
Protilátky proti složkám komplementového systému a systémový lupus erythematodes
Antibodies directed against complement components and systemic lupus erythematosus
SLE is a disease characterized by overproduction of various types of autoantibodies. Under certain circumstances, it is possible to detect the presence of autoantibodies directed against neoepitopes of complement system, as well. Neoepitopes are not present in native proteins, but develop with their structural alterations. The following antibodies come under known anticomplement auoantibodies: C3 nephritic factor, antibody directed against C1 inhibitor or antibodies directed against C1R (C1 receptor). Anti-C1q antibodies are concededly the most significant autoantibodies directed against complement system. They are present in about one third of patients with SLE and there is a correlation with clinical activity and the presence of lupus glomerulonephritis. High titers of anti-C1q antibodies are accompanied by decreased levels of C1, as well as C3 and especially C4 complement component. The presence of anti-C1q antibodies is not limited or specific for lupus only. They were described for the first time in case of HUVS (Hypocomplementemic Urticarial Vasculitis Sydrome), later in Felty’s syndrome, rheumatoid vasculitis, hepatitis C and in senescent population, as well. The association of anti-C1q antibodies, complement consumption and presence of renal affection in case of SLE calls into question, whether and how do these autoantibodies participate in organ affection pathogenesis. As shown in animal models of disease, the presence of both anti-dsDNA and anti-C1q antibodies is necessary for the genesis of lupus nephritis. Their mutual interaction triggers the mechanisms of development of renal affection mediated by immune complexes.
Key words:
systemic lupus erythematosus, complement system, anti-C1q antibodies
:
P. Horák 1; H. Ciferská 1; J. Zadražil 1; Z. Heřmanová 2
:
III. interní klinika, FN a LF UP Olomouc, 2 Oddělení klinické imunologie, FN Olomouc
1
:
Čes. Revmatol., 16, 2008, No. 1, p. 16-22.
:
Overview Reports
SLE je onemocnění charakterizované nadprodukcí různých typů autoprotilátek. Za jistých okolností lze zjistit přítomnost autoprotilátek rovněž proti neoepitopům komplementového systému. Neoepitopy nejsou přítomny v nativních proteinech, ale objevují se při jejich strukturálních změnách. Mezi známé antikomplementové autoprotilátky patří: C3 nefritický faktor, protilátka proti C1 inhibitoru či protilátky proti C1R (C1 receptor). Bezesporu nejvýznamnější autoprotilátkou proti komplementu jsou anti-C1q protilátky. Jsou přítomny asi u třetiny nemocných se SLE, korelují s klinickou aktivitou a s přítomností lupusové glomerulonefritidy. Vysoké titry anti C1q protilátek jsou doprovázeny sníženými hladinami C1 ale také C3 a zejména C4 složky komplementu. Přítomnost anti-C1q protilátek není omezena či specifická pouze pro lupus. Poprvé byly popsány v případě syndromu HUVS (Hypocomplementemic Urticar Vasculitis Sydrome), později také při Feltyho syndromu, revmatoidní vaskulitidě, u hepatitidy C a ve stárnoucí populaci. Asociace mezi přítomností protilátek anti C1q, spotřebou komplementu a přítomností renálního postižení v případě SLE vzbuzuje otázku, zda a jak se tyto autoprotilátky podílejí na patogenezi orgánového postižení. Jak ukazují zvířecí modely choroby, pro vznik lupusové nefritidy je potřebná přítomnost jak anti-dsDNA, tak anti-C1q protilátek, jejichž vzájemná interakce spouští mechanizmy rozvoje imunokomplexového renálního postižení.
