
Trombotická trombocytopenická purpura u nemocných se systémovým lupus erythematodes
Thrombotic thrombocytopenic purpura in patients with systemic lupus erythematosus
Thrombotic thrombocytopenic purpura (TTP) is a rare, however life threatening syndrome, which is characterized by thrombocytopenia with increased turnover and by loss of peripheral platelets. It belongs to so-called MAHA or MAS disorders (microangiopathic heamolytic syndromes). Combination of microangiopathy, thrombocytopenia and intravascular haemolysis is observed. Clinical picture is also accompanied by fever, alteration of renal functions and variable neurological symptoms which are associated with impaired brain perfusion. The increase of relative amount of highly multimeric (ultra large-UL) von Willebrand factor (vWF) plays a crucial role in the pathogenesis of the disease. The multimer cumulates in the circulation as a result of insufficient concentration of ADAMTS 13 protease that share vWF metalloprotease activity and cleave UL-vWF under normal conditions. Treatment option is either substitution of frozen plasma that contains vWF metaloprotease or plasmapheresis that is obviously more effective and has a potential to eliminate autoantibodies against ADAMTS 13 protease, which usually cause the functional ADAMTS 13 protease deficit. TTP also occurs in patients with systemic connective tissue diseases, mostly with SLE. The authors describe 3 patients with SLE that developed TTP. SLE was defined in all patients according to the ARA criteria. All three patients were hospitalized for rather acute onset of haemolytic anemia, thrombocytopenia and neurological symptoms. They were treated with plasmapheresis, and furthermore with high doses of frozen plasma and intensive immunosuppressive agents. One patient flared with the necessity of rehospitalization, and one died of multiorgan failure during the attack of TTP. TTP may accompany systemic connective tissue diseases as a secondary syndrome or can develop after the treatment with some drugs. In the case of acute decrease of blood elements, particularly thrombocytes, it is necessary to be aware of this possibility because unrecognized and insufficiently treated disease results very often in death.
Key words:
SLE, thrombotic thrombocytopenic purpura
Authors:
Z. Fojtík; Z. Kořístek; L. Červinek; M. Navrátil; M. Obrovská
Authors‘ workplace:
Interní hematoonkologická klinika, FN Brno Bohunice
Published in:
Čes. Revmatol., 15, 2007, No. 2, p. 121-126.
Category:
Case Report
Overview
Trombotická trombocytopenická purpura (TTP) je vzácný, ale život ohrožující syndrom, který je charakterizován trombocytopenií ze zvýšeného obratu a zánikem destiček na periferii. Patří k tzv. MAHA či MAS onemocněním (mikroangiopatické hemolytické syndromy). U těchto stavů se kombinuje mikroangiopatie, trombocytopenie a intravaskulární hemolýza, klinický obraz pak doplňuje horečka, alterace renálních funkcí a neurologická symptomatologie, která má měnlivý charakter a jsou spojeny s poruchou perfuze mozku. V etiopatogenezi stojí na počátku nárůst relativního zastoupení velkých multimerů (ultra large-UL) von Willebrandova faktoru (vWF), které se v oběhu koncentrují v důsledku nedostatku vWF metaloproteinázové aktivity tzv. ADAMTS 13 proteázy, která za normálních okolností štěpí UL-vWF. Léčba spočívá v prosté substituci vWF metaloproteinázy, která je obsažena v mrazené plazmě, výměnná plazmaferéza je samozřejmě účinnější a navíc má potenciál odstraňovat z oběhu autoprotilátku proti ADAMTS 13 proteáze, která obvykle funkční deficit ADAMTS 13 proteázy způsobuje. TTP se vyskytuje rovněž u nemocných se systémovými chorobami pojiva, nejčastěji se SLE. Autoři popisují tři nemocné se SLE, u kterých došlo k rozvoji TTP. U všech byla stanovena diagnóza systémového lupus erythematodes na podkladě ARA kritérií. Všechny 3 nemocné ženy byly hospitalizovány pro poměrně náhle vzniklou symptomatologii hemolytické anémie, trombocytopenie a neurologických příznaků. Byly léčeny výměnnými plazmaferézami, dále pomocí vysokých dávek mrazených plazem a intenzivní imunosupresí. U jedné pacientky došlo k recidivě s nutností rehospitalizace, jedna nemocná se SLE zemřela během ataky TTP na multiorgánové selhání. TTP je onemocnění, které se může vyskytovat jako sekundární syndrom u systémových onemocnění pojiva, eventuálně může vznikat jako následek léčby po některých lécích. V případě náhlejšího poklesu krevních elementů, zejména trombocytů je potřeba na toto onemocnění myslet, protože nepoznané a neadekvátně léčené velmi často končí smrtelně.
