
Těhotenství a porod u pacientky s čistým karyotypem 46,XY
Souhrn dosavadních poznatků o XY ženách
Pregnancy and delivery in a patient with pure 46,XY karyotype
Summary of actual knowledge about XY women
Type of study:
Summary review and a case report.
Settings:
GEST IVF, Centre of Reproductive Medicine, Prague.
Introduction:
In scientific literature there two syndroms have been described in the presence of pure 46,XY karyotype when an individual is phenotypically and psychosexually identified as a woman. Androgen insensitivity syndrom (AIS) and pure gonadal dysgenesis XY (GD XY, Swyer syndrom). Thanks to the presence of a uterus in Swyer syndrom we can treat this type of sterility with donated oocytes.
Method:
The paper describes both syndromes from prenatal, genetical, endocrinological, oncological, reproductive and perinatological points of view. A case study corncerning a patient with pure gonadal dysgenesis XY, who successfully became pregnant through a donated oocytes programme, is also described. The pregnancy progressed physiologically, and a healthy boy, 3820g/52cm, was delivered in term by ceasarean section.
Discussion:
In world scientific literature at least fifteen successful pregnancies with pure gonadal dysgenesis XY have been described. In spite of the expectation of diminished uterine capacity, children are born to term with a normal delivery weight.
Conclusion:
This article should be considered as a summary of all actual knowledge about these patients. This article should be available and usefull for clinicians who come across XY females. The case study provides evidence that even an individual with male genetic gender can be pregnant and deliver a healthy child.
Keywords:
pure gonadal dysgenesis – androgen insensivity syndrom – Swyer syndrom – primary ovarian failure
:
M. Poláková 1
![]() ; D. Alexander 1; J. Šulc 1; L. Zetová 1; R. Vlk 2; A. Křepelová 3; D. Žantová 4
; D. Alexander 1; J. Šulc 1; L. Zetová 1; R. Vlk 2; A. Křepelová 3; D. Žantová 4
:
GEST IVF, Centrum reprodukční medicíny, Praha
1; Gynekologicko-porodnická klinika FN Motol, Praha, přednosta prof. MUDr. L. Rob, CSc.
2; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol, Praha, přednosta prof. MUDr. M. Macek Jr., DrSc.
3; Gynekologická ambulance, Praha
4
:
Ceska Gynekol 2013; 78(5): 443-447
:
Original Article
Typ studie:
Souhrnný článek a kazuistika.
Název a sídlo pracoviště:
GEST IVF, Centrum reprodukční medicíny, Praha.
Úvod:
V odborné literatuře jsou popisovány dva syndromy s čistým karyotypem 46,XY, kdy jedinec je fenotypicky i psychosexuálně identifikován jako žena. Syndrom testikulární feminizace (androgen insensivity syndrom – AIS) a čistá dysgeneze gonád XY (pure gonadal dysgenesis – GD XY – Swyerův syndrom). Díky přítomnosti dělohy u syndromu čisté dysgeneze gonád XY můžeme tento typ neplodnosti léčit pomocí darovaných oocytů.
Metodika:
Práce se zamýšlí nad oběma syndromy z různých hledisek: prenatálního, genetického, endokrinologického, onkologického, reprodukčního a perinatologického. Dále práce popisuje kazuistiku pacientky se syndromem čisté dysgeneze gonád XY, která úspěšně otěhotněla v programu darovaných oocytů. Těhotenství proběhlo fyziologicky, císařským řezem byl porozen zdravý hoch 3820 g/52 cm.
Diskuse:
Ve světové odborné literatuře již bylo popsáno nejméně patnáct úspěšně donošených těhotenství u čisté dysgeneze gonád XY. I přes předpokládanou malou kapacitu dělohy se rodí děti v termínu porodu a s normální porodní hmotností.
Závěr:
Práce by měla sloužit jako shrnutí dosavadních poznatků o ženách s karyotypem 46,XY. Měla by být k dispozici a užitečná pro všechny specialisty, kteří se s těmito pacientkami setkají ve své praxi. Popsaná kazuistika je důkazem, že i jedinec, jehož genetické pohlaví je mužské, je schopen donosit a porodit zdravé dítě.