Klíčová slova:
systémový lupus erythematodes, komplementový systém, anti C1q protilátky
Úvod
SLE (systémový lupus erythematodes) je choroba charakteristická největším známým počtem různých typů autoprotilátek. Jejich produkce může být řízena antigenem, je však také výsledkem aktivace B buněk a poruch apoptózy a výsledkem dysregulace cytokinové sítě. U SLE bylo popsáno více než sto autoprotilátek, které cílí nukleární, cytoplazmatické, membránové či fosfolipidové antigeny, dále krevní a endoteliální buňky, antigeny v nervovém systému, plazmatické či matrixové proteiny včetně CRP a různé další antigeny (1).
V patogenezi SLE hraje komplement ambivalentní roli (2). Aktivace komplementu přispívá k tkáňovému postižení, komplementový systém však současně zvyšuje solubilitu imunokomplexů zejména při jejich transportu k fagocytárním buňkám při odstraňování apoptotického materiálu z cirkulace a tím působí proti autoimunitním mechanismům (3). Jak se pokusí ukázat tato práce, komplement se může rovněž stát cílem autoprotilátek, což může mít pro rozvoj SLE důležité patogenetické i klinické relevantní důsledky
2. Komplementový systém
Komplement je fylogeneticky starou součástí imunitního systému a spolu s interferonem, cytokiny a chemokiny představuje jednu ze základních složek nespecifické odpovědi. Komplementový systém je tvořen více než dvaceti membránovými a sérovými proteiny (enzymy), které jsou tvořeny jaterní buňkou, v menší míře pak monocytomakrofágovým systémem či jinými buňkami.
Komplement představuje hlavní efektorový mechanismus zánětlivé odpovědi, hraje důležitou roli v fyziologickém odstranění imunokomplexů a rovněž má pomocnou roli v indukci protilátkové odpovědi. Může být aktivován třemi různými cestami, klasickou, alternativní a lektinovou. Aktivace probíhá formou kaskádovitého štěpení (cleavage) jednotlivých součástí komplementu, při kterém vznikají jeho aktivní formy.
Klasická cesta aktivace komplementu je závislá na protilátkách a je aktivována agregovanými IgG či IgM protilátkami v imunokomplexech. Lektinová cesta aktivace komplementu je nezávislá na protilátkách a je aktivovaná vazbou lektinu vážícího manózu (mannose-binding lectin, MBL) na bakteriální povrchy. Alternativní cesta je vývojově starší, je nezávislá na protilátkách či imunokomplexech a je aktivována potenciálními patogeny. Tři cesty aktivace komplementu se protínají v tvorbě C3b konvertázy, která přetváří nativní C3 do aktivní formy C3b. Aktivace komplementu pokračuje tvorbou trimolekulárního komplexu C3b konvertázy, C3b a C5. Po kaskádě aktivace dochází na buněčné stěně k vytvoření komplexu atakujícího membránu ze složek C5b-C9, který způsobí osmotickou lýzu buňky (membrane attacking complex, MAC). Z hlediska správné funkce je důležité přesné pořadí aktivace jednotlivých složek, chybí-li některý z proteinů, je celá sekvence přerušena (3, 4). Tradičně jsou přisuzovány komplementu čtyři biologické role – opsonizace, aktivace zánětu, clearance imunokomplexů a osmotická cytolýza infekčních agens. V posledních dekádách byla popsána řada dalších funkcí komplementu, který zřejmě slouží rovněž jako pojítko mezi vrozenou a adaptivní imunitou (5, 6).
2.1. Klasická cesta aktivace komplementu
Úvodním krokem v klasické cestě aktivace komplementu je vazba dvou či více globulárních domén C1q složky na Fc fragment protilátek vázaných na antigen. Samotná IgG či IgM protilátka je pouze slabým aktivátorem této složky. Dvě a dvě molekuly C1r a C1s vytvoří tetramer, který se naváže na tento komplex, což vede k jeho strukturální změně a rozštěpení C1s na aktivní enzymy (7). Jeho hlavní funkcí je rozštěpení neaktivní složky C4 na C4b a C4a fragmenty. Zejména pak C4b složka hraje klíčovou roli v tvorbě C3 konvertázy vznikající jako komplex C4b s C2a. Tento enzym rozštěpí C3, což vede v tvorbě velkého množství C3b složky pokrývající aktivované povrchy, tento fenomén se nazývá opsonizace. C4bC2a komplex váže také aktivní C3b, čímž vzniká další enzym v kaskádě, C5 konvertáza.