Klíčová slova:
SLE, trombotická trombocytopenická purpura
Úvod
Trombotická trombocytopenická purpura (TTP) je vzácný, ale život ohrožující syndrom, který je charakterizován trombocytopenií ze zvýšeného obratu a zánikem destiček na periferii. Patofyziologicky se jedná o mikroangiopatické okluzivní onemocnění charakterizované systémovou či renální agregací destiček s tvorbou destičkových trombů s následnou konzumpční trombocytopenií a s mechanickým postižením erytrocytů (1). První případ nemocné s obdobnými klinickými příznaky byl poprvé popsán Eli Moschcowitzem v roce 1924. Mikrovaskulární destičkové tromby mohou být přítomny v cévách většiny orgánů, dominantně v mozku a v ledvinách. Mikrotromby obsahují nerozštěpené multimery vWF (von Willebrandova faktoru). Tyto multimery jsou tvořeny v megakaryocytech a endoteliálních buňkách a uskladňovány v α granulích trombocytů a Weibel-Paladeho tělíscích endoteliálních buněk (1). Multimery vWF mají schopnost vázat glykoprotein Ibα – komponentu destičkového receptoru pro vWF (1–3). Za fyziologických podmínek jsou multimery vWF štěpeny štěpící metaloproteinázou obsahující molekuly zinku a vápníku. Tato proteáza štěpí peptidové můstky v monomerních jednotkách vWF, je označována ADAMTS 13 a je produkována hepatocyty (1, 4, 5). Zajímavá v rámci patofyziologie tohoto syndromu je skutečnost, že měření hladin, respektive ADAMTS 13 aktivity, nemusí korelovat s tíží ataky, tedy pro rozhodovací proces o diagnóze a léčbě TTP není nutné měření hladin této proteázy (6).
Standardními léčebnými postupy u nemocných s atakami TTP jsou podání čerstvé mrazené plazmy či výměnná velkoobjemová plazmaferéza (2, 7, 8). Tyto léčebné postupy jsou založeny na substituci ADAMTS 13 proteázy, která je obsažena v mrazené plazmě, a při plazmaferéze navíc dochází k eliminaci protilátek neutralizujících ADAMTS 13 proteázu. K dalším možnostem ovlivnit autoprotilátky patří podání vysokodávkovaných kortikosteroidů, u nemocných refrakterních na standardní léčebné postupy lze zvažovat a využít i podání agresivnějších imunosupresivních přístupů, jako je léčba cyklofosfamidem (9), vinkristinem (10) či rituximabem (11). Před érou výměnných plazmaferéz byla mortalita až 90 % a hlavní příčinou úmrtí byly projevy mikrovaskulární trombózy s následnými cerebrálními a myokardiálními infarkty a známkami renálního selhání (7).
Autory jsou popsány 3 nemocné s diagnózou SLE a rozvojem atak mikroangiopatické hemolytické anémie, podle klinických kritérií TTP. Diagnóza systémový lupus erythematodes byla stanovena na podkladě revidovaných ARA kritérií pro SLE. Všechny 3 nemocné byly sledovány na revmatologické ambulanci Interní hematoonkologické kliniky FN Brno. Dvě nemocné po zvládnutí akutní ataky TTP jsou sledovány dále, jedna nemocná zemřela pod obrazem multiorgánového selhávání do 16 hodin od stanovení diagnózy.
Popis 1. případu
Nemocná, 35letá žena, je sledována pro SLE od roku 1997, kdy ataka klinických příznaků základního onemocnění vznikla asi za 2 měsíce po porodu 4. dítěte. Z klinických kritérií byl přítomen obličejový motýlovitý exantém, artralgie a neerozivní artritidy drobných kloubů rukou, dle echokardiografického vyšetření perikardiální výpotek. Laboratorně byla u nemocné zjištěna pozitivita ANA 1 : 320 homogenního typu, pozitivita anti ds DNA a ENA vše nepřímou imunofluorescencí, lupus antikoagulant byl negativní. V základním biochemickém vyšetření dominovala proteinurie 1690 mg/24 hodin, proto byla provedena renální biopsie s nálezem lupus nefritidy IV dle WHO. Byla zahájena léčba prednisonem a cyklofosfamidem s následnou stabilizací stavu a poklesem proteinurie na 320 mg/24 hodin. Na podzim roku 1999 po poštípání hmyzem na dolních končetinách došlo k akceleraci základního onemocnění se vzestupem FW na 95/120, poklesem složek komplementu (C3 0,63 g/l, C4 0,08 g/l) a zejména vzestupem proteinurie na 11 040 mg za 24 hodin. Vzhledem k tomuto patologickému nálezu byla opět zahájena intenzivní imunosupresivní léčba (prednison 1 mg/kg, cyklofosfamid i.v. v pulzech 1000 mg) s následným poklesem proteinurie na 5 520 mg/24 hodin. V říjnu 2000 pro přetrvávající středně vysokou proteinurii byl přidán k léčbě cyklosporin A v dávce 2,5 mg/kg/den, poté proteinurie klesla na 1900 mg. Vzhledem ke stabilizaci klinického stavu a laboratorních parametrů byly intervaly podávání cyklofosfamidu prodlouženy na několik měsíců a ponechán pouze cyklosporin A s prednisonem 10 mg/den.