Klíčová slova:
čistá dysgeneze gonád XY – Swyerův syndrom – primární ovariální selhání –androgen insensitivity syndrom
ÚVOD
V odborné literatuře jsou popisovány dva syndromy s čistým karyotypem 46,XY, kdy jedinec je fenotypicky i psychosexuálně identifikován jako žena. Známější syndrom testikulární feminizace (androgen insensivity syndrom – AIS) popsal v roce 1953 Morris, ale označení jako Morrisův syndrom se nevžilo. Méně známou čistou dysgenezi gonád XY (pure gonadal dysgenesis – GD XY) objevil v roce 1955 Swyer a název Swyerův syndrom se běžně používá.
Výskyt těchto syndromů je velmi vzácný (asi 1–5 : 100 000 živě narozených dětí) [8] a v praxi se s nimi setkáme zcela ojediněle.
Oba syndromy jsou velmi zajímavé nejen z hlediska prenatálního a postnatálního vývoje, ale také z hlediska genetického a endokrinologického. Na nich lze demonstrovat a pochopit principy genetické, vývojové i endokrinologické a jejich odraz ve fenotypu jedince.
PRENATÁLNÍ VÝVOJ
K diferenciaci testes u embryí mužského pohlaví je zapotřebí aktivace série genů na Y chromozomu. Embryonální gonády tak začnou produkovat androgeny a antimülleriánský hormon (AMH). Androgeny přes androgenní receptory umožní vývoj mužských pohlavních orgánů a díky nim dojde také k diferenciaci mezonefrického (Wolffova) vývodu ve vývodný mužský pohlavní systém s přídatnými žlázami. Funkcí AMH v raném embryonálním období je blokování dalšího vývoje paramezonefrických (Müllerových) vývodů, ze kterých se vyvíjejí u žen vejcovody, děloha a většina pochvy. Ovaria a estrogeny nejsou k primárnímu sexuálnímu vývoji potřebné. V nepřítomnosti androgenů či androgenního receptoru dojde primárně k vývoji organismu ve smyslu ženském. V nepřítomnosti AMH dojde i k vývoji ženských pohlavních orgánů, které se vyvíjejí z Müllerova vývodu.
GENETICKÉ POZADÍ
Syndrom testikulární feminizace (AIS) je gonozomálně recesivní onemocnění. Dochází k mutacím v genu na X chromozomu, který je zodpovědný za androgenový receptor. I když lze tuto mutaci potvrdit molekulárně genetickým vyšetřením, ke klinické diagnóze nám mnohdy postačí vyšetření karyotypu, hormonální profil a klinický nález. Dochází k vývoji testes, které produkují androgeny i AMH, ale s nefunkčním androgenovým receptorem tkáně na tyto androgeny nereagují. Vyvíjí se proto jedinec ve smyslu ženském. Vzhledem k tomu, že gonády však produkují funkční AMH (má jiné receptory), dojde k potlačení vývoje Müllerových vývodů, a proto se nevyvinou vejcovody, děloha a proximální část pochvy. Vyvíjí se pouze ženský zevní genitál s pahýlem distální části pochvy. Je popisována také částečně funkční va-rianta genové mutace, kdy fenotyp jedince je odvislý od stupně androgenizace.
Patogeneze Swyerova syndromu (GDXY) je zcela odlišná. Zde nedojde k přeměně tzv. indiferentní embryonální gonády v testes. Y chromozom má na svém krátkém raménku oblast SRY (Sex Region of Y), která je zodpovědná za vývoj testes. Pokud dojde k mutaci (většinou deleci) tohoto úseku na Y chromozomu, testes se nevyvinou. Literatura však udává, že pouze 15 % jedinců s GDXY má potvrzenou ztrátu úseku SRY [21]. Do vývoje testes se pravděpodobně zapojuje více genů, a tyto geny dokonce nemusí být ani přítomné na Y chromozomu [1, 8, 10, 16]. Australští vědci v roce 2011 [22] podrobili 23 jedinců s DGXY podrobnému zkoumání k nalezení konkrétní mutace. Kauzální příčinu tak našli u méně než 50 % jedinců, a to v genech SOX9, SF1, DAX1, GATA4 atd. Svou úlohu zde jistě hrají další mechanismy na genové úrovni, jako je genová regulace, aktivace transkripce a další. Zdá se, že jde o poměrně heterogenní příčinu jednoho projevu – vývoje dysgenetické gonády (varlete), a to se všemi důsledky, které chybění gonády s sebou přináší.
