
Rizikové faktory maligní lymfoproliferace u Sjögrenova syndromu
Risk factors for malignant lymphoproliferation in Sjögren’s syndrome
Sjögren’s syndrome is a chronic autoimmune disease characterized by lymphoproliferative infiltration of exocrine glands. Infiltration leads to deterioration of gland and alteration of its function such as loss of tears and decreased salivary excretion. Course of the disease is characterized by either local autoimmune process or systemic disease affecting many internal organs. Regarding the fact that Sjögren’s syndrome is mostly associated with malignant transformation to lymphoproliferative disease, it is considered as a borderline disease between autoimmunity and malignant lymphoproliferation. The results of several studies evaluating risk factors for malignant transformation are inconsistent. The enlargement of parotid gland, lymphadenopathy, palpable purpura, cutaneous vasculitis and neuropathy belong to the most frequently referred clinical risk factors. Lymphocytopenia, decline in CD4 positive lymphocytes, hypogammaglobulinemia, monoclonal gammapathy, cryoglobulinemia and low levels of complement C3 and C4 represent the most frequently referred laboratory risk factors. Presented case report describes most of abovementioned risk factors in a patient who demonstrated transformation from primary Sjögren’s syndrome to diffuse large B-cell non-Hodgkin lymphoma with kappa positive plasmocellular differentiation.
Key words:
Sjögren’s syndrome, lymphoma, risk factors for malignant transformation
:
H. Dejmková 1; R. Bečvář 1; J. Střítecký 2; M. Hnátková 3; E. Žikešová 3; M. Trněný 3
:
Revmatologický ústav, Praha, 2Ústav patologie, Praha, 3I. interní klinika UK, Praha
1
:
Čes. Revmatol., 15, 2007, No. 3, p. 158-161.
:
Case Report
Sjögrenův syndrom je chronické autoimunitní onemocnění charakterizované lymfocytární infiltrací exokrinních žláz. Infiltrát vede k poškození žláz a k poruše funkce – ztrátě slz a snížení slinné sekrece. Onemocnění probíhá buď ve formě lokálního autoimunitního procesu nebo ve formě systémového onemocnění s postižením celé řady orgánů. Vzhledem k tomu, že Sjögrenův syndrom je chorobou s nejčastějším přechodem do maligního lymfoproliferativního onemocnění, je řazen mezi nemoci na pomezí autoimunity a maligní lymfoproliferace. Výsledky studií zabývajících se rizikovými faktory přechodu nejsou zcela jednotné. Mezi nejčastěji uváděné rizikové klinické faktory patří zvětšení příušních žláz, lymfadenopatie, hmatná purpura, kožní vaskulitida a neuropatie. Mezi nejčastěji uváděná laboratorní rizika patří lymfopenie, pokles CD4 lymfocytů, hypogamaglobulinemie, monoklonální gamapatie, kryoglobulinemie a nízká hladina C3 a C4 složky komplementu. Kazuistika demonstruje většinu uvedených rizikových faktorů na případu pacientky, u které došlo k přechodu primárního Sjögrenova syndromu do difuzního velkobuněčného ne-Hodgkinova B lymfomu s kappa pozitivní plazmocelulární diferenciací.
Klíčová slova:
Sjögrenův syndrom, lymfom, rizikové faktory přechodu
Úvod
Sjögrenův syndrom (SS) je chronické autoimunitní onemocnění charakterizované lymfocytární infiltrací slzných a slinných žláz (1). Infiltrát způsobuje poškození žláz, které se projeví poruchou funkce – suchostí očí a suchostí v ústech (2). Rozsah SS kolísá od místních projevů až po těžké systémové onemocnění s poškozením celé řady orgánů. SS může existovat samostatně (primární SS) nebo může provázet jiné systémové autoimunitní onemocnění (sekundární SS) (3, 4). Charakteristickým znakem SS je polyklonální aktivace B buněk, která vede ke vzniku celé řady autoprotilátek jak orgánově specifických, tak orgánově nespecifických (5–7). Choroba bývá spojena s celou řadou hematologických abnormalit, z nichž je nejzávažnější přechod do maligního lymfoproliferativního onemocnění. Existuje řada studií, která se rizikovými faktory tohoto přechodu zabývá (8–11).
Na příkladu nemocné budou demonstrovány nejčastěji uváděné rizikové faktory vývoje maligního, většinou ne-Hodgkinova lymfomu.