2.2. Alternativní cesta aktivace komplementu
Alternativní cesta aktivace komplementu zahrnuje šest plazmatických proteinů: C3, faktory B, D, H, I a properdin. Základním rysem alternativní cesty je možnost aktivace mechanismem nezávislým na protilátkách, a to působením patogenů jako jsou bakterie, houby, viry, paraziti či abnormální vlastní tkáně – nádorové buňky či infikované buňky. Tato cesta aktivace je ve stálém stavu mírné aktivity vyúsťující do trvalé tvorby nižších hladin aktivovaného C3b. Prvním krokem je interakce faktoru B s C3i v tekuté fázi nebo s C3b na cílovém povrchu. Faktor B je proteolyticky rozštěpen na aktivní Bb a neaktivní Ba. Komplex C3bBb vykazuje silnou C3 štěpící aktivitu a je také známý jako C3 konvertáza alternativní cesty. Faktor H a I působí jako regulační proteiny. Pro osud C3b je velmi důležitý charakter aktivovaného povrchu, ke kterému je enzym vázaný kovalentní vazbou. Pokud se jedná o „cizí“ povrchy (například bakterie), mohou tyto působit jako akceptory faktoru B a tím spouštět amplifikační kličku alternativní cesty. Jelikož faktory H a I, potažmo membránové regulační molekuly CD35 a CD46 činí vazbu C3b na vlastní povrchy nestabilní, k aktivaci C3 konvertázy nedochází. Tzv. amplifikační klička alternativní cesty aktivace komplementu působí prostřednictvím C3 konvertázy produkci velkého množství C3b složky, která má opět schopnost vazby na cizorodé povrchy a celý systém pozitivní zpětnou vazbou posiluje (3).
2.3. Lektinová cesta aktivace komplementu
Aktivace lektinové cesty je vyvolána vazbou MBL (mannose binding lectin) na terminální skupiny manózy na površích některých bakterií. Tato vazba vede k aktivaci dvou proteáz MASP-1 a MASP-2 vykazujících blízkou příbuznost k molekulám C1r a C1s. MASP-1 pak štěpí C4 a C2 velmi podobným způsobem jako C1s, což vede k tvorbě C3 konvertázy. Fyziologická funkce MASP-1 není známá (3).
2.4. Terminální cesta
Všechny tři cesty aktivace komplementu se protínají v oblasti štěpení C3 složky komplementu. Konečnou fází aktivace komplementu je tvorba komplexu atakujícího membrány (membrane attacking complex, MAC). C3 konvertázy všech cest aktivace komplementu váží další volné C3b molekuly za vniku C5 konvertáz (C4b2aC3b a C3bBbC3b). Rozštěpení C5 složky komplementu C5 konvertázou vytvoří C5a a C5b fragmenty. C5b reaguje s C6, následně s C7 a C8 složkami komplementu. Konečným krokem je pak vazba komplexu C5b-C8 s C9 složkou. Tím je tvorba MAC komplexu dokončena (3).
2.5 Regulace aktivace komplementu
Aktivace komplementu je ve všech fází regulovaná, což ochraňuje vlastní tkáně před vedlejšími efekty aktivace komplementu při obraně proti cizorodým organismům. Evoluce komplementového systému šla ruku v ruce s evolucí složitých regulačních mechanismů, které se uplatňují na několika úrovních v fázi „tekuté“ i povrchové aktivace komplementu.