Dochází k vývoji jedince ve smyslu ženském v důsledku absence androgenů. Vzhledem k tomu, že není produkován ani AMH, dochází k další diferenciaci Müllerových vývodů – jedinec má přítomné vejcovody, dělohu a celou pochvu (obr. 1).
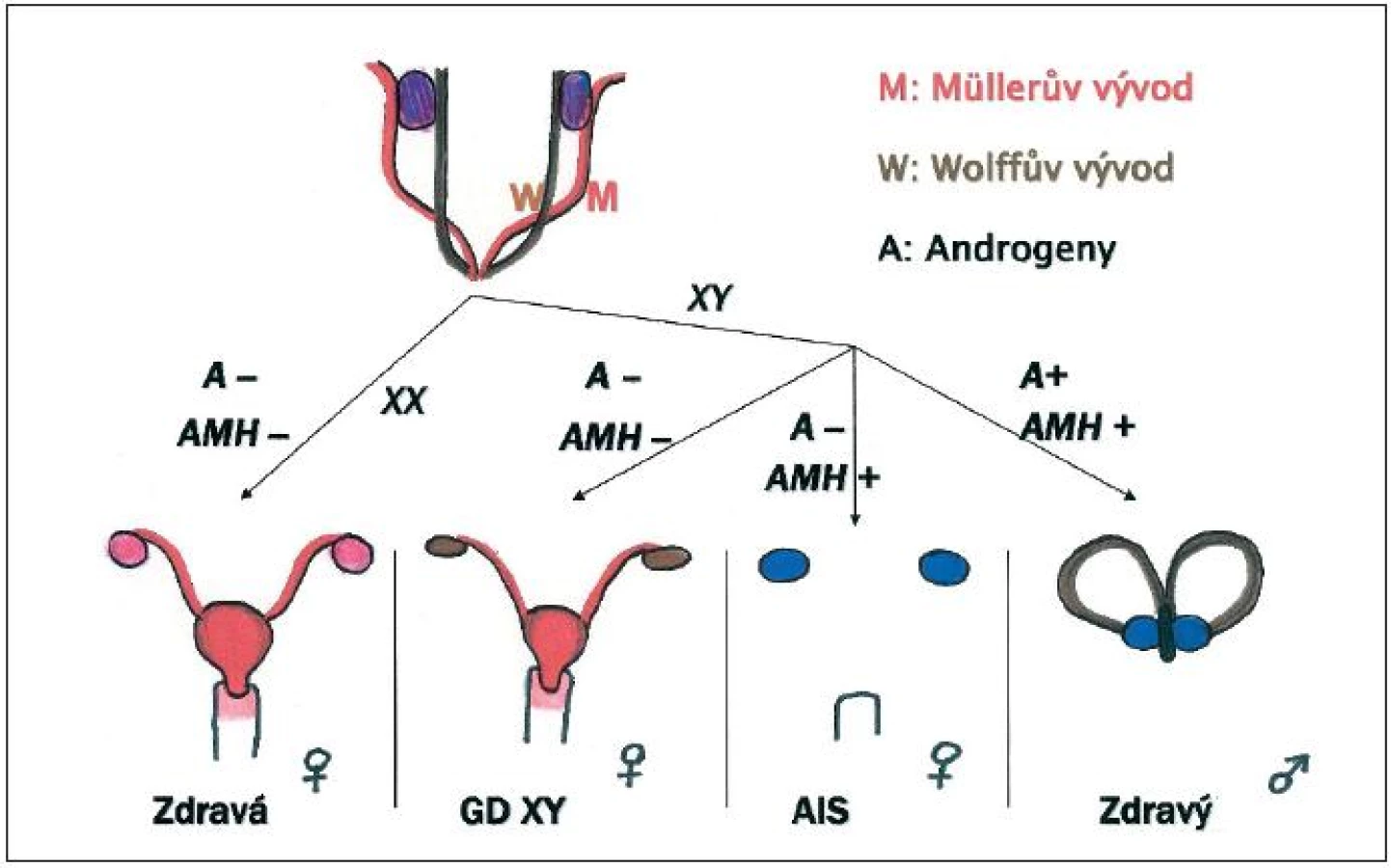
U úplné dysgeneze gonád je vždy nutné udávat v názvu gonozomy XY, aby nedošlo k záměně s úplnou dysgenezí gonád X0 – Turnerovým syndromem. Existují i syndromy s úplnou dysgenezí gonád XX, kdy porucha vývoje gonád je pak často součástí syndromů se závažným mentálním a fyzickým postižením, jako např. Perraultův syndrom [18].
