
Molekulárně genetická příčina a klinický nález u dvou probandů se Stargardtovou chorobou
The Molecular Genetic and Clinical Findings in two Probands with Stargardt Disease
Purpose:
The aim of our study was to describe the phenotype and to perform molecular genetic investigation in two probands of Czech origin diagnosed with Stargardt disease (STGD).
Methods:
Both males underwent ocular examination including assessment by high-resolution spectral domain optical coherence tomography (SD-OCT). DNA was isolated from venous blood. Mutation detection was performed using the ABCA4 genotyping microarray (Asper Ophthalmics, Estonia).
Results:
The best corrected visual acuity in proband 1 (aged 39 years) was 0.1 bilaterally, and 0.05 in proband 2 (aged 26 years). Fundus examination showed typical multiple yellow-white lesions and macular atrophy. Alterations of retinal pigment epithelium, retinal thinning and disruption of the photoreceptor inner segment ellipsoid band were detected with an SD-OCT. Two known disease-causing mutations in ABCA4 were identified in proband 1; c.4234C>T, p.(Gln1412*) in exon 28; and c.5882G>A, p.(Gly1961Glu) in exon 42. Only one pathogenic change was detected in proband 2; c.1988G>A, p.(Trp663*) in exon 14. A second change, anticipated because of the recessive status of the disease, was not identified.
Conclusion:
The frequency and full spectrum of ABCA4 mutations in Czech patients with inherited retinal disorders is yet to be established. The inability to detect a second pathogenic change in ABCA4 coding sequences in proband 2 warrants further investigation.
Key words:
Stargardt disease, ABCA4, mutation, SD-OCT
:
B. Kousal 1,5; J. Záhlava 2; Š. Vejvalková 3; M. Hejtmánková 4; P. Lišková 1,5
:
Oãní klinika, 1. lékafiská fakulta, Univerzita
Karlova v Praze a V‰eobecná fakultní
nemocnice v Praze, pfiednostka
doc. MUDr. Bohdana Kalvodová, CSc.
1; Oãní klinika JL, s. r. o., primáfi MUDr.
Ján Le‰ták, CSc., FEBO, MBA, LL. A
FAOG
2; Ústav biologie a lékafiské genetiky, 2.
lékafiská fakulta, Univerzita Karlova
v Praze a Fakultní nemocnice v Motole
pfiednosta prof. MUDr. Milan Macek jr.
DrSc.
3; GENNET, Praha, vedoucí MUDr.
David Stejskal
4; Laboratofi biologie a patologie oka
Ústav dûdiãn˘ch metabolick˘ch poruch
1. lékafiská fakulta, Univerzita Karlova
v Praze a V‰eobecná fakultní nemocnice
v Praze, pfiednosta prof. MUDr.
Viktor KoÏich, CSc.
5
:
Čes. a slov. Oftal., 70, 2014, No. 6, p. 228-233
:
Original Article
Cíl:
Popsat klinický nález a provést molekulárně genetickou analýzu u dvou probandů českého původu s diagnózou Stargardtovy choroby. Poskytnout souhrn praktických poznatků plynoucích z mutační analýzy genu ABCA4 a poukázat na některé problematické aspekty spojené se screeningem tohoto genu.
Metody:
U obou mužů ve věku 39 a 26 let jsme provedli oční vyšetření včetně optické koherenční tomografie se spektrální doménou (SD-OCT). DNA byla izolována z venózní krve. Identifikace patogenních sekvenčních variant byla provedena pomocí genotypovacího mikročipu pro gen ABCA4, verze 11.0 (Asper Ophthalmics, Estonsko) detekující 558 známých mutací a jednonukleotidových polymorfismů.
Výsledky:
U prvního probanda byla nejlepší korigovaná zraková ostrost obou očí 0,1, u druhého probanda 0,05. Klinické vyšetření prokázalo typický nález atrofie makuly s přítomností žlutavých skvrn na očním pozadí. Vyšetření SD-OCT odhalilo oboustranně chybění linie elipsoidů vnitřních segmentů fotoreceptorů, také v literatuře označovaná jako linie junkce vnitřních a zevních segmentů fotoreceptorů, ztenčení neuroretiny a nepravidelnosti pigmentového epitelu sítnice. V souladu s autozomálně recesivním typem přenosu byla rodinná anamnéza pro Stargardtovu chorobu v obou případech negativní. Molekulárně genetickým vyšetřením bylo zjištěno, že první proband je nositelem mutace c.4234C>T; p.(Gln1412*) v exonu 28 a c.5882G>A; p.(Gly1961Glu) v exonu 42. U druhého probanda byla detekována pouze jedna známá patogenní mutace c.1988G>A p.(Trp663*) v exonu 14.