C1 inhibitor blokuje C1 složku komplementu a reguluje podobně jako C4b vázající protein klasickou cestu aktivace C3 konvertázy. Faktory H a I se zásadní měrou účastní regulace alternativní cesty. Vitronektin interaguje s reaktivními místy C5b-7 komplexu, což omezuje konečnou schopnost MAC komplexu přilnout k buněčné membráně a chrání tak zdravé hostitelské buňky. Tyto molekuly se řadí mezi regulační proteiny tekuté fáze. Na membránových površích se uplatňují regulátory tvorby C3 a C5 konvertáz (MCP, DAF a CR1) a protein inhibující inserci MAC k membránám, CD59 (3).
3. Role komplementu v patogenezi SLE
SLE může sloužit jako prototyp onemocnění, u kterého se vyskytuje široká škála manifestací spojených s aktivací či dysfunkcí komplementového systému hrajících důležitou roli v patogenezi této choroby (3, 8, 9). Se SLE se pojí řada vrozených i získaných deficiencí komplementu. Dominantní cestou aktivace komplementu je v případě SLE klasická cesta iniciovaná interakcí C1q složky s imunokomplexy. Složky klasické cesty (C1, C2, C4) jsou u aktivního lupusu většinou nízké. Hladiny C3 jsou typicky na dolní hranici fyziologické normy, ale mohou být i signifikantně snížené. Aktivaci komplementu zahajuje jeho interakce s imunokomplexy, které aktivují hlavní cestu. Mechanismus, kterým imunokomplexy zprostředkovávají poškození tkání, je předmětem intenzivního studia. Do nedávna převládal lehce zjednodušený názor, že poškození tkání je důsledkem produkce imunokomplexů aktivujících komplement, který pak iniciuje zánětlivou reakci prostřednictvím aktivace enzymatické kaskády jednotlivých složek komplementu, koagulace a cytokinové sítě. Tyto děje jsou pak následovány přílivem zánětlivých buněk, zejména polymorfonukleárů a monocytů. Vývoj myších kmenů s chybějícím geny pro jednotlivé proteiny zánětlivé kaskády vytvořil několik zvířecích modelů pro studium mechamismů tkáňového poškození mediovaného imunokomplexy (8, 10, 11), které ukazují roli komplementu v těchto dějích v nové perspektivě. Důležitou se jeví zejména vazba komplementu prostřednictvím jeho receptoru pro Fc fragment protilátek k imunokomplexům, která pak aktivuje a udržuje zánětlivou reakci na jejich přítomnost. Je zřejmé, že význam jednotlivých typů imunokomplexů pro patogenezi SLE není identický a že je odvislý od místa jejich tvorby a typu zúčastněného antigenu (12). V mnoho zvířecích modelech byla tak potvrzena dominantní funkce Fc receptorů v tkáňovém poškození. Jiné modely pak prokázaly zásadní vliv aktivace anafylatoxinu C5a pro plnou expresi tkáňového poškození mediovaného imunokomplexy (13).
Výskyt vrozených deficiencí komplementu je v populaci nízký, ale jsou velmi významně spojeny s rozvojem různých chorob (14). Homozygotní deficience každého z ranných proteinů aktivace klasické cesty (C1q, C1r, C1s, C2, C4) je silně spojena s rozvojem SLE (15-23). Tyto deficience jsou jedny z nejsilnějších genetických vazeb chorobných stavů popsaných u lidí (24). Existuje jednoznačná hierarchie prevalence a závažnosti SLE dle pozice, kterou daný protein v kaskádě zaujímá. Nejčastější a nejzávažnější jsou deficience proteinů C1 komplexu a C4 složky komplementu (25, 26).
Nejlépe definovaným genetickým defektem souvisejícím se SLE je selektivní chybění C1q složky komplementu (27–29). Tato vzácná genetická abnormalita byla intenzivně studovaná v postižených rodinách (viz Pickering pro detailní review, 30). Postižení jedinci trpí často závažnou formou lupusu od dětského věku. Homozygotní deficience samotné C4a složky či heterozygotní deficience C4a a/nebo C4b je častější a je rovněž popsána vazba k SLE (19, 30).
