoch vyššie popísaných pacientiek k stanoveniu diagnózy SOD. V súčasnosti preto u všetkých troch pacientiek realizujeme molekulárno-genetickú analýzu HESX1 génu.
Diskusia
Napriek tomu, že SOD je klinickou jednotkou popísanou v medicínskej literatúre pred vyše 50 rokmi [7] a predstavuje jedno z množstva tzv. medziodborových ochorení, v diagnostických záveroch z očných, neurologických, resp. endokrinologických vyšetrení sa nachádza iba výnimočne. Po identifikácii prvých pacientov s mutáciami génu HESX1 sa predpokladalo, že SOD môže predstavovať jedno z monogénne podmienených ochorení. S narastajúcim počtom prípadov SOD však pribúdali pacienti bez identifikácie mutácií v kódujúcej sekvencii génu HESX1 a dnes je zrejmé, že ide o multifaktoriálne podmienené ochorenie. V nedávnej štúdii s pravdepodobne najväčším počtom pacientov (celkovo 927 pacientov s prítomným minimálne jedným príznakom z triády SOD) boli mutácie génu HESX1 identifikované v menej ako 1 % prípadov [6]. Z literárnych údajov vyplýva, že ochorenie sa častejšie vyskytuje u detí mladých matiek, resp. matiek rodiacich v teenegerskom veku [8, 11]. Okrem genetických faktorov [14] sa uvažuje o environmentálnych vplyvoch (užívanie liekov, fajčenie matky, abúzus alkoholu a drôg počas tehotenstva) [12].
Na príklade troch pacientiek s fenotypovo typicky vyjadrenou SOD autori prezentujú túto v literatúre síce uvádzanú, ale v praxi opomínanú klinickú jednotku. V našej klinickej praxi sa iba zriedkavo stretávame s pacientmi odosielanými na endokrinologické vyšetrenie oftalmológmi pre ozrejmenú dyspláziu optických nervov, iba výnimočne s pacientmi odosielanými priamo od rádiológov po realizácií zobrazovacieho vyšetrenia s nálezom anomálií stredočiarových štruktúr mozgu. Na druhej strane, diagnóza SOD bola u všetkých vyššie popísaných pacientiek stanovená ex post až v dospelosti, po ich prevzatí od pediatrických endokrinológov a po zhodnotení pridružených (zdanlivo nesúvisiacich) ochorení.
Fenotypové spektrum SOD je pestré, čo potvrdzujú aj nami prezentované kazuistiky. U pacientov s miernou formou môže byť prítomný iba izolovaný deficit rastového hormónu, zatiaľ čo pre plne vyjadrenú formu SOD je charakteristický panhypopituitarizmus, aplázia/hypoplázia adenohypofýzy, ektopia neurohypofýzy, agenéza corpus callosum, absencia septum pellucidum a hypoplázia optických nervov. Variabilným je aj čas manifestácie a poradie nástupu deficitov jednotlivých hormónov hypofýzy. V štúdii 55 pacientov s hypopláziou optických nervov Birkebaek a spol. zistili, že 49 % pacientov malo abnormálne septum pellucidum a 64 % pacientov malo abnormality hypotalamo-hypofyzárnej osi v MR obraze [2]. Frekvencia endokrinných porúch, ako aj závažnosť endokrinopatie (kombinovaný deficit hormónov hypofýzy) boli vyššie u pacientov s abnormalitami septum pellucidum a anomáliami hypotalamo-hypofyzárneho komplexu, v porovnaní s pacientmi s normálnym MR obrazom uvedených štruktúr. Tieto sa preto pokladajú za prediktory pravdepodobnosti a závažnosti spektra deficitu hormónov hypofýzy [2].
Vzájomné odosielanie pacientov medzi špecialistami (oftalmológ, neurológ, rádiológ, endokrinológ) po diagnostikovaní prvej zo súčastí je cestou k identifikácii ďalších zložiek klinickej triády SOD. U pacientov s hypopláziou optických nervov, resp. závažnou poruchou zraku, ako aj u pacientov s anomáliami stredočiarových štruktúr mozgu je indikované detailné a opakované endokrinologické vyšetrenie, vice versa u pacientov s „idiopatickým“ izolovaným deficitom rastového hormónu alebo s „idiopatickým“ kombinovaným deficitom hormónov hypofýzy je nutné v rámci diferenciálnej diagnózy uvažovať o SOD a pomocou armamentária vyšetrovacích metodík identifikovať prípadné dve ostávajúce súčasti syndrómu. Iba včasná diagnostika a adekvátna liečba môžu zabrániť fatálnym následkom (napr. neliečeného hypokorticizmu), resp. zlepšiť kvalitu života pacientov so SOD (napr. elimináciou sociálneho handicapu nízkeho vzrastu po liečbe rastovým hormónom). Realizácia molekulárno-genetických štúdií u pacientov so SOD je cestou k prehĺbeniu nášho poznania o včasných fázach vývoja hypofýzy a zodpovedaniu aktuálne nastolených otázok, ako je napr. potvrdenie predpokladanej centrálnej úlohy HESX1 v supresii PROP1-mediovanej aktivácie transkripcie [6].