Závěr:
Poprvé byly u českých pacientů zjištěny mutace zodpovědné za vznik Stargardtovy choroby. Nemožnost nalézt jednu nebo obě patogenní sekvenční varianty je u této choroby za použití současných metod poměrně častým jevem. Budoucí studie by se měly zaměřit na stanovení spektra a frekvence jednotlivých mutací v ABCA4 u rozsáhlejšího souboru pacientů českého původu. Znalost příčinných mutací zlepšuje klinické poradenství pacientům a v indikovaných případech umožňuje preimplantační diagnostiku.
Klíčová slova:
Stargardtova choroba, ABCA4, mutace, SD-OCT
ÚVOD
Stargardtova choroba a fundus flavimaculatus jsou varianty dědičného onemocnění, které postihuje pigmentový epitel sítnice (retinální pigmentový epitel, RPE) a fotoreceptory. Charakteristickým klinickým nálezem je přítomnost žlutavých skvrn v makule na úrovni RPE spolu s atrofií makuly. Stargardtova choroba se typicky projevuje jako ireverzibilní snížení centrální zrakové ostrosti, které vzniká již v dětství nebo v rané dospělosti, známky onemocnění se ale mohou dostavit i později. Při úvodních projevech může mít makula normální vzhled, poté se mohou objevit drobné hyperpigmentace, následně nabývá vzhledu tepaného bronzu a postupně vzniká atrofie, někdy se žlutavými skvrnami při okrajích nebo i difuzně po celém fundu. Jejich vzhled a velikost bývá extrémně různorodý. V periférii sítnice se mohou později objevit také přesuny pigmentu. V konečném důsledku může choroba vést až k úplné slepotě. Prevalence onemocnění je relativně vysoká a odhaduje se 1 postižený na 8-10 000 obyvatel (10).
Za nejspolehlivější metodu, jak rozpoznat Stargardtovu chorobu a zvláště rozsah patologických změn, je dnes považována autofluorescence fundu (7, 22, 47). V diagnostice se také používají optická koherenční tomografie se spektrální doménou (SD-OCT), fluorescenční angiografie, vyšetření zorného pole a elektroretinografie (23, 25, 28, 40, 41). SD-OCT umožňuje zobrazit změny již od časných stadií onemocnění. Typický je nález nepravidelností RPE, dezorganizace až ztráta linie elipsoidů vnitřních segmentů fotoreceptorů, v literatuře také označovaná jako linie junkce vnitřních a zevních segmentů fotoreceptorů, ztráta zevní nukleární vrstvy a ztenčování neuroretiny (32).
Stargardtova choroba se dědí nejčastěji autozomálně recesivně (STGD1). Onemocnění je podmíněné mutacemi v genu ABCA4 (ATP-binding cassette, subfamily A, member 4) lokalizovaném na chromozómu 1p22, který kóduje sítnicově specifický ATP-vázající transportní protein nalézající se na okrajích fotoreceptorových disků ve vnějších segmentech tyčinek a čípků (6). ABCA4 pomáhá odstraňovat potenciálně toxické sítnicové sloučeniny z fotoreceptorů po excitaci. Narušením tohoto transportu dochází po fagocytóze vnějších segmentů fotoreceptorů k jejich akumulaci v RPE a k vytváření lipofuscinových depozit. Hromadící se toxické sloučeniny pak vedou k apoptóze RPE, a protože přežití fotoreceptorů je na RPE závislé, dochází následně i k jejich degeneraci (29).
Vzácně mohou onemocnění podobající se Stargardtově chorobě vyvolat mutace v genech ELOVL4 (ELOVL fatty acid elongase 4) (STGD3) a PROM1 (prominin 1) (STGD4) (14, 45, 49, 52).
METODY
Vyšetřili jsme dva nepříbuzné pacienty s klinickými známkami Stargardtovy choroby. Nejlepší korigovaná zraková ostrost byla zjišťována pomocí ETDRS optotypů. Vidění do blízka bylo testováno pomocí Jägerových tabulek s rozsahem od nejmenšího k největšímu textu 1–12. Provedli jsme biomikroskopické vyšetření fundu v mydriáze, barevnou fotografii očního pozadí, snímek v bezčerveném světle a vyšetření autofluorescence fundu (Visucam 200, Carl Zeiss Meditec AG, Německo), která využívá fluorescenční schopnosti lipofuscinu ke znázornění jeho distribuce a akumulace a k odlišení poškozených receptorů sítnice a poškozeného RPE. Dále bylo provedeno vyšetření zorného pole statickým perimetrem (M-700, Medmont International Pty Ltd., Vermont, Austrálie), vyšetření kontrastní citlivosti na tabuli Pelli-Robson (HS Clement Clarke International, Essex, Velká Británie), vyšetření SD-OCT s rozlišením axiálním 5 μm a transverzálním 15 μm (RTVue, Optovue, Inc, Fremont, USA) a přístrojem, který je kombinací SD-OCT s axiálním rozlišením 6 μm a skenovacího laserového oftalmoskopu (Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Kanada).