Slabší asociace s lupusem je pozorována také při defektech proteinů alternativní či MBL cesty aktivace komplementu. Genetické poruchy terminální společné cesty aktivace jsou spojeny spíše než s autoimunitními projevy se zvýšeným rizikem bakteriálních infekcí včetně meningokokové sepse (30).
Existují tři hlavní, navzájem se však nevylučující hypotézy, které se pokoušejí vysvětlit roli vrozených i získaných defektů komplementu v patogenezi SLE (11). První teorie vidí chybění komplementu jako podmínku vedoucí k nedostatečné clearanci imunokomplexů, jelikož vazba komplementu (zejména C1q, C4b, C3b) ovlivňuje kovalentní vazbou jejich velikost a rozpustnost a poskytuje další vazebné místo potřebné pro jejich efektivní odstranění (28). Tato hypotéza je v souladu s další teorií, která ji vidí zejména v porušené clearanci apoptotického jaderného materiálu. Ten se může stát, pokud není efektivně odstraněn, zdrojem autoantigenů – nukleozomů (10). Nukleozomy pak vyvolávají protilátkovou odpověď a tvorbu antinukleozomálních protilátek, které se stávají prostřednictvím rozšířených epitopů při humorální odpovědi zdrojem dalších antigenních komponent (dsDNA, histony). Defektní clearance apoptotického materiálu hraje zjevně roli rovněž při vrozené deficienci C1q (24, 30). Dále bylo prokázáno, že C1q složka komplementu se váže k apoptotickým keratocytům (31) a v případě C1q deficientní myši byla pozorována zvýšená apoptóza renálních buněk (24). Rovněž monocyty patientů s C1q defektem vykazují nedostatečnou schopnost odstraňovat apoptotické buňky, což lze upravit in vitro přidáním purifikované C1q složky (30). Tímto způsobem se může defekt komplementu podílet na rozvoji imunitní odpovědi spouštěné autoantigenem. Nicméně souslednost dalších změn, které vedou od poruchy clearance apoptotických buněk až k plnému rozvoji autoimunitní reakce, není znám (11). Je třeba brát do úvahy řadu dalších genetických faktorů i vlivů zevního prostředí. Alternativní hypotéza postuluje možnou pozitivní roli normální fungujícího komplementu v indukci autotolerance prostřednictvím determinace aktivačního prahu B a T lymfocytů. Defekty komplementu by dle této teorie mohly ovlivnit fyziologický mechanismus negativní selekce autoreaktivních lymfocytů (32–34).
4. Protilátky proti proteinům komplementové kaskády
Jak již bylo řečeno výše, deficience komplementu jsou silně spojeny s rozvojem SLE a aktivace komplementu hraje zásadní roli v zánětu. Komplement se však může sám stát cílem adaptivní imunitní od-povědi, v literatuře byly popsány protilátky proti různým komponentám komplementového systému (35, 36). Za určitých okolností se vyskytují rovněž protilátky, které cílí neoepitopy komplementového systému. Neoepitopy nejsou přítomny v nativních komplementových proteinech, ale mohou se vyskytnou při strukturálních změnách komplementového systému.
4.1 Anti - C1q protilátky
Většina anti-komplementových protilátek je namířena proti C1q složce. První popis C1q precipitátů o nízké molekulární hmotnosti v některých sérových vzorcích nemocných se SLE pochází z roku 1971 (37). Přítomnost těchto protilátek byla prokázána rovněž mnoha dalšími autory v následujících letech (38,39). Protilátky proti C1q cílí kolagenu podobnou oblast molekuly a reagují v solidní fází s C1q díky expozici skrytých neoepitopů při strukturálních změnách C1q při vazbě na její ligandy, jako jsou imunokomplexy (40–42).