MUDr. Mikuláš Pura
Endokrinologické oddelenie,
Národný endokrinologický a diabetologický ústav,
034 91 Ľubochňa, Slovensko
tel.: +421-44-4306 214
fax: +421-44-4306 322
e-mail: mikulas.pura@nedu.sk
Sources
1. Arslanian, S. A., Rothfus, W. E., Foley jr., T. P.: Hormonal, metabolic, and neuroradiologic abnormalities associated with septo-optic dysplasia. Acta Endocrinol. Copenh. 107, 1984; 2 : 282–288.
2. Birkebaek, N. H., Patel, L., Wright, N. B. et al.: Endocrine status in patients with optic nerve hypoplasia: relationship to midline central nervous system abnormalities and appearance of the hypothalamic-pituitary axis on magnetic resonance imaging. J. Clin. Endocrinol. Metab. 88, 2003; 11 : 5281–5286.
3. Dattani, M. T., Martinez-Barbera, J.-P., Thomas, P. Q., et al.: Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nature Genet. 19, 1998; 2 : 125–133.
4. Harrison, I. M., Brosnahan, D., Phelan, E. et al.: Septo-optic dysplasia with digital anomalies – a recurrent pattern syndrome. Am. J. Med. Genet. 131, 2004; 1 : 82–85.
5. Hermesz, E., Mackem, S., Mahon, K. A.: Rpx: a novel anterior-restricted homeobox gene progressively activated in the prechordal plate, anterior neural plate and Rathke‘s pouch of the mouse embryo. Development. 122, 1996; 1 : 41–52.
6. McNay, D. E. G., Turton, J. P., Kelberman, D. et al.: HESX1 mutations are an uncommon cause of septo-optic dysplasia and hypopituitarism. J. Clin. Endocrinol. Metab. 92, 2007; 2 : 691–697.
7. de Morsier, G.: Études sur les dysraphies, crČnioencéphaliques. III. Agénésie du septum palludicum avec malformation du tractus optique. La dysplasie septo-optique. Schweizer Archiv für Neurologie und Psychiatrie, Zurich, 77, 1956; 267–292.
8. Murray, P. G., Paterson, W. F., Donaldson, M. D.: Maternal age in patients with septo-optic dysplasia. J. Pediatr. Endocrinol. Metab. 18, 2005; 5 : 471–476.
9. Orrico, A., Galli, L., Zappella, M.: Septo-optic dysplasia with digital anomalies associated with maternal multidrug abuse during pregnancy. Eur. J. Neurol. 9, 2002; 6 : 679–682.
10. Pagon, R. A., Stephan, M. J.: Septo-optic dysplasia with digital anomalies. J. Pediatr. 105, 1984; 6 : 966–968.
11. Patel, L., McNally, R. J., Harrison, E. et al.: Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J. Pediatr. 148, 2006; 1 : 85–88.
12. Rainbow, L. A., Rees, S. A., Shaikh, M. G. et al.: Mutation analysis of POUF-1, PROP-1 and HESX-1 show low frequency of mutations in children with sporadic forms of combined pituitary hormone deficiency and septo-optic dysplasia. Clin Endocrinol (Oxf). 62, 2005; 2 : 163–168.
13. Vosáhlo, J., Krásný, J., Srp, A. et al.: Septooptická dysplazie: morfologické, oftalmologické a endokrinní nálezy u 11 pacientů. Čes.-slov. Pediat. 5, 2003; 5 : 287–290.
14. Wales, J. K., Quarrell, O. W.: Evidence for possible Mendelian inheritance of septo-optic dysplasia. Acta Paediatr. 85, 1996; 3 : 391–392.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2009 Issue 2
Most read in this issue
- Central Serous Choroidopathy as Rare Complication of the Corticosteroid Treatment
- nfluence of Haemorheopheresis in the Dry Form of the Age Related Macular Degeneration
- Septo-optic Dysplasia – Omitted Interdisciplinary Clinic Entity: Report on Three Patients
- Statistical Analysis of the Nerve Fiber Layer in Color Digital Images of the Retina