Odebrali jsme 3 ml venózní krve do EDTA zkumavek a izolovali genomovou DNA za použití systému FLEX STAR (AutoGen, USA) podle instrukcí výrobce. K identifikaci mutací jsme použili genotypovací mikročip pro gen ABCA4, verze 11.0 (Asper Ophthalmics, Estonsko) detekující 558 známých mutací a jednonukleotidových polymorfismů.
Nomenklatura popisu mutací vycházela z doporučení Společnosti pro variace v lidském genomu (Human Genome Variation Society, http://www.hgvs.org/rec.html), kdy první adenin inicializačního kodonu ATG v kódující DNA je vždy označen číslem 1. NM_000350.2 byla použita jako referenční sekvence. Výzkum byl proveden v souladu s Helsinskou deklarací. Oba pacienti podepsali písemný informovaný souhlas.
VÝSLEDKY
Rozsah klinického vyšetření probandů závisel na dostupnosti výše uvedených metod na jednotlivých pracovištích.
Proband 1 v dětství viděl na obě oči stejně dobře, ale od 15 let věku pozoroval pomalé postupné zhoršování centrální zrakové ostrosti a světloplachost při ostrém osvětlení. Známky Stargardtovy choroby byly při vyšetření fundu poprvé popsány ve věku 25 let. Zraková ostrost byla 0,16 na obou očích bez korekce. Vidění do blízka bylo oboustranně sníženo na čtení textu s Jägerovým číslem 6. Ve věku 30 let zůstala zraková ostrost na obou očích do dálky s korekcí -1,5 D zachována, ale do blízka byla snížena na čtení textu s Jägerovým číslem 8. Při posledním vyšetření ve věku 39 let byla zraková ostrost snížena na 0,1 oboustranně, vpravo s korekcí -2,75 D = -1,0 Dcyl ax 20° a vlevo s korekcí -3,0 D. Do blízka četl pacient text s Jägerovým číslem 10. V makulách obou očí bylo přítomno horizontálně oválné ložisko nepravidelné poruchy RPE vzhledu tepaného bronzu, biomikroskopicky velikosti 1,7 průměru terče zrakového nervu, chyběl foveální reflex a při nazálním okraji makuly pravého oka pak bylo drobné, síťovitě pigmentované okrouhlé ložisko velikosti 0,5 průměru terče zrakového nervu. Kolem makul při cévních arkádách i nazálně od papil byla přítomna žlutavá ložiska na úrovni RPE, sítnicové cévy se jevily normální (obr. 1a-d). Nález na SD-OCT odhalil oboustranně ztenčení neuroretiny v makulární oblasti s atrofií zevní nukleární vrstvy, nepřítomnost linie elipsoidů vnitřních segmentů fotoreceptorů a atrofii RPE s výraznějším znázorněním struktur cévnatky (obr. 1e, f). Nejmenší tloušťka neuroretiny ve foveách obou očí byla 20 μm. Vyšetření zorného pole prokázalo oboustranně paracentrální skotomy nahoře, větší vpravo, validita vyšetření byla snížena ztrátou fixace (obr. 1g, h).