4.1.1 Role anti-C1q protilátek v patogenezi SLE
Asociace mezi přítomností anti-C1q protilátek a spotřebou komplementu u SLE a HUVS (hypokomplementemická urtikární vaskulitida) nabízí otázku, zda tyto protilátky zapřičiňují či pouze zesilují aktivaci klasické cesty komplementu (13). Trouw et al. vyvinuli murinní monoklonální protilátku JL-1, která je identifikovatelná ELISA metodami díky své schopnosti rozpoznat „tail“ doménu myší C1q. Pokud byla anti-C1q (JL-1) podána samostatně, byla vázána v glomerulu k C1q složce, která se zde i za normálních okolností vyskytuje v malém množství. Nicméně tato interakce nebyla schopna sama o sobě vyvolat signifikantní glomerulární poškození. Pokud byla ale JL-1 podána myším s vysokou expresí C1q v glomerulech, zvířata vykazovala významné glomerulární poškození, sníženou ledvinnou funkcí a zvýšené pronikání bílkovin do moči (43, 44).
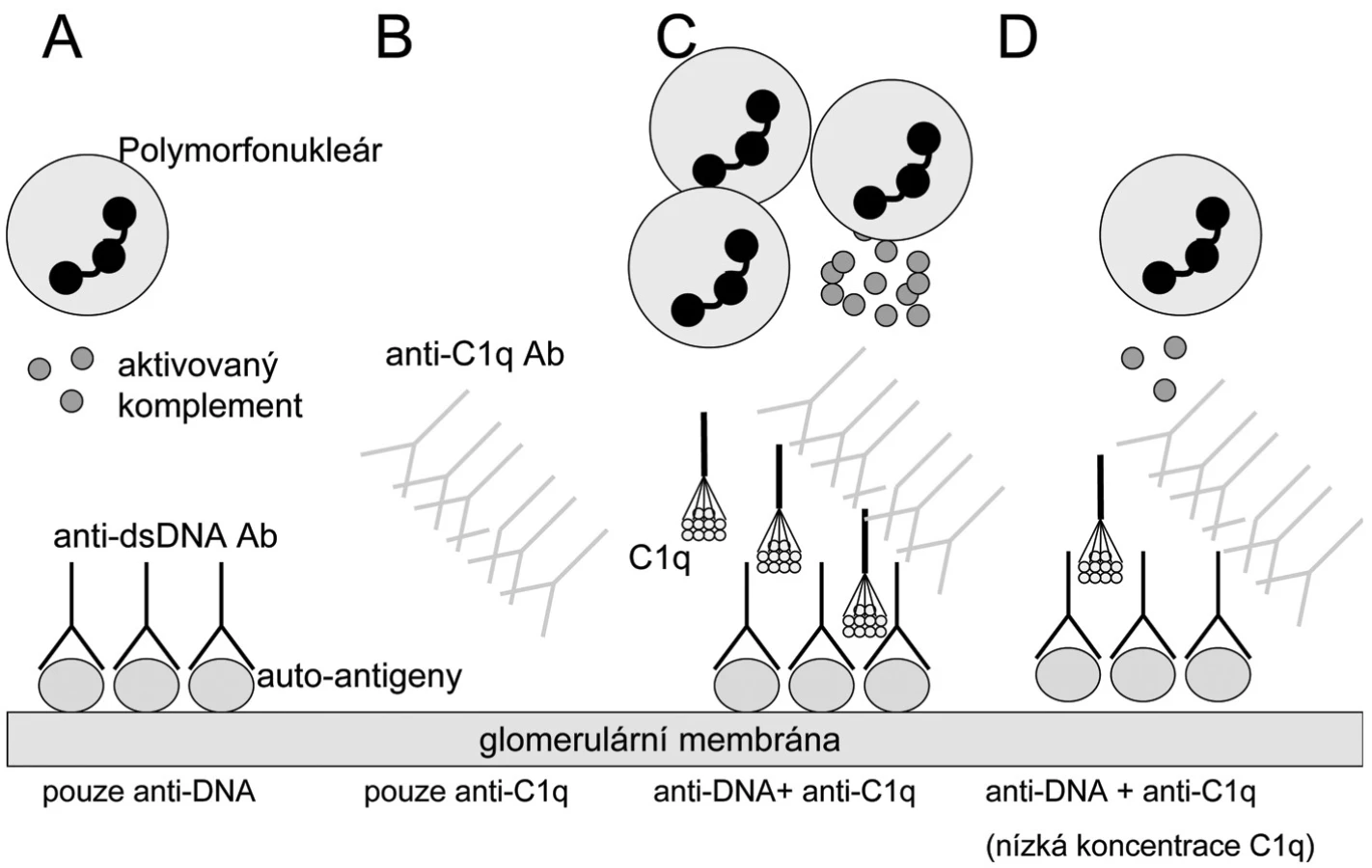
Patogenetická role anti-C1q protilátek v rozvoji lupusové nefritidy je předmětem hypotézy navržené Fliermanem (45). Vychází z nálezu spojení zejména vysokých titrů anti-C1q protilátek s renální chorobou a flary aktivity u SLE (44, 46–51). Samotná přítomnost anti-C1q protilátek však není dostatečná podmínka pro rozvoj nefritidy. Další nutnou podmínkou je přítomnost depozit imunokomplexů obsahujících anti-dsDNA protilátky na glomerulární bazální membráně. Jejich rozpoznání C1q složkou vede dle této teorie k expozici neoepitopů C1q molekuly cirkulujícím anti-C1q protilátkám. Při dostatečně vysokých hladinách anti-C1q a dostatečné lokální přítomnosti C1q složky pak dochází k plné aktivaci klasické komplementové cesty prostřednictvím C1q + anti-C1q komplexů, což vede k rozvoji plné formy lupusové nefritidy (obr. 1). Alternativní hypotéza považuje anti-C1q protilátky pouze za vedlejší produkt aktivace komplementu, která sama vytváří neoepitopy v molekule C1q (15).