O rok později při pravidelné kontrole nemocná pozorovala mírnou únavu a ojedinělé hematomy na kůži. Laboratorně byla zjištěna anémie a trombocytopenie. Bylo doporučeno vysadit cyklosporin A a zvýšit prednison na 20 mg/den. O týden později si nemocná při kontrole stěžovala na únavnost, pocity brnění půlky těla a hematomy. V krevním obrazu byl zaznamenán výrazný pokles v hodnotách erytrocytů a trombocytů (trombocyty 5 x 109/l, Hb 62 g/l, erytrocyty 2,46 x 1012/l, leukocyty 7,54 x 109/). Nemocná byla ihned hospitalizována a vzhledem k přítomnosti intravaskulární hemolýzy charakteru MAHA (schistocyty v periferním krevním nátěru, zvýšená koncentrace volného hemoglobinu) byla pracovní diagnóza uzavřena jako TTP, což vedlo k neodkladnému zahájení léčby podáváním čerstvých mrazených plazem (ČMP). Přes tento postup se v noci, pouhých několik hodin po přijetí, začali rozvíjet kvalitativní poruchy vědomí a ataky paroxysmů křečí. Provedeným CT vyšetřením nebylo prokázáno krvácení do CNS. Stav rychle progredoval a během dalších několika hodin vyústil do obrazu šokového stavu s multiorgánovým selháním: srdeční insuficience s difúzní poruchou kontraktility a s poklesem ejekční frakce levé komory na 40%, hemodynamicky významný perikardiální výpotek (400 ml) se symptomatologií srdeční tamponády, plicní edém kombinované etiologie rezistentní na léčbu, poškození jater (ALT 49 μkat/l, AST 121 μkat/l, LD 131 μkat/l) a ledvin (urea 19,4 mmol/l, kreatinin 368 μmol/l, anurie), kvantitativní porucha vědomí na úrovni kómatu. Pro rezistentní plicní edém bylo nutné zavedení řízené plicní ventilace. Stav nemocné byl kritický, jednalo se o šok s generalizovaným postižením mikrocirkulace s nutností podávání vazoaktivních katecholaminů pro udržení systémového tlaku krve alespoň pro základní perfúzi CNS. Zároveň s progresí stavu byly zahájeny velkoobjemové výměnné plazmaferézy (4600 ml/den) pomocí přístroje COBE Spectra s kontinuálním průtokem krve. Současně byla zahájena léčba metylprednisolonem 1000 mg/den i.v. 3 dny, následně 125 mg i.v. další 3 dny a přechod na 50 mg prednisonu p.o. Po přibližně 72 hodinách od přijetí došlo k výraznému zlepšení zdravotního stavu nemocné včetně výrazného poklesu enzymů jaterního souboru, obnovy diurézy a zlepšení renálních parametrů, což umožnilo ukončení umělé plicní ventilace, extubaci a postupné vysazení katecholaminů. Byl pozorován i vzestup hodnot v krevním obrazu a po 14 dnech léčby došlo k restituci erytrocytů a trombocytů. Nemocná byla bez reziduálních neurologických příznaků a bez subjektivně vnímaných potíží propuštěna do domácího ošetřování. O 10 dnů později došlo k recidivě ataky TTP s poklesem krevních elementů, tentokrát bez neurologické symptomatologie a renálního postižení, ale protože při komplexní léčbě nedocházelo k plné restituci trombocytů, byla elektivně provedena splenektomie. Promptně po ní dochází ke zlepšení v parametrech krevního obrazu a nemocná byla propuštěna do domácího ošetřování bez rozvoje dalších atak trombocytopenie či známek hemolytické anémie. V dnešní době, tedy 5 let po atace TTP, je nemocná bez výrazných výkyvů v krevním obrazu, přetrvává pouze proteinurie, která je řešena dávkami glukokortikoidů a azathioprinem.