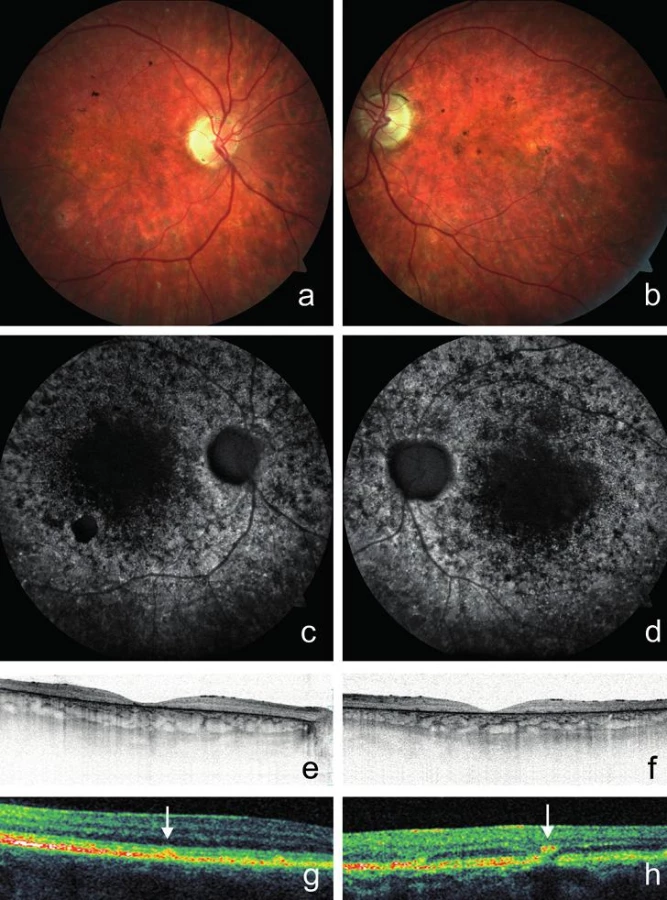
U probanda 2 se onemocnění sítnice projevilo poklesem vidění do blízka již v 6 letech. Šeroslepý ani světloplachý nebyl. Při našem vyšetření ve věku 26 let byla zraková ostrost obou očí 0,05, do blízka nečetl ani největší Jägerovu tabulku. Klinicky jsme nalezli pokročilé projevy onemocnění s atrofií neuroretiny a pigmentového epitelu sítnice v makule. Kolem makuly i zevně cévních arkád byla rozmístěna žlutobílá ložiska (obr. 2a, b). Autofluorescence fundu vykazovala poruchu distribuce. V oblastech atrofie RPE zcela chyběla, zatímco v jejich okolí jsme pozorovali oblasti s nepravidelně zrnitou autofluorescencí. Naopak v místech hromadění lipofuscinových depozit byla autofluorescence zvýšena (obr. 2c, d). Při vyšetření SD-OCT byla oboustranně zaznamenána rozsáhlá atrofie zevní nukleární vrstvy provázená chyběním linie elipsoidů vnitřních segmentů fotoreceptorů, atrofie RPE a ztenčení neuroretiny (obr. 2e, f). Také byla dokumentována hyperreflexivní ložiska uložená v různé hloubce sítnice a odpovídající žlutobílým skvrnám (obr. 2g, h). Minimální výška neuroretiny ve fovee pravého oka byla 35 μm a levého oka 20 μm. Vyšetření zorného pole statickou počítačovou perimetrií bylo nespolehlivé pro neschopnost fixovat.
Molekulárně genetická analýza
U probanda 1 byly detekovány dvě příčinné mutace v genu ABCA4: záměna cytosinu za tymin (c.4234C>T) v exonu 28 vedoucí ke vzniku terminačního kodonu na pozici 1412 p.(Gln1412*). Druhá patogenní mutace c.5882G>A se nalézala v exonu 42 a její přítomnost znamená záměnu aminokyseliny glycinu za glutamin na pozici 1961 p.(Gly1961Glu).
U probanda 2 byla v genu ABCA4 prokázána pouze jedna známá mutace c.1988G>A v exonu 14 způsobující zařazení terminačního kodonu na pozici 663 p.(Trp663*).
DISKUSE
Naše práce poprvé popisuje molekulárně genetickou příčinu Stargardtovy choroby u pacientů českého původu. Podrobně byl zároveň zdokumentován také oční nález pomocí moderních zobrazovacích metod, jako jsou SD-OCT a vyšetření autofluorescence fundu.
Stargardtova choroba byla poprvé popsána v roce 1909 Karlem Stargardtem a patří mezi nejčastější dědičná onemocnění makuly. U této choroby je popisován oboustranný pokles zrakové ostrosti s počátkem obvykle v dětství nebo rané dospělosti, jak jsme pozorovali i u našich pacientů. Obvykle pacienti trpí postupnou ztrátou zrakové ostrosti až na hodnotu okolo 0,1, avšak míra ztráty zrakové ostrosti závisí na věku, ve kterém nastaly první projevy onemocnění. U probanda 2 se objevily první příznaky onemocnění dříve než u probanda 1 a v souladu s tím měl i větší ztrátu zrakové ostrosti, která byla zároveň pod průměrem zrakové ostrosti obvyklé v konečném stadiu.
Také nález na očním pozadí byl u probanda 2 pokročilejší než u probanda 1, přestože byl pacient v době našeho vyšetření mladší. V makulách byla zjištěna u probanda 1 ložiska nepravidelné atrofie RPE a částečně choriocapillaris vzhledu tepaného bronzu, u probanda 2 již byla přítomna atrofie sítnice včetně RPE. U obou probandů jsme v oblasti zadního pólu i v periferii pozorovali typické žlutavé až žlutobílé skvrny různého tvaru. Obecně ve shodě s publikovanými údaji jsou při biomikroskopickém vyšetření a na barevné fotografii fundu skvrny často málo kontrastní, lépe jsou viditelné v bezčerveném světle, nejlépe a nejzřetelněji jsou však patrné při vyšetření autofluorescence fundu, kdy na podkladě hromadění lipofuscinu v RPE vykazují hyperautofluorescenci (46, 47). U probanda 1 byly skvrny větší, vytvářely síťovité vzory a měly žlutavou barvu, u probanda 2 byly menší velikosti, více izolované a okrouhlé, barvy žlutobílé.