4.1.2 Anti-C1q protilátky u různých chorob
Přítomnost anti-C1q protilátek není omezena či specifická pouze pro SLE či lupusovou nefritidu. Poprvé byla klinická asociace anti-C1q protilátek popsána v případě HUVS (hypokomplementemická urtikární vaskulitida), choroby charakterizované chronickou kopřivkou, kožní leukocytoklastickou vaskulitidou, angioedémem, glomerulonefritidou a bronchiální obstrukcí (52). Jiné protilátky nebyly u této jednotky dosud popsány. Vztah této nozologické jednotky a SLE je podobný vztahu mezi lupusem a antifosfolipidovým syndromem. HUVS může být součástí SLE (zejména v přítomnosti anti-C1q protilátek), ale může se rovněž vyskytnou jako samostatná primární jednotka (53). Anti-C1q protilátky byly zovněž nalézány u Feltyho syndromu a u revmatoidní vaskulitidy (38) stejně jako u zdravých lidí s narůstajícím trendem v seniu (48). Anti-C1q protilátky se rovněž objevují u 26 % infikovaných virem hepatitidy C (HCV). Ačkoliv prevalence anti-C1q protilátek je vyšší u nosičů HCV viru s kryoglobulinemií III. typu (50 %), nebyla popsána asociace mezi výskytem a hladinami protilátek a HCV genotypem, závažností jaterní choroby či specifickými projevy HCV vaskulitidy (54, 55).
4.1.3 Prevalence anti-C1q protilátek u SLE
V rámci SLE se prevalence anti-C1q protilátek pohybuje mezi 30–50 % a je spojena s vysokou klinickou aktivitou a zejména s lupusovou nefritidou (31, 38, 56–61). Asociace s jiným orgánovým postižením nebyla popsána. Navíc při absenci anti-C1q protilátek se neobjevuje zpravidla lupusová nefritida, zatímco nejméně u 50% nemocných s pozitivními protilátkami lupusová nefritidy vznikne (56, 59).