Popis 2. případu
Nemocná, 34letá žena, byla sledována pro SLE od roku 1997. V úvodu onemocnění se vyskytovaly obličejový rash, artralgie, podle echokardiografického vyšetření perikardiální výpotek. Labo-ratorně byla u nemocné zjištěna pozitivita ANA v titru 1 : 320, kontrolně až 1 : 1280, homogenního typu, pozitivita anti ds DNA, anti Sm, anti U1RNP, anti Ro a ENA vše nepřímou imunofluorescencí, lupus antikoagulant a aCl protilátky byly negativní. Nemocná i přes vysokou autoprotilátkovou aktivitu včetně aktivity komplementového systému (CIK 220, C3 0,78, C4 0,16) byla po několik let stabilizována na malé dávce kortikosteroidů (prednison 2,5–5 mg) a antimalarik (hydroxychlorochin 200 mg). Dále byla sledována na psychiatrii a užívala Lithium. Při tomto vcelku stabilizovaném stavu se v průběhu několika dnů rozvíjí nevysvětlitelná únava, subfebrilie, výraznější artralgie, což nemocná přičítala možnému virovému infektu. Anamnesticky uvedla asi 3 dny nechutenství a nízký příjem tekutin. Při takto uváděném stavu se náhle objevuje ataka kvantitativní poruchy vědomí s tonickými záškuby končetin, která trvala asi 1 hodinu. Byla přivezena na neurologické oddělení, kde bylo provedeno neurologické vyšetření a CT mozku bez průkazu krvácení či jiné patologické formace. Pro výraznou trombocytopenii byla nemocná přeložena na Interní hematoonkologickou kliniku Brno, kde byla do několika hodin stanovena diagnóza TTP na podkladě přítomnosti schistocytů v nátěru periferní krve, těžké trombocytopenie a MAHA (trombocyty 3x109/l, Hb 74 g/l, erytrocyty 2,46x1012/l, leukocyty 5,39x109/l). V biochemickém vyšetření byl nález kreatininu 273 μmol/l, ALT 1,26 μkat/l, AST 2,32 μkat/l, LD 20,74 μkat/l, bilirubin 51 μmol/l, v koagulaci aPTT 103,6 sekund, fibrinogen 3,46 g/l, tedy známky postižení jater a ledvin. Bylo zahájeno podávání ČMP a byl aplikován metylprednisolon 250 mg i.v. a hydratace krystaloidy. Přesto došlo v průběhu několika hodin k progresi hemolýzy (bili 173 μmol/l, LD 15,9 μkat/l,) a celkově k horšení stavu nemocné, byla pozorována 2 krát ataka kvantitativní poruchy vědomí s tonicko-klonickými křečemi horních končetin. Pro rychlou progresi stavu přes podání 6 jednotek plazmy byla po 12 hodinách od přijetí zahájena výměnná plazmaferéza. V průběhu plazmaferézy však bohužel došlo k zástavě dechu a oběhu, kardiopulmonální resuscitací včetně intubace se nepodařilo obnovit životní funkce a nemocná zemřela. V rámci pitevního nálezu byly patrné výrazné projevy hemoragické diatézy na vnitřních orgánech.
Popis 3. případu
Nemocná, 26letá žena, byla přijata pro aktivitu SLE s dominujícími kožními a kloubními projevy, subfebriliemi a s celkovou únavou, které progredovaly po dobu asi 6 měsíců. Diagnóza SLE byla stanovena v roce 2001 (tedy před 5 lety od vzniku ataky) na podkladě motýlovitého erytému, artritid drobných kloubů rukou, pozitivity ANA v titru 1 : 320 granulárního typu a anti ds DNA, silně pozitivních ENA a anti-La protilátek. Nemocná byla dlouhodobě léčena prednisonem v malých dávkách, hydroxychlorochinem 200 mg/den a lokální léčbou na kůži.
Vzhledem ke klinické aktivitě onemocnění i přes podávané kortikosteroidy v dávce 40 mg/den po dobu 3 měsíců, byla po dohodě s dermatologem přidána imunosuprese cyklosporinem A v dávce 3 mg/kg hmotnosti. Pro teplotní špičku přes 38 stupňů Celsia byla empiricky zahájena antibiotická léčba cefuroximem. Přes zavedenou p.o. imunosupresivní léčbu (prednison 40 mg/den, cyklosporin A 3 mg/kg hmotnosti) dochází k progresi exkoriace kůže v oblasti plosek a dlaní až typu exfoliativní dermatitidy, ke tvorbě krust v oblasti krku a obličeje. Při kontrolním dermatologickém vyšetření bylo vysloveno podezření i na alergickou kožní reakci na podané antibiotikum, a proto byla zahájena parenterální léčba kortikosteroidy (metylprednisolon 250 mg i.v.) a lokální léčba. Přes celkovou intenzivní