Při vyšetření SD-OCT jsme u obou probandů zaznamenali ztenčení neuroretiny, chybění linie elipsoidů vnitřních segmentů fotoreceptorů a hyperreflexivní ložiska umístěná v zevních vrstvách neuroretiny. Podle lokalizace se dělí na dvě skupiny. Ložiska typu 1 mají tvar kupole a nacházejí se v úrovni nebo těsně nad úrovní vnitřní strany linie RPE. Reflexivita obou objektů může být obdobná a jejich odlišení při SD-OCT vyšetření obtížné. Ložiska typu 2 se jeví jako malé lineární hyperreflexivní léze zasahující do zevní nukleární vrstvy sítnice a mohou být odděleny od linie RPE (46). Léze obou typů jsme pozorovali u obou našich probandů.
Snížení zrakové ostrosti odpovídá úbytku receptorů, který také koreluje s rozsahem změn patrných při vyšetření autofluorescence fundu. Naopak snížení výšky sítnice nemusí být nutně provázeno ztrátou receptorů a i přes progresivní atrofii intraretinálních vrstev může být zraková ostrost lepší, než by se mohlo očekávat (15).
Vyšetření zorného pole statickou perimetrií bylo u probanda 1 méně validní pro ztrátu fixace, u probanda 2 nešlo ze stejného důvodu provést vůbec. Typickým nálezem při perimetrickém vyšetření je u Stargardtovy choroby centrální skotom.
Ani u jednoho z pacientů nebyla pozitivní rodinná anamnéza pro Stargardtovu chorobu, což je v souladu s autozomálně recesivní dědičností a tudíž se STGD1, podmíněnou mutacemi v genu ABCA4. Mutace v tomto genu způsobují řadu autozomálně recesivních sítnicových degenerací s postižením makuly, kromě Stargardtovy choroby i autozomálně recesivní čípko-tyčinkové dystrofie a retinitis pigmentosa (9, 12, 17, 26). Některé varianty v tomto genu jsou spojovány i se zvýšeným rizikem vzniku věkem podmíněné makulární degenerace (3, 5), nicméně ne všechny práce tuto asociaci potvrzují (4, 42).
Gen ABCA4 má 50 exonů, kóduje protein o velikosti 2273 aminokyselin, a je tedy relativně veliký. Provedení screeningu konvenční mutační analýzou pomocí Sangerova sekvenování by bylo náročné jak časově, tak i finančně, a proto byla zvolena levnější metoda detekce mutací pomocí genotypovacího mikročipu, který využívá technologii APEX („arrayed primer extension“) (24). V době vyšetření obou probandů tento mikročip obsahoval 558 různých patogenních sekvenčních variant a benigních polymorfismů (51) a k systematickému screeningu onemocnění sítnice asociovaných s mutacemi v genu ABCA4 byl použit již v řadě předchozích studií pro příznivý poměr ceny k výkonu (20, 34, 44). Obě mutace jsou touto metodou nalezeny v průměru u 40 % pacientů s diagnostikovanou klasickou STGD1, u dalších 40 % pacientů je zjištěna mutace jedna a u 20 % mutace detekována není (19). Falešná pozitivita či negativita je při použití genotypovacího mikročipu menší než 2%, nicméně pokud to podmínky dovolují, je vhodné prokázat přítomnost zjištěných mutací ještě jinou metodou, nejčastěji Sangerovým sekvenováním (24, 34). Nevýhodou genotypovacího mikročipu je, že lze vyšetřit přítomnost pouze známých sekvenčních variant, nové mutace tak nejsou detekovány. V současné době se při zjišťování mutací přechází na technologii sekvenování nové generace, která vykazuje příznivý poměr ceny a výkonu a umožňuje i detekci dosud nepopsaných sekvenčních variant (19, 51).
Obě mutace v genu ABCA4 prokázané u probanda 1 byly již dříve popsány v souvislosti se STGD1 u případů z různých populací. Zatímco je poměrně vzácná a byla nalezena pouze v několika v rodinách z USA a v dánské populaci (28, 35), je v řadě populací nejčastěji detekovanou patogenní sekvenční variantou (1, 11, 20, 24). Mutace přítomná u probanda 2 byla zjištěna u jednoho pacienta německého původu a v jedné rodině původem z USA (10, 33). Nenalezení druhé patogenní varianty může být dáno právě výskytem nové, dosud nepopsané mutace. Nicméně sekvence genu ABCA4 je značně variabilní a ani použití nejnovějších technologií nevede u řady pacientů k detekci jedné či obou mutací. Dosud neodhalené změny v ABCA4 se mohou totiž nacházet v regulatorních sekvencích či hluboko v intronech s obtížně prokazatelnou patogenitou (8).