Popis případu
43letá pacientka, s primárním SS diagnostikovaným v r. 1998, byla přijata na oddělení RÚ v V/2006 pro tříměsíční zhoršení zdravotního stavu. Nemocná udávala progresi chronické únavnosti, otok pravé příušní žlázy a vyrážku na dolních končetinách. Dále si stěžovala na brnění a změnu citlivosti dolních končetin a na bolest v prstech rukou a nohou provázenou promodráváním a tvorbou vřídků na konečcích prstů.
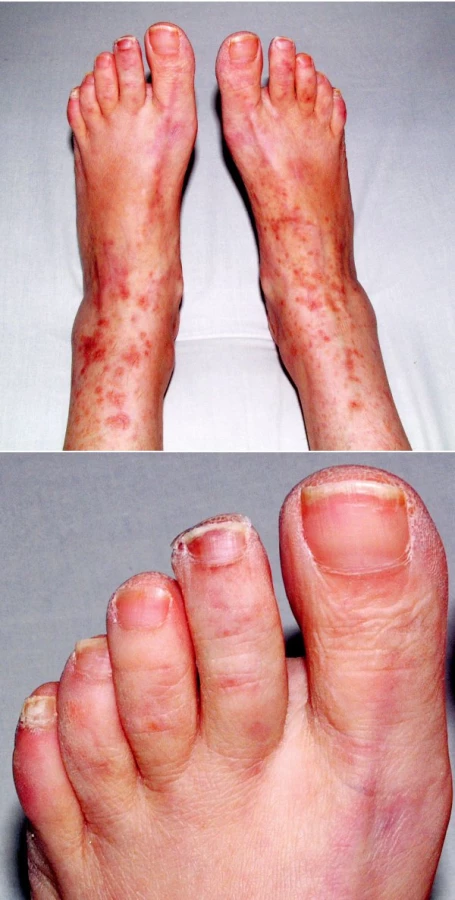
V objektivním nálezu bylo přítomno zvětšení pravé příušní žlázy a zvětšení krčních lymfatických uzlin. Na dolních končetinách byla hmatná purpura, prsty horních i dolních končetin byly chladné a cyanotické (obr. 1). Na 3. a 4. prstu pravé ruky a na 3. a 5. prstu levé ruky byly již defekty (obr. 2). V laboratorních nálezech byla zvýšená sedimentace (86/h) a CRP (145,98 mg/l). V krevním obraze byla přítomna anémie (HB 89 g/l, HCT 0,28), trombocytóza (558) a lymfopenie (0,058) s poklesem B lymfocytů a s převahou CD4 lymfocytů. V elektroforéze byla hypogamaglobulinemie (5,68 g/l), v imunoelektroforéze byl zjištěn monoklonální paraprotein IgM typu kappa. V imunologickém vyšetření byly zvýšeny hodnoty antinukleárních protilátek (160) a byly pozitivní protilátky proti La, Ro, Ro 52 a Ro 60. C4 složka komplementu byla neměřitelná (0,00). Ostatní laboratorní nálezy byly ve fyziologických mezích. Z dalších patologických nálezů byla zjištěna absence signálu digitálních arterií na třetím a čtvrtém prstě pravé ruky a na třetím a pátém prstě levé ruky. Pletysmografie prokázala chybějící signál pulsové vlny na druhých prstech dolních končetin. Klinické podezření na periferní neuropatii bylo prokázáno elektromyograficky. Klinický obraz kožní vaskulitidy byl podložen histologickým nálezem svědčícím pro obliterační vaskulitidu. Počítačovou tomografií bylo prokázáno zvětšení krčních a mediastinálních uzlin. Histologický a imunohistochemický nález ve vyjmuté krční uzlině prokázal difuzní velkobuněčný B-lymfom s kappa pozitivní plazmocelulární diferenciací (obr. 3). Nemocná byla přeložena na I. interní kliniku VFN, kde byla podrobena odpovídající chemoterapii.