imunosupresi byl kožní nález stále v progresi, nově se objevily laboratorní známky intravaskulární hemolýzy s poklesem trombocytů (tromb 20x109/l, Hb 82 g/l, erytrocyty 2,88x1012/l, leukocyty 6,3x109/ l) a dochází k vzestupu kreatininu (161 μmol/l). V rámci koagulace byly zaznamenány přítomnost D dimerů (2,17 μg/l) a vyšší fibrinogen (4,4 g/l). Při takto zavedené léčbě po 5 dnech dochází náhle u nemocné k recidivujícím epileptickým záchvatům typu grand mall s tonicko-klonickými křečemi, následně i ke kvalitativním změnám vědomí ve smyslu dezorientace. Provedenými vyšetřeními (lumbální punkce, neurologické vyšetření) nebyla prokázána neuroinfekce, ale CT vyšetření mozku ukázalo nález mnohočetných hypodenzních ložisek. Pro zpřesnění nálezu bylo CT doplněno o MR vyšetření mozku, kde v T1 a T2 obrazech byl nález hyperintenzních ložisek v bílé hmotě bilaterálně okcipitálně, jejichž původ byl pravděpodobně postischemický, přičemž postkontrastně nedocházelo k patologickému sycení. Klinicky se onemocnění projevovalo kvalitativními poruchami vědomí ve smyslu změn psychického stavu s dezorientacemi. Po diferenciálně diagnostické rozvaze byl stav uzavřen jako mikroangiopatická hemolytická anémie typu TTP na podkladě přítomnosti neurologických projevů, progrese renálního selhávání, hemolytické anémie s trombocytopenií. Byly zahájeny velkoobjemové výměnné plazmaferézy, po kterých regreduje neurologická symptomatologie a kvalitativní poruchy vědomí, ale přetrvává výrazná intravaskulární hemolýza a těžká trombocytopenie. Následně byly podány intravenózní imunoglobuliny v dávce 2 g/kg hmotnosti a bylo navázáno dvěma pulzy cyklofosfamidu v dávce 1100 mg i.v. ve čtrnáctidenním intervalu. Došlo postupně ke klinické stabilizaci a k normalizaci laboratorních ukazatelů hemolýzy s normalizací počtu trombocytů. Renální parametry se upravily do 2 týdnů, kožní nález regredoval velmi pozvolna v průběhu několika týdnů. V současné době je nemocná rok po atace v uspokojivém stavu na malé dávce imunosupresiv (prednison 5 mg a azathioprin 50 mg denně).
Diskuse
TTP je mikroangiopatické okluzivní onemocnění charakterizované systémovou či renální agregací destiček s tvorbou destičkových trombů s následnou konzumpční trombocytopenií a s mechanickým postižením erytrocytů (1). Klinická diagnostika TTP je založena na přítomnosti nálezů trombocytopenie, mikroangiopatické hemolytické anémie se schistocyty a na postižení CNS většinou přechodného rázu. TTP a SLE mají obdobné klinické příznaky, jako jsou teplota, postižení CNS, renální postižení a hemolytickou anémii, avšak přítomnost mikroangiopatické hemolytické anémie se schistocyty významně podporuje diagnózu TTP syndromu (12). Rozlišení, zda o jde ataku aktivity SLE či TTP je důležité vzhledem k možnostem léčby, kdy lékem volby u ataky TTP je výměnná plazmaferéza se substitucí čerstvou mraženou plazmou doplněná o podávání plazmy i v době mezi jednotlivými plazmaferézami, zatímco v případě aktivity SLE je lékem první volby intenzivní imunosuprese (12).
TTP může být pozorována jako komplikace probíhajícího SLE a podle literatury se asi u 1–4 % SLE nemocných v průběhu jejich života může objevit ataka TTP (13). U SLE nemocných byly v průběhu 20 let pozorovány ataky TTP s incidencí 3,8 % (14). Avšak v jedné starší studii bylo zjištěno, že 23 % ze 151 dospělých nemocných s TTP mělo symptomy SLE (15). Je však nutné připomenout, že zjištění TTP u SLE nemocných je všeobecně podhodnocováno z důvodů velmi obdobných příznaků a symptomů, jako jsou trombocytopenie a neurologické abnormality u aktivních stavů SLE. U většiny nemocných je SLE diagnostikován před atakami TTP, u 6,8–15 % TTP předchází diagnózu SLE a v 12–26 % jsou oba syndromy diagnostikovány současně (13, 16, 17). V našem případě byly všechny tři nemocné sledovány již několik let pro SLE s různou klinickou i laboratorní aktivitou.