Pokud nejde o čistou formu tohoto genetického postižení, ale o mozaiku, dostáváme se do úplně jiné problematiky. Pak velmi záleží na procentu a typu mozaiky. Je důležité, jaké buněčné linie obsahují patologický genom a jaké gonády vlastně byly vytvořeny. Toto všechno se pak odráží v klinickém obrazu postižených jedinců. Malá mozaika se nemusí projevit vůbec, u závažných forem se dostáváme do problematiky tzv. intersexuálních malformací.
PUBERTA A ENDOKRINOLOGICKÉ ASPEKTY
Společným znakem obou syndromů je primární amenorea. Tento projev vede většinou k dalšímu vyšetřování a ke stanovení diagnózy. Diagnózu lze stanovit i prenatálně, je-li indikována karyotypizace plodu. Jde o více méně náhodný nález. Je zde však riziko, že po narození děvčátka se na tyto syndromy nemyslí a výsledek může být interpretován jako záměna vzorků v laboratoři!
U syndromu AIS se retinovaná testes mohou nacházet v malé pánvi, v tříselném kanále nebo sestouplá až do labia major. Tyto gonády v období pohlavního dospívání začnou produkovat androgeny i estrogeny. Androgeny dosahují fyziologických hodnot jako u mužského jedince, ale jak již bylo řečeno, kvůli mutaci androgenového receptoru nejsou účinné. Androgeny u muže i u ženy jsou mimo jiné zodpovědné za vývoj pubického a axiálního ochlupení. Pubické ochlupení je sporé a axiální zcela chybí, a proto se někdy tomuto syndromu říká „hairless woman“. Hladina estrogenů dostačuje k tomu, aby došlo k vývoji sekundárních ženských pohlavních znaků – dochází k normálnímu vývoji prsů, vývoji ženského typu postavy a estrogenizaci distálního zbytku pochvy. Díky zpětnovazebným mechanismům hladina FSH a LH odpovídá normálnímu reprodukčnímu rozmezí u muže(je normogonadotropní).
Jaká je situace u Swyerova syndromu? Otázkou zůstává, zda jsou dysgenetické gonády schopny v období pohlavního dospívání produkovat alespoň minimální množství steroidních hormonů. Z dosud popsaných kazuistik se tyto údaje překvapivě liší. Je popisován úplný sexuální infantilismus, ale i částečný rozvoj sekundárních ženských pohlavních znaků [7, 8, 9, 22]. Rozdíl bude pravděpodobně způsoben mírně se lišícím histologickým složením gonády díky různému genetickému původu dysgeneze. Vždy však, na rozdíl od AIS, u Swyerova syndromu dojde alespoň k částečnému vývinu pubického a axilárního ochlupení, a to díky produkci slabých nadledvinových androgenů, jako je dihydroepiandrostendion (DHEA) a androstendion. Po dozrání hypotalamo-hypofyzární osy budou hladiny FSH a LH odpovídat hypergonadotropnímu „ovariálnímu“ selhání (tab. 1). V období pohlavního dospívání je nutno postupnou estrogenizací dosáhnout plného rozvoje sekundárních pohlavních znaků a cyklickým rozpisem gestagenů navodit v přítomné děloze menarche. Přecházíme na cyklickou hormonální substituční terapii (HRT), která nám po celý „reprodukční věk“ zajišťuje dostatečnou osifikaci kostní tkáně, má však dobře známý pozitivní vliv i na jiné tkáně. V hormonální substituci dáváme přednost cyklické HRT před cyklickou orální kontracepcí. Upřednostňujeme přirozený estrogen s menším rizikem nežádoucích účinků.