Diskuse
SS, zejména jeho primární forma, bývá v literatuře označován jako hraniční onemocnění pohybující se na pomezí autoimunity a lymfoproliferace (12). Toto označení vyplývá z výsledku studií, které prokazují 16–43, 8x vyšší výskyt lymfomu u nemocných se SS oproti zdravé populaci (13, 14). Pacient se SS má přibližně 1–10 % riziko, že v průběhu choroby lymfomem onemocní (3, 15). Jedná se většinou o ne-Hodgkinův lymfom, převážně B-buněčného typu (7, 16). T buněčné lymfomy jsou vzácné (7, 17). Malignita lymfomů kolísá od lymfomů s nízkým stupněm malignity po lymfomy vysoce maligní. Přechod původní benigní lymfoepiteliální léze do maligní lymfoproliferace je pozvolný proces, v jehož průběhu je polyklonální aktivace B buněk nahrazena aktivací monoklonální, která může postupně přejít v lymfom (18). Zprvu vzniká lymfom s nízkým stupněm agresivity, který může přejít v lymfom s vysokým stupněm agresivity. Na vzniku lymfomu se pravděpodobně uplatňuje dlouhodobá antigenní stimulace B buněk autoimunitou nebo viry. Význam virů, zejm. viru Epstein Barrové, viru hepatitidy C, lidského T lymfotropního viru je nejasný. Studie zaměřující se na úlohu virů v etiopatogenezi vývoje lymfomu u nemocných se SS jsou zatím sporadické a jejich výsledky jsou nejednotné (12). Na přechodu do agresivní formy lymfomu se podílí řada onkogenních vlivů, například genetická alterace včetně genových mutací a translokací (12). Histologicky se jedná většinou o lymfomy vycházející z B buněčné okrajové zóny lymfatické tkáně a proto jsou označovány jako marginální lymfomy (MZL). Tyto lymfomy se vyznačují značnou morfologickou variabilitou. Lymfomy jsou nejčastěji lokalizovány v exokrinních žlázách nebo v lymfatických uzlinách, mohou se však vyvinout v kůži, v žaludku i v jiných tkáních (19).
Řada studií se zabývá rizikovými klinickými a laboratorními znaky přechodu SS do lymfomu. K nejčastěji uváděným klinickým rizikovým faktorům patří zvětšení příušních žláz, splenomegalie a lymfadenopatie. K dalším rizikovým faktorům patří hmatná purpura, vaskulitida s kožními defekty a periferní neuropatie (20, 21) (tab 1).

Laboratorní rizikové faktory zahrnují hematologické, biochemické a imunologické abnormality. Mezi hematologická rizika patří anémie a lymfocytopénie, zejména nízké hodnoty CD4 lymfocytů (12, 22). Mezi biochemické rizikové faktory patří pokles dříve zvýšených gamaglobulinů až k hodnotám odpovídajícím hypogamaglobulinemii (22). Dalším výrazným rizikovým faktorem je monoklonální gamapatie a přítomnost kryoglobulinů (12, 22). V imunologickém vyšetření bývá nápadné snížení C 3 nebo C4 složky komplementu. Alarmujícím znakem je sérový pokles IgM a vymizení původně přítomných autoprotilátek (8–12) (tab. 2).

V případě naší pacientky byla přítomna celá řada rizikových faktorů uváděných v literatuře. Z klinických rizik to byl nález zvětšené příušní žlázy, lymfadenopatie, vaskulitidy s vývojem defektů prstů a hmatná purpura na dolních končetinách. Nemocná měla rovněž neuropatii. Z laboratorních rizik byla přítomna anémie, lymfocytopenie, hypogamaglobulinemie, monoklonální gamapatie a neměřitelná hodnota C4 složky komplementu. Další v literatuře uváděné rizikové faktory (např. kryoglobulinemie a nízké hodnoty CD4 lymfocytů) u pacientky prokázány nebyly.
Závěr
Vzhledem k podstatně vyššímu riziku maligní lymfoproliferace u nemocných se SS, a to zejména u pacientů s primární formou, je nutné provádět podrobné klinické a laboratorní kontroly v šestiměsíčních intervalech. Laboratorní vyšetření by mělo kromě běžného programu obsahovat vyšetření gamaglobulinů, imunoglobulinů, imunofixaci a vyšetření kryoglobulinů. Dále je vhodné doplnit vyšetření CD4 lymfocytů, vyšetřit autoprotilátky a komplement. Jsou-li přítomny rizikové faktory přechodu je nutné program rozšířit o další komplementární vyšetření ve spolupráci s hematologem.
Podporováno výzkumnými záměry MZO 00023728
MUDr. Helena Dejmková
Revmatologický ústav
Na Slupi 4
128 50 Praha 2
e-mail: dejm@revma.cz
Sources
1. Vitali C, Bombardieri S, Jonnsson R, et al. European Study Group on Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Concensus Group. Ann Rheum Dis 2002; 61 : 554–8.
2. McAvoy BA, et al. Sjögren’s syndrome II. Clinical associations and immunological phenomena.Q J Med 1973; 42 : 513–48.
3. Moutsopoulos HM, Chused TM, Mann DL, et al. Sjögren’s syndrome (sicca syndrome): current issues. Ann Intern Med 1980; 92 : 212–26.