Na začátku ataky hemolytického syndromu je nutná diferenciální diagnostika stavů, které by mohly vést k obdobnému klinickému a laboratornímu obrazu (18–20). Takto jsme postupovali i v případě našich tří SLE nemocných. Stav prvních dvou nemocných byl kritický při selhávání kardiálním, renálním a hepatálním, s neurologickými projevy. V laboratorních parametrech byla zaznamenána výrazná trombocytopenie a anémie bez významných odchylek v koagulogramu. Byly zvýšeny schistocyty v periferním nátěru, dále pokles haptoglobinu, u první nemocné byl přímý Coombsův test negativní. Diferenciálně diagnosticky jsme zvažovali několik onemocnění s možným fulminantním průběhem (viz tab. 1). U poslední nemocné ataka vznikla při několik měsíců progredujícím kožně-kloubním nálezu a k rozvoji mohla přispět i některá medikace (cyklosporin, antibiotika). I tato nemocná měla schistocyty v periferním krevním nátěru, byly ale také přítomny silně pozitivní antigranulocytární a slabě reagující antitrombocytární protilátky a pozitivní přímý Coombsův test. U první a třetí nemocné došlo k významnému a promptnímu ústupu kvalitativních poruch vědomí po zahájení velkoobjemových výměnných plazmaferéz, ale u třetí nemocné se projevy mikroangiopatické hemolýzy podařilo stabilizovat až po podání cyklofosfamidu. I přes tuto skutečnost se domníváme, že u nemocné se jednalo o souběh dvou syndromů, určité aktivity SLE představované jako kožně-kloubní projevy s jistými znaky hemolýzy (pozitivita přímého Coombsova testu) a ataky TTP, zastoupenou nálezem schistocytů, neurologickými projevy a nálezem renálního postižení. Tuto skutečnost lze podpořit i faktem, že neurologické a renální projevy výrazně regredovaly po zahájení velkoobjemových výměnných plazmaferéz, zatímco projevy hemolýzy se začaly zlepšovat po přidání imunosupresivní léčby pomocí cyklofosfamidu. V případě druhé nemocné došlo k fulminantnímu projevu mikroangiopatické hemolytické anémie s významnou trombocytopenií, s renálním selháváním a tonicko-klonickými neurologickými projevy s krátkodobými kvalitativními poruchami vědomí, typickými pro syndrom TTP. Nemocná umírá pod obrazem multiorgánového selhávání i přes zahájenou léčbu čerstvými mrazenými plazmami a plazmaferézou do 16 hodin od přijetí a v podstatě okamžitém stanovení diagnózy TTP.
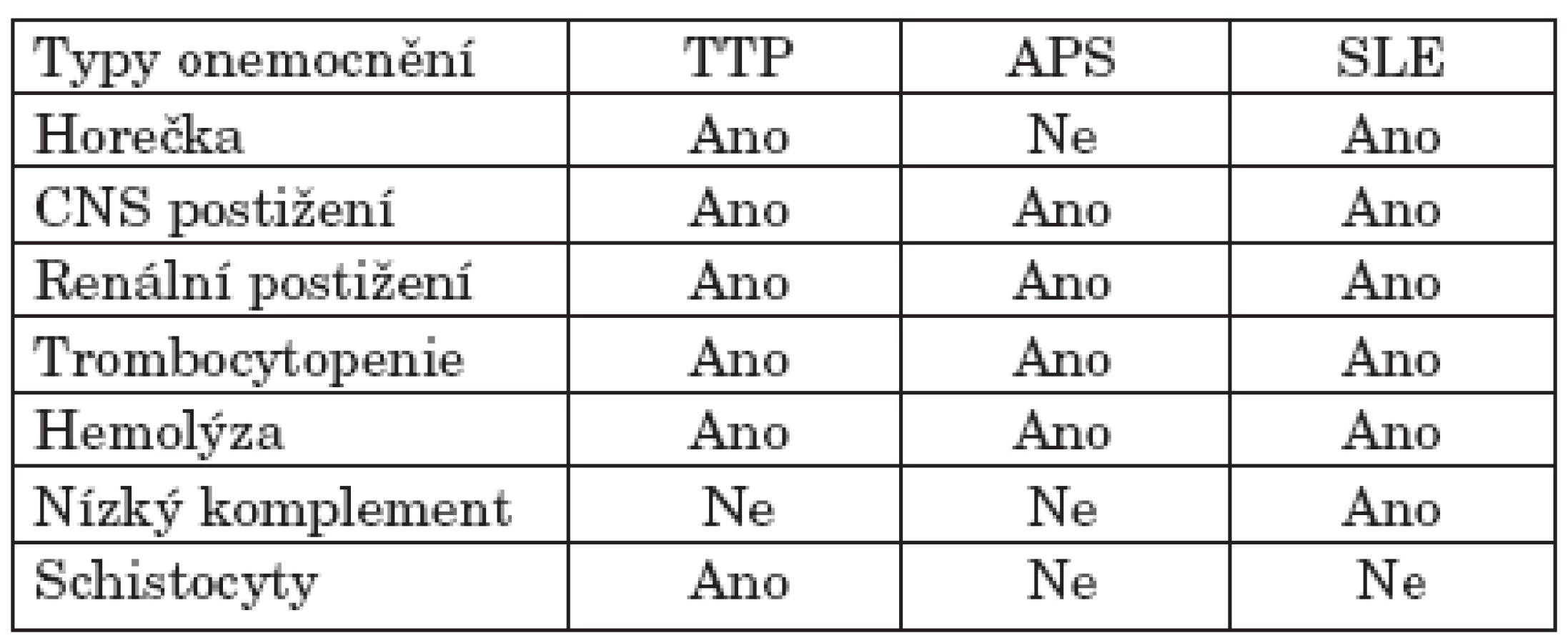
Akutní vzplanutí SLE je další syndrom, u kterého se klinická charakteristika a projevy mohou překrývat s TTP. Přesto přítomnost mikroangiopatické hemolytické anémie a schistocyty jsou více charakteristické pro TTP. U námi sledovaných nemocných spíše dominovaly nepřítomnost muskuloskeletálních příznaků, laboratorně bez známek významného poklesu ve složkách komplementu a v krevním obrazu bez významné leukopenie, což při splnění pentády typické pro TTP vedlo k diagnóze právě tohoto syndromu. Precizní klinické i laboratorní rozlišení mezi příznaky TTP a akutním vzplanutí SLE je v současnosti hůře definovatelné i právě pro skutečnost, že hladiny ADAMTS 13 proteázy mohou kolísat jak při atakách TTP, tak i SLE (21) a navíc u čistých TTP atak plazmatické hladiny ADAMTS 13 kolísají nemocný od nemocného v rozmezí méně než 10 % normálních hodnot až po normální hodnoty (22). Tyto nálezy mohou vést ke spekulacím, že tyto dva syndromy (tedy určitý souhrn velmi obdobných symptomů) ve skutečnosti mohou náležet k jedné nosologické jednotce, nebo že jsou tyto syndromy společně asociovány více, než se dříve předpokládalo.