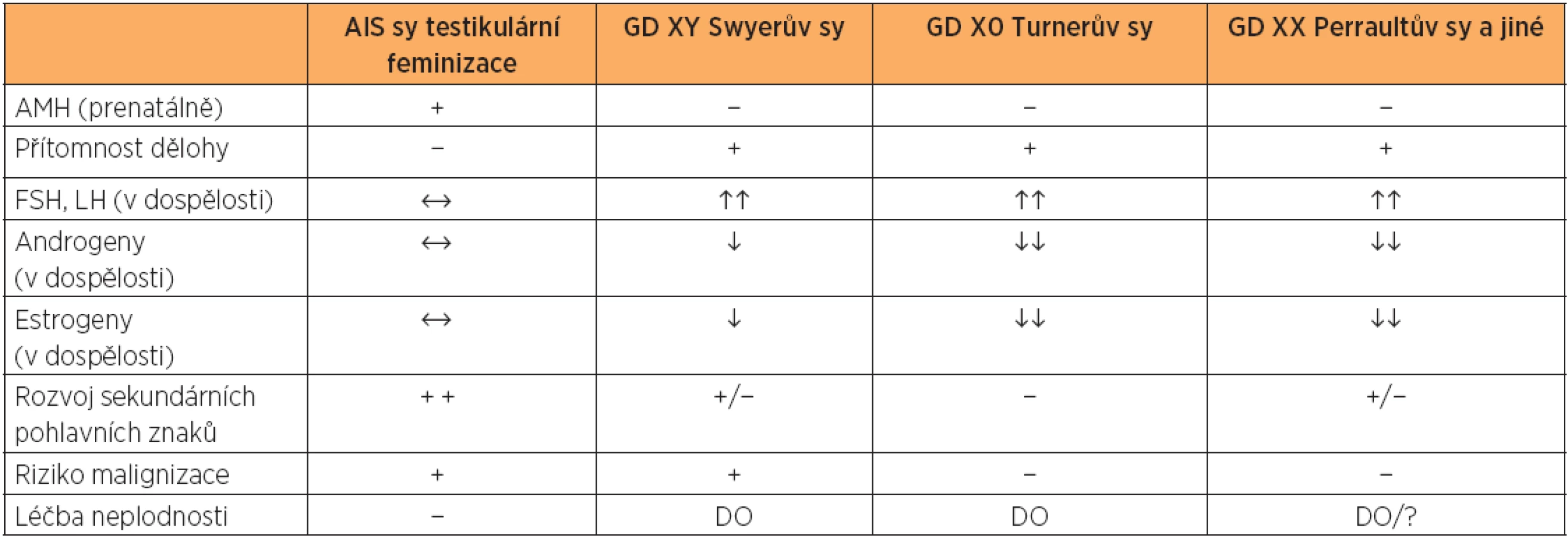
ONKOLOGICKÉ ASPEKTY
Gonády syndromů s Y chromozomem je nutno považovat za dysgenetická retinovaná varlata, ve kterých existuje zvýšené riziko nádoru. Toto riziko je v dětství nízké, ale po pubertě narůstá na 20–30 %. Jde především o maligní nádory germinální – dysgerminom, gonadoblastom [2, 5, 6]. Proto takové gonády po pubertě odstraňujeme. AIS je nutno pak také zajistit hormonální substituční terapií.
PSYCHOLOGICKÉ ASPEKTY
Od narození jsou tito jedinci okolím považováni bezpochyby za ženy (ženské jméno, rodné číslo). Sami se od narození také jako ženy cítí. Po stanovení diagnózy – obvykle v pubertě – je otázkou, jak s touto informací naložit. Objasnění přesné podstaty nemoci by mohlo narušit identitu jedince a mít dalekosáhlé následky v jeho dalším životě. Doporučuje se ve zprávě klinického genetika neuvádět chromozomální nález a uvádět diagnózu pouze ve zkratce.
Hovoříme obecně o „genetické poruše způsobující neplodnost“ a jejich „mužský“ genetický původ jim nezdůrazňujeme. Pacientky samozřejmě vedeme celoživotně ve smyslu ženském.
REPRODUKČNÍ MOŽNOSTI
Pacientka s AIS nemá dělohu a netvoří pohlavní buňky. Použití náhradního mateřství s darovanými oocyty etická komise sekce asistované reprodukce ČGPS JEP v současné době nedoporučuje. V léčbě neplodnosti ji bohužel nemůžeme nic nabídnout.
U GD XY je přítomna děloha a těhotenství lze dosáhnout pomocí darovaných oocytů. Je pozoruhodné, že složitý proces embryo-maternálního dialogu přítomností Y chromozomu není narušen. Děloha je schopna fyziologicky reagovat na exogenní estrogen-gestagenní přípravu endometria a vykazuje schopnost implantace embrya. Navíc tato schopnost implantace u jedince s genotypem XY dokazuje nepřítomnost pohlavně specifických protilátek, které by celý proces mohly znemožňovat.