Efekt mutací na funkci ABCA4 je různorodý, dochází např. ke snížení exprese, nenavázání substrátu, retenci v endoplazmatickém retikulu, snížení ATPázové aktivity, což vše vede ve svém důsledku ke snížení transportní aktivity tohoto proteinu. Tyto různé funkční důsledky se pravděpodobně podílí na širokém spektru fenotypového vyjádření onemocnění sítnice na podkladě mutací v ABCA4 (29). První patogenní varianta prokázaná u probanda 1 vede ke vzniku terminačního kodonu ve druhé polovině genu, jehož důsledkem je předčasné ukončení translace, a tedy zkrácení proteinu ABCA4 se závažným narušením jeho funkce. Druhá mutace p.(Gly1961Glu) zjištěná u probanda 1 je svojí povahou missense, tj. vede k záměně jedné aminokyseliny za jinou. Předpokládá se, že způsobuje alteraci funkce proteinu snížením vazby ATP a ATPázové aktivity (43). Tato mutace se nalézá mimo funkční domény ABCA4 a bývá spojována s mírnějším klinickým vyjádřením onemocnění (18, 38), což by odpovídalo i nálezu u probanda 1 v porovnání s probandem 2. V důsledku sekvenční patogenní varianty nalezené u probanda 2 vzniká terminační kodon již v první čtvrtině genu a kódovaný protein je tak pravděpodobně zcela nefunkční, alternativně může docházet k degradaci mRNA na podkladě obranného buněčného mechanismu nonsense mediated decay (21).
Frekvence výskytu změn v ABCA4 nebyla v běžné české populaci stanovena. O tomto genu je známo, že obsahuje extrémní množství polymorfismů, často s nejasným vlivem na funkci proteinu (37, 48). Odhaduje se, že zhruba každý 20. jedinec by mohl být nositelem nějaké potenciálně patogenní změny v ABCA4 a je tudíž možné, že prevalence onemocnění sítnice asociovaných s tímto genem je vyšší, než je v současné době odhadováno (10, 24, 28, 50). V rámci plánování rodičovství pacientů s dystrofií sítnice podmíněnou mutacemi v ABCA4 není tedy žádost o screening ABCA4 u zdravého partnera zcela neopodstatněná. Právě kvůli vysoké frekvenci patogenních změn se mohou recesivní onemocnění vznikající na podkladě mutací v tomto genu vyskytovat i ve více generacích jedné rodiny (tzv. pseudodominance), což ztěžuje diferenciální diagnostiku a genetické poradenství (10, 34, 36).
Průkaz příčinných mutací umožňuje nabídnout nositelům patogenních mutací (např. rodičům již postiženého dítěte) zabránění přenosu onemocnění na další generaci pomocí preimplantační diagnostiky (13). V roce 2010 byla publikována práce popisující tento postup právě u muže se Stargardtovou chorobou a jeho partnerky, která byla prokázanou nositelkou patogenní alely v ABCA4. Jejich společný potomek by měl tedy při spontánním početí 50% riziko onemocnět Stargardtovou chorobou (39).
Pro Stargardtovu chorobu zatím neexistuje účinná léčba. Na základě studia zvířecích modelů prokazujícího zvýšenou tvorbu toxických sloučenin ve vnějších segmentech fotoreceptorů při expozici světlu je pacientům doporučeno chránit oči slunečními brýlemi a nedoplňovat stravu o vitamín A (16, 27, 31, 34). Pilotní studie, v rámci které byli pacienti s diagnózou sítnicové degenerace asociované s ABCA4 suplementováni luteinem po dobu 6 měsíců, vedla u dvou třetin očí ke zvýšení optické hustoty makulárního pigmentu, ke zlepšení centrální zrakové ostrosti ale nedošlo (2).
Znalost přesné molekulární diagnózy může být jedním z požadavků pro zařazení do klinických zkoušek genové terapie nebo terapie kmenovými buňkami, které byly v nedávné době započaty. Od roku 2011 probíhá první klinická studie na lidech zkoumající bezpečnost lentivirového vektoru vyvinutého pro genovou terapii Stargardtovy choroby. Léčebné intervence v budoucnosti se také mohou zaměřit na odstraňování toxických derivátů vitamínu A (30), zkouší se i účinek neuroprotektiv (www.clinicaltrials.gov). Do doby, než budou známy výsledky těchto klinických zkoušek, jsou však pacienti se sítnicovým onemocněním podmíněným mutacemi v genu ABCA4 odkázáni pouze na různé korekční pomůcky pro zrakově postižené.