Dalším syndromem s obdobnými symptomy je katastrofický antifosfolipidový syndrom. Pro vznik a rozvoj katastrofického antifosfolipidového syndromu jsou charakteristické i kožní projevy typické pro toto onemocnění, tedy livedo reticularis, a laboratorně přítomnost získaného inhibitoru koagulace, tedy aCL protilátky a lupus antikoagulant. Při poklesu hladiny fibrinogenu a antitrombinu III by stav mohl být hodnocen jako akutní diseminovaná intravaskulární koagulopatie, což jsme u našich nemocných rovněž nepozorovali. Hemolyticko-uremický syndrom rovněž patří k mikroangiopatickým hemolytickým syndromům, pro které je typická střevní infekce v úvodu onemocnění a známky renálního postižení v časných fázích. Diskutovaná je také otázka podání cyklosporinu v léčbě systémových onemocnění pojiva jako možného etiopatogenetického spouštěcího mechanismu rozvoje TTP (7).
Vzhledem k přítomnosti v podstatě všech složek pentády typické pro TTP, tedy teplota, neurologické postižení, mikroangiopatická hemolytická anémie, akutní renální insuficience a konzumpční trombocytopenie, byly tyto stavy hodnoceny jako akutní ataky TTP. Laboratorně byly dále přítomny schistocyty v periferním krevním nátěru a vysoká hladina LDH. Ačkoli přítomnost klasické pentády usnadňuje diagnózu TTP, u většiny nemocných nebývají tato kritéria splněna. Někteří autoři doplňují charakteristiku TTP jako syndrom mikroangiopatické hemolytické anémie s negativním Coombsovým testem a s trombocytopenií, u kterého se primárně nevyskytují patologické změny v koagulačních testech (2). Je běžně akceptovaným přístupem zahájit léčbu standardními postupy v přítomnosti trombocytopenie, MAHA a nepřítomnosti jiných onemocnění, která mohou způsobovat tyto laboratorní známky (8). Bylo zjištěno, že prodleva v zahájení léčby TTP výměnnými plazmaferézami často způsobuje rozvoj renálního selhání (8). Rovněž podání glukokortikoidů ve vysokých dávkách vedlo k lepším výsledkům zejména u nemocných s mírnou formou TTP bez neurologických příznaků (8). V případě nemocných se SLE a s rozvojem TTP lze při selhání standardní léčby pomocí velkoobjemových plazmaferéz využít imunosuprese pomocí alkylačních cytostatik, která lze použít i na udržení remise (23, 24). Splenektomie jako další krok v případě relabující TTP je častým krokem v případě selhání výměnných plazmaferéz a imunosupresivní léčby (2, 25).
Závěry
Z případů tří nemocných léčených na našem pracovišti je zřejmé, že u jedinců se systémovými chorobami pojiva ve stabilizovaném stavu při léčbě imunosupresivy v běžných dávkách lze pozorovat rozvoj hemolytických syndromů, které nemusí být způsobeny přímo základní chorobou. Diferenciální diagnostika těchto hemolytických syndromů vyskytujících se často v kombinaci se selháváním životně důležitých orgánů je někdy složitý rozhodovací proces, podle kterého volíme strategii léčby. Tyto hemolytické syndromy, často TTP, vyžadují bezprostřední léčbu první volby, tedy podání čerstvé mrazené plazmy či výměnnou velkoobjemovou plazmaferézu vzhledem k bezprostřednímu ohrožení života.
MUDr. Zdeněk Fojtík, PhD.
Revmatologická ambulance
Interní hematoonkologická klinika FN Brno
Jihlavská 20
625 00 Brno
e-mail: zfojtik@fnbrno.cz
Sources
1. Moake JL. Thrombotic microangiopathies. N Engl J Med 2002; 347 : 589–600.
2. Rock GA. Management of thrombotic thrombocytopenic purpura. Brit J Haematol 2000; 109 : 496–507.
3. Allford SL, Machin SJ. Current understanding of the pathophysiology of thrombotic thrombocytopenic purpura. J Clin Pathol 2000; 53 : 497–501.