PERINATOLOGICKÉ ASPEKTY GD XY
Ve světové odborné literatuře již bylo popsáno nejméně 15–20 úspěšně donošených těhotenství [3, 5, 7, 17, 19]. U syndromu GD XY jsme v podobné situaci s darovanými oocyty jako u Turnerova syndromu (GD X0). Oproti Turnerovu syndromu jsou však pacientky s GDXY vyšší, netrpí vrozenými vadami kardiovaskulárního systému apod. a nemají výrazně hypoplastickou dělohu. Těhotenství probíhá srovnatelně fyziologicky jako u zdravé populace, děti se rodí v termínu s normální porodní hmotností. Bylo popsáno i donošené těhotenství dvojčat [7, 9]. Nedochází však ke spontánnímu nástupu děložní činnosti, děloha nereaguje na indukci porodu a těhotenství je nutno ukončit císařským řezem. Otázkou zůstává jistě mužský typ pánve u těchto jedinců.
KAZUISTIKA
Pacientka ve věku 38 let byla odeslána na naše pracoviště registrujícím gynekologem k léčbě neplodnosti pomocí darovaných oocytů pro primární ovariální selhání. Z anamnézy: RA: bratr zdráv, OA: s ničím se neléčí, výška 185 cm, váha 101 kg, GA: spontánní rozvoj sekundárních pohlavních znaků již zpětně nelze bohužel posoudit, pro primární amenoreu byla nasazena v 18 letech HAK. Hormonální vyšetření: FSH 41,1 .. 79,7 IU/l, LH 18,0 IU/l. Bylo provedeno genetické vyšetření se stanovením karyotypu: 46,XY – bez mozaiky. Molekulárně genetické vyšetření nepotvrzuje mutaci androgenového receptoru, nepotvrzuje však ani mutaci v oblasti SRY na Y chromozomu. Klinickým genetikem byl nález uzavřen jako čistá dysgeneze gonád 46,XY. Při gynekologickém vyšetření nacházíme normální zevní ženský genitál s pubickým ochlupením, pochvu bez odchylek. Vaginální ultrazvukové vyšetření zobrazuje dělohu v AVF 67×35×29 mm, endometrium 6 mm (při HRT), ovaria se nezobrazují. Pro riziko vzniku maligního nádoru gonád byla pacientka odeslána k laparoskopické adnexektomii. Při laparoskopii byly popsány proužkovité gonády, histologicky tvořené tukovou a vazivovou tkání s drobnými cystickými formacemi vystlanými mezotelem. Dále jsou popsány normální vejcovody a drobnější děloha normální tvaru.
Terapie: Pacientka vstupuje do programu darovaných oocytů v nesynchronním cyklu. Oocyty od dárkyně byly oplozeny spermiemi partnera a vzniklá embrya jsou kryalizována ve stadiu prvojader. Po standardní estrogen-gestagenní přípravě endometria, po rozmrazení a 24hodinové kultivaci byla transferována do dělohy dvě embrya. S odstupem byla potvrzena jednočetná gravidita. Fyziologický průběh gravidity. Vaginální vyšetření po termínu porodu prokazuje nezralý porodnický nález. Byla provedena zkusmá preindukce porodu prostaglandiny, na které děloha nereaguje. Šest dní po exaktně stanoveném termínu porodu bylo těhotenství ukončeno císařským řezem. Obtížné bylo vybavení plodu pro zcela nerozvinutý dolní děložní segment! Porozen byl plod mužského pohlaví 3820 g / 52 cm, zdráv. Šestinedělí bylo bez komplikací. Dochází k předpokládanému normálnímu nástupu laktace. HRT nasazujeme až po skončení laktace.
ZÁVĚR
Práce by měla sloužit jako shrnutí dosavadních poznatků o ženách s karyotypem 46,XY. Měla by být k dispozici a je užitečná pro všechny specialisty, kteří se s těmito pacientkami setkají ve své praxi. Popsaná kazuistika je důkazem, že i jedinec, jehož genetické pohlaví je mužské, je schopen donosit a porodit zdravé dítě.
MUDr. Monika Poláková
GEST IVF, Centrum reprodukční medicíny
Nad Buďánkami II/24
150 00 Praha 5
e-mail: monika.polakova@gest.cz
Sources
1. Bashamboo, A., McElreavey, K. NR5A1/SF-1 and development and function of the ovary. Ann Endocrinol, 2010, 71, 3, p. 177–182.