Do redakce doručeno dne 2. 7. 2014
Do tisku přijato dne 14. 11. 2014
MUDr. P. Lišková, Ph.D.
Laboratoř biologie a patologie oka, Ústav dědičných metabolických poruch,
1. lékařská fakulta, Univerzita Karlova v Praze a
Všeobecná fakultní nemocnice v Praze
Ke Karlovu 2
128 00 Praha 2
petra.liskova@lf1.cuni.cz
Sources
1. Aguirre-Lamban, J., Riveiro-Alvarez, R., Maia-Lopes, S., et al.: Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: identification of 21 novel variants. Br J Ophthalmol, 2009; 93(5): 614–21.
2. Aleman, TS., Cideciyan, AV., Windsor, EA., et al.: Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest Ophthalmol Vis Sci, 2007; 48(3): 1319–29.
3. Allikmets, R.: Further evidence for an association of ABCR alleles with age-related macular degeneration. The International ABCR Screening Consortium. Am J Hum Genet, 2000; 67(2): 487–91.
4. Allikmets, R.: Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet, 2000; 67(4): 793–9.
5. Allikmets, R., Shroyer, NF., Singh, N., et al.: Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science, 1997; 277(5333): 1805–7.
6. Allikmets, R., Singh, N., Sun, H., et al.: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet, 1997; 15(3): 236–46.
7. Bellmann, C., Holz, FG., Schapp, O., et al.: Topography of fundus autofluorescence with a new confocal scanning laser ophthalmoscope. Ophthalmologe, 1997; 94(6): 385–91.
8. Braun, TA., Mullins, RF., Wagner, AH., et al.: Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet, 2013; 22(25): 5136–45.
9. Briggs, CE., Rucinski, D., Rosenfeld, PJ., et al.: Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci, 2001; 42(10): 2229–36.
10. Burke, TR., Tsang, SH., Zernant, J., et al.: Familial discordance in Stargardt disease. Mol Vis, 2012; 18 : 227–33.
11. Cella, W., Greenstein, VC., Zernant-Rajang, J., et al.: G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp Eye Res, 2009; 89(1): 16–24.
12. Cremers, FP., Van de Pol, DJ., Van Driel, M., et al.: Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet, 1998; 7(3): 355–62.
13. Downs, K., Zacks, DN., Caruso, R., et al.: Molecular testing for hereditary retinal disease as part of clinical care. Arch Ophthalmol, 2007; 125(2): 252–8.
14. Edwards, AO., Donoso, LA., Ritter, R., 3rd. A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest Ophthalmol Vis Sci, 2001; 42(11): 2652–63.
15. Ergun, E., Hermann, B., Wirtitsch, M., et al.: Assessment of central visual function in Stargardt’s disease/fundus flavimaculatus with ultrahigh-resolution optical coherence tomography. Invest Ophthalmol Vis Sci, 2005; 46(1): 310–6.
16. Fishman, GA.: Historical evolution in the understanding of Stargardt macular dystrophy. Ophthalmic Genet, 2010; 31(4): 183–9.
17. Fishman, GA., Stone, EM., Eliason, DA., et al.: ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol, 2003; 121(6): 851–5.
18. Fishman, GA., Stone, EM., Grover, S., et al.: Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol, 1999; 117(4): 504-10.
19. Fujinami, K., Zernant, J., Chana, RK., et al.: ABCA4 gene screening by next-generation sequencing in a British cohort. Invest Ophthalmol Vis Sci, 2013; 54(10): 6662–74.
20. Hargitai, J., Zernant, J., Somfai, GM., et al.: Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2005; 46(12): 4402–8.
21. Hentze, MW., Kulozik, AE.: A perfect message: RNA surveillance and nonsense-mediated decay. Cell, 1999; 96(3): 307–10.
22. Holz, FG.: Autofluorescence imaging of the macula. Ophthalmologe, 2001; 98(1): 10-8.
23. Itabashi, R., Katsumi, O., Mehta, MC., et al.: Stargardt’s disease/fundus flavimaculatus: psychophysical and electrophysiologic results. Graefes Arch Clin Exp Ophthalmol, 1993; 231(10): 555–62.
24. Jaakson, K., Zernant, J., Kulm, M., et al.: Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat, 2003; 22(5): 395–403.
25. Lachapelle, P., Little, JM., and Roy, MS.: The electroretinogram in Stargardt’s disease and fundus flavimaculatus. Doc Ophthalmol, 1989; 73(4): 395–404.
26. Martinez-Mir, A., Paloma, E., Allikmets, R., et al.: Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet, 1998; 18(1): 11–2.