4. Tsai HM. Deficiency of ADAMTS 13 in thrombotic thrombocytopenic purpura. Int J Hematol 2002; 76 : 132–8.
5. Fujikawa K, Suzuki H, McMullen B, Chung D. Purification of human von Willebrand factor – cleaving protease and its identification as a new member of the metalloproteinase family. Blood 2001; 98 : 1662–6.
6. George JN. Thrombotic thrombocytopenic purpura. N Engl J Med 2006; 354 : 1927–35.
7. George JN. How I treat patients with thrombotic thrombocytopenic purpura – hemolytic uremic syndrome. Blood 2000; 96 : 1223–9.
8. Kwaan HC, Soff GA. Management of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Sem Haematol 1997; 34 : 159–66.
9. Allan DS, Kovacs MJ Clakr WF. Frequently relapsing thrombotic thrombocytopenic purpura treated with cytotoxic immunosuppressive therapy. Haematologica 2001, 86, 844–850.
10. Bobbio-Pallavicini E, Porta C, Centurioni R, et al. Vincristine sulfate for the treatment of thrombotic thrombocytopenic purpura refractory to plasma-exchange. Eur J Haematol 1994; 52 : 222–6.
11. Reddy PS, Deauna-Limayo D, Cook JD, et al. Rituximab in the treatment of relapsed thrombotic thrombocytopenic purpura. Am J Hemato 2005; 84 : 232–5.
12. Cheung WY. Thrombotic thrombocytopenic purpura and systemic lupus erythematosus – distinct entities or overlapping syndromes? Transfusion Apheresis Science 2006; 34 : 263–6.
13. Starck M, Abedinpour F, Dendorfer U, et al. Acquired thrombotic thrombocytopenic purpura as the presenting symptom of systemic lupus erythematosus. Successful treatment with plasma exchange and immunosuppression – report of two cases. Eur J Haematol 2005; 75 : 436–40.
14. Porta C, Bobbio-Pallavicini E, Centurioni R, et al. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus. Italian cooperative group for TTP. J Rheumatol 1993; 20 : 1625–6.
15. Levine S, Shearn MA. Thrombotic thrombocytopenic purpura and systemic lupus erythematosus. Arch Int Med 1964; 113 : 826–36.
16. Caramaschi P, Riccetti MM, Pasini AF, et al. Systemic lupus erythematosus and thrombotic thrombocytopenic purpura. Report of three cases and review of the literature. Lupus 1998; 7 : 37–41.
17. Hunt L, Li X, James J, et al. Thrombotic thrombocytopenic purpura (TTP) and systemic lupus erythematosus (SLE): distinct but potentially overlapping syndromes. Blood 2004; 104 : 245a.
18. Asherson RA. The catastrophic antiphospholipid syndrome, 1998. A review of the clinical features, possible pathogenesis and treatment. Lupus 1998; 7, Suppl 2 : 55–62.
19. Vaidya S, Abul-ezz S, Lipsmeyer E. Thrombotic thrombocytopenic purpura and systemic lupus erythematosus. Scand J Rheumatol 2001; 30 : 308–10.
20. Fojtík Z., Kořístek Z., Klabusay M., Navrátil M. Recidivující trombotická trombocytopenická purpura u nemocné se systémovým lupus erythematodes. Čes Revmatol 2003; 11 : 157–60.
21. Mannucci PM, Vanoli M, Forza I, et al. Von Willebrand factor cleaving protease (ADAMTS-13) in 123 patients with connective tissue disease (systemic lupus erythematosus and systemic sclerosis). Haematologica 2003; 88 : 914–8.
22. Peyvandi F, Ferrari S, Lavoretano S, et al. Von Willebrandt factor cleaving protease (ADAMTS-13) and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura. Br J Haematol 2004; 127 : 433–9.
23. Perez-Sanchez I, Anguita J, Pintado T. Use of cyclophosphamide in the treatment of thrombotic thrombocytopenic purpura complicating systemic lupus erythematosus: report of two cases. Ann Hematol 1999; 78 : 285–7.
24. Vasoo S, Thumboo J, Fong KY. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus: disease activity and the use of cytotoxic drugs. Lupus 2002; 11 : 443–50.
25. Crowther MA, Heddle N, Hayward CP, et al. Splenectomy done during hematologic remission to prevent relapse in patients with thrombotic thrombocytopenic purpura. Ann Intern Med 1996; 125 : 294–6.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2007 Issue 2
Most read in this issue
- Osteonekróza asociovaná se systémovou kortikoterapií
- Trombotická trombocytopenická purpura u nemocných se systémovým lupus erythematodes
- Doporučení České revmatologické společnosti pro léčbu revmatoidní artritidy. Účinnost a strategie léčby
- Zobrazovací metody v hodnocení strukturální progrese u ankylozující spondylitidy