2. Ben, TR., et al. 46,XY pure gonadal dysgenesis with gonado-blastoma and dysgerminoma. Tunis Med., 2008, 86, 7, p. 710–713.
3. Dirnfeld, M., et al. Sebsequent successful pregnancy and delivery after intracytoplasmatic sperm injection in a patient with XY donadal dysgenesisms. EJOG, 2000, 88, p. 101–102.
4. Dumic, M., et al. Report of fertility in a woman with a predominantly 46,XY karyotype in a family with multiple disorders of sexual development. JCEM, 2008, 93, 1, p. 182–189.
5. Eunice, M., et al. Familiar pure gonadal dysgenesis with 46,XY karyotype in three siblings and gonadoblastoma in the youngest siblings. Int J Hum Genet, 2009, 9, 2, p. 123–126.
6. Hořejší, J. Dětská gynekologie. Praha: Avicenum, 1990.
7. Chen, M., et al. Successful pregnancy in a gonadectomised woman with 46,XY gonadal dysgenesis and gonadoblastoma. Fertil Steril, 2005, 84, 1, p. 217–219.
8. Jorgensen, PB., Kjartansdóttir, KR., Fedder, J. Care of women with XY karyotype: a clinical practice guideline. Fertil Steril, 2010, 94, 1, p. 105–113.
9. Kan, AKS., Abdalla, HI., Oskarsson, T. Two successful pregnancies in a 46,XY patient. Human Reprod, 1997, 12, 7, p. 1434–1435.
10. Knower, KC., et al. Failure of SOX9 regulation in 46XY disorders of sex development with SRY, SOX9 and SF1 mutations. PLoS One, 2011, 6,3, e17751.
11. Kohler, B., et al. Five novel mutations in steroidogenic factor 1 (SF1,NR5A1) in 46,XY patiens with severe underandrogenisation but without adrenal insuficiency. Hum Mutat, 2008, 29, 1, p. 59–64.
12. Kokcu, A., et al. Pure gonadal dysgenesis and spontaneous pregnancy: A case report. Gynecol Endocrinol, 2010, 26, 2, p. 103–104.
13. McKnight, KK., Bates, GW. Jun. Atypical presentation and management dilemma of mixed gonadal dysgenesis. Fertil Steril, 2011, 95, 1, p. 324–326.
14. Moore, KL., Persaud, TVN. Zrození člověka – Embryologie s klinickým zaměřením. Praha: ISV, 2002.
15. Nussbaum, R., McInnes, R., Willard, H. Klinická genetika. 6. vyd. Thompson & Thompson, 2004.
16. Palival, P., et al. Identification of novel SRY mutations and SF1 (NR5A1) changes in patiens with pure gonadal dysgenesis and 46,XY karyotype. Mol Hum Reprod, 2011, 17, 6, p. 372–378.
17. Plante, BJ., Fritz, MA. A case report of successful pregnancy in a patient with pure 46,XY gonadal dysgenesis. Fertil Steril, 2008, 90, 5, p. 43–44.
18. Pierce, SB., et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovaria dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet, 2010, 87, 2, p. 282–288.
19. Selvaraj, K., Ganesh, V., Selvaraj, P. Successful pregnancy in a patient with a 46,XY karyotype. Fertil Steril, 2002, 78, 2, p. 419–420.
20. Siddique, H., Daggett, P., Artley, K. Successful term vaginal delivery in a 46,XY woman. PLoS One, Brief comunications, 2007, p. 298–299.
21. Yen, SSC., Jaffe, RB. Reproductive endocrinology. Philadelphia: WB Saunders, 2009.
22. White, S., et al. Copy number variation in patiens with disorders of sex development due to 46,XY gonadal dysgenesis. PLoS One, 2011, 6, 3, e17793.
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Czech Gynaecology

2013 Issue 5
Most read in this issue
- Incarcerated uterus in pregnancy – pitfalls of diagnosis, clinical course and therapy: two case reports
- Risk factors for endometrial cancer
-
Pregnancy and delivery in a patient with pure 46,XY karyotype
Summary of actual knowledge about XY women - Doctor’s specialty training in obstetrics and gynecology at university medical schools: 2012–2013