4. Mičeková D, Rovenský J, Lukáč J, et al. Sjögrenův syndrom In Pavelka K, Rovenský J. Klinická revmatologie. Praha: Galén, 2003, 303–9.
5. Scofield RH, Bruner GR, Harley JB, et al. Autoimmune thyroid disease is associated with a diagnosis of secondary Sjögrenś syndrome in familial systemic lupus. Ann Rheum Dis 2007; 66 : 410–3.
6. Scofield RH. Autoimmune thyreoid disease in systemic lupus erythematodes and Sjögren’s syndrome. Clin Exp Rheumatol 1996; 14 : 321–30.
7. Saito M, Fukuda T, Shiohara T, et al. Angioimmunoblastic T-cell lymphoma: A relatively common type of T-cell lymphoma in Sjögren’s syndrome. Clin Exp Rheumatol 2005; 23 : 888–90.
8. Voulgarelis M, Dafni UG, Isenberg DA, et al. Malignant lymphoma in primary Sjögren’s syndrome. A multicenter, retrospective, clinical study by the European Concerted action on Sjögren’s syndrome. Arthritis Rheum 1999; 8 : 1765–72.
9. Suitcliffe N, Inanc M, Speight P, et al. Predictors of lymphoma development in primary Sjögren’s syndrome. Semin Arthritis Rheum 1998; 28 : 80–7.
10. Tzioufas GA. B-cell lymphoproliferation in primary Sjögren’s syndrome. Clin Exp Rheumatol 1996;14 (Suppl)14 : 65–70.
11. Valesini G, Priori R, Bavoillot D, et al. Differential risk of non-Hodgkin’s lymphoma in Italian patients with primary Sjögren’s syndrome. J Rheumatol 1997; 24 : 2376–80.
12. Manganelli P, Fietta P, Quaini F. Hematologic manifestations of primary Sjögren’s syndrome. Clin Exp Rheumatol 2006; 24 : 438–48.
13. Kassan SS, Thomas TL, Mounsopoulos HM, et al. Increased risk of lymphoma in sicca syndrome. Ann Intern Med 1978; 89 : 888–92.
14. Whaley K, Webb J, Theander E, Henriksson G, Ljunberg O, et al. Lymphoma and other malignancies in primary Sjögren’s syndrome: a cohort study on cancer incidence and lymphoma predictors. Ann Rheum Dis 2006;65 : 796–803
15. Masaki Y, Sugai S. Lymphoproliferative disorders in Sjögren’s syndrome. Autoimun Rev 2004;3 : 175–82.
16. Kruize AA, Hené RJ, Van Der Heide A, et al. Long-term follow-up of patiens with Sjögren’s syndrome. Arthritis Rheum 1996;39 : 297–303.
17. Seror R, Sordet CH, Guillevin L, et al. Tolerance and efficacy of rituximab and changes in serum B cell biomarkers in patiens with systemic complications of primary Sjögrenś syndrome. Ann Rheum Dis 2007; 66 : 351–7.
18. Mortlock AM, Lim CSE, Morgan H, et al. Renal MALToma: an unusual lymphoma in a patient with lupus. Lupus 2006; 15 : 613–5.
19. Tzioufas AG, Bomba DS, Skopouli FN, et al. Mixed monoclonal cryoglobulinemia and monoclonoal rheumatoid factor cross - reactive idiotypes as predictive factors for the development of lymphoma in primary Sjögren’s syndrome. Arthritis Rhem 1996; 39 : 767–72.
20. Klener P. Myeloproliferativní onemocnění in Klener P., et al. Vnitřní lékařství. Praha: Galén, 2006, 458–483.
21. Ionnidis JAP, Vassiliou VA, Moutsopoulos M. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren’s syndrome. Arthritis Rheum 2002; 46 (3): 741–7.
22. Anaya JM, McGuff HS, Banks PM, et al. Clinicopathological factors relating malignant lymphoma with Sjögren’s syndrome. Semin Arthritis Rheum 1996;25 : 337–46.
23. Ramos-Casals M, Font J, Garcia-Carrasco M, et al. Primary Sjögren’s syndrome. Hematologic patterns of disease expression. Medicine 2002; 81 : 281–92.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2007 Issue 3
Most read in this issue
- Arthroscopic synovectomy of the wrist
- Risk factors for malignant lymphoproliferation in Sjögren’s syndrome
- Possibilities of early detection of severe cardiovascular manifestations of SLE
- The role of S100A4 protein in rheumatoid arthritis