27. Mata, NL., Weng, J., Travis, GH.: Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A, 2000; 97(13): 7154–9.
28. Maugeri, A., van Driel, MA., Van de Pol, DJ., et al.: The 2588G—>C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet, 1999; 64(4): 1024–35.
29. Molday, RS., Zhang, K.: Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog Lipid Res, 2010; 49(4): 476–92.
30. Radu, RA., Han, Y., Bui, TV., et al.: Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci, 2005; 46(12): 4393–401.
31. Radu, RA., Yuan, Q., Hu, J., et al.: Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci, 2008; 49(9): 3821–9.
32. Rencová, E., Studnička, J., Marák, J., et al.: Koincidence lokalizace defektů vrstvy junkce IS/OS fotoreceptorů na SD OCT s funkčními poruchami v případě Stargardtovy choroby. Čes a slov Oftal, 2012; 68(2): 84–88.
33. Rivera, A., White, K., Stohr, H., et al.: A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet, 2000; 67(4): 800–13.
34. Roberts, LJ., Ramesar, RS., Greenberg, J.: Clinical utility of the ABCR400 microarray: basing a genetic service on a commercial gene chip. Arch Ophthalmol, 2009; 127(4): 549–54.
35. Rosenberg, T., Klie, F., Garred, P., et al.: N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol Vis, 2007; 13 : 1962–9.
36. Shroyer, NF., Lewis, RA., Lupski, JR.: Complex inheritance of ABCR mutations in Stargardt disease: linkage disequilibrium, complex alleles, and pseudodominance. Hum Genet, 2000; 106(2): 244–8.
37. Schindler, EI., Nylen, EL., Ko, AC., et al.: Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum Mol Genet. 2010; 19(19): 3693–701.
38. Simonelli, F., Testa, F., Zernant, J., et al.: Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res, 2005; 37(3): 159–67.
39. Sohrab, MA., Allikmets, R., Guarnaccia, MM., et al.: Preimplantation genetic diagnosis for stargardt disease. Am J Ophthalmol, 2010; 149(4): 651–655 e2.
40. Stanga, PE., Downes, SM., Ahuja, RM., et al.: Comparison of optical coherence tomography and fluorescein angiography in assessing macular edema in retinal dystrophies: preliminary results. Int Ophthalmol, 2001; 23(4-6): 321-5.
41. Stavrou, P., Good, PA., Misson, GP., et al.: Electrophysiological findings in Stargardt’s-fundus flavimaculatus disease. Eye (Lond), 1998; 12 ( Pt 6): 953–8.
42. Stone, EM., Webster, AR., Vandenburgh, K., et al.: Allelic variation in ABCR associated with Stargardt disease but not age-related macular degeneration. Nat Genet, 1998; 20(4): 328–9.
43. Sun, H., Nathans, J.: ABCR: rod photoreceptor-specific ABC transporter responsible for Stargardt disease. Methods Enzymol, 2000; 315 : 879–97.
44. Valverde, D., Riveiro-Alvarez, R., Aguirre-Lamban, J., et al.: Spectrum of the ABCA4 gene mutations implicated in severe retinopathies in Spanish patients. Invest Ophthalmol Vis Sci, 2007; 48(3): 985–90.
45. Vasireddy, V., Wong, P., Ayyagari, R.: Genetics and molecular pathology of Stargardt-like macular degeneration. Prog Retin Eye Res, 2010; 29(3): 191–207.
46. Voigt, M., Querques, G., Atmani, K., et al.: Analysis of retinal flecks in fundus flavimaculatus using high-definition spectral-domain optical coherence tomography. Am J Ophthalmol, 2010; 150(3): 330–7.
47. Von Ruckmann, A., Fitzke, FW., Bird, AC.: In vivo fundus autofluorescence in macular dystrophies. Arch Ophthalmol, 1997; 115(5): 609–15.
48. Webster, AR., Heon, E., Lotery, AJ., et al.: An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci, 2001; 42(6): 1179–89.
49. Yang, Z., Chen, Y., Lillo, C., et al.: Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest, 2008; 118(8): 2908–16.
50. Yatsenko, AN., Shroyer, NF., Lewis, RA., et al.: Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet, 2001; 108(4): 346–55.
51. Zernant, J., Schubert, C., Im, KM., et al.: Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci. 2011; 52(11): 8479–87.
52. Zhang, K., Kniazeva, M., Han, M., et al.: A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet, 2001; 27(1): 89–93.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2014 Issue 6
Most read in this issue
- Orbital Complications of Sinusitis
- Actual State of the One Day Simultaneous Bilateral Cataract Surgery Issue
- Diagnostic Pitfalls of Pseudo-Foster Kennedy Syndrome – A Case Report
- The Molecular Genetic and Clinical Findings in two Probands with Stargardt Disease