V přítomnosti vysokých titrů anti-C1q protilátek jsou většinou také nízké C1q a C4 složky komplementu (29). C3 složka bývá dle některých jen lehce snížena či se pohybuje v normálním rozmezí (29), dle jiných koreluje rovněž s hladinou anti-C1q protilátek (62).
4.2 Další protilátky proti komplementovému systému
Data získaná in vitro jakož také in vivo prokázala časté chybění receptoru pro komplement 1 (CR1, CD35) na erytrocytech nemocných s určitými autoimunitními chorobami, zejména se SLE a také u chronických infekcí typu HIV, HCV či lepry (63–66). U nemocných s lupusem byla prokázána vyšší prevalence protilátek proti CR1 (46 %). Tyto protilátky jsou nalézány také u jaterní cirhózy (36 %), HIV infekce (30 %) a u nemocných s antikardiolipinovými protilátkami (19 %), roztroušenou sklerózou (14 %), mnohočetným myelomem (14 %), avšak s chybějící korelací s hladinami anti-C1q protilátek (67). Rovněž reaktivita anti-CR1 protilátek s CR1 receptory byla in vivonízká (s výjimkou dvou pacientů s SLE), takže všeobecně přijímaná teorie předpokládá, že CR1 se ztrácí při předávce imunokomplexů v játrech a slezině z erytrocytů na fixované makrofágy (68). Role anti-CR1 protilátek není zřejmě významná. Rovněž jejich případné klinické korelace nejsou známé.
Protilátky proti iC3bse váží na aktivovaný produkt štěpení C3 zvaný iC3b; nicméně jejich případná účast v patogeneze jakékoliv choroby není známá (69).
C3 nefritický faktor je na hořčíku závislá IgG protilátka přítomná v séru některých nemocných s chronickou mesangioproliferativní hypokomplementemickou glomerulonefritidou. Inaktivuje C3 složku prostřednictvím jejího rozštěpení do dvou neúčinných fragmentů, C3c a C3d, místo vzniku normální C3b složky. C3 nefritický faktor je spojený s parciální lipidystrofií a s určitým typem mesangioproliferativní glomerulonefritidy (70). Tento faktor se rovněž může vzácně vyskytnout u nemocných se SLE.
Další protilátkou proti proteinům komplementové kaskády je protilátka proti C1 inhibitoru. Je přítomna u některých nemocných s lymfomy či angioedémy, ale nebyla zatím nalezena u SLE (71).
Závěr
Anti-C1q protilátky jsou nejvýznamnější protilátky proti komplementovému systému u SLE, jsou důležité v patogenezi poškození ledvin, mohou sloužit jako serologický ukazatel přítomnosti lupusové glomerulonefritidy či jejího rizika nebo jako možný indikátor aktivity choroby. Další známé protikomplementové autoprotilátky nebyly tak extenzivně studovány a jejich praktický význam je velmi omezený.
Tato práce byla podpořena grantem IGA MZdr ČR NR8406
Doc. MUDr. Pavel Horák, CSc.
III. interní klinika, FN a LF UP Olomouc
I. P. Pavlova 6
772 00 Olomouc
e-mail: horak@pfnol.cz
Sources
1. Sherer Y, Gorstein A, Fritzler M, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: More than 100 different antibodies found in SLE patients. Sem Arthritis Rheum 2004; 34 : 501–537.
2. Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res 2002; 4, Suppl.3 : 279–293.
3. Pickering MC, Walport MJ. The complement system. In.: Hochberg MC, Silman AJ, Smolen JS, et al.: Rheumatology, Third Edition, Mosby, 2003, 1323–1336.
4. Cooper NR. The classical complement pathway: activation and regulation of first complement component. Adv Immunol 1985; 37 : 151–216.
5. Fearon DT, Locksley RM. The instructive role of innate immunity in acquired immune response. Science 1996; 272 : 50–53.
6. Carrol MC. The role of complement and complement receptors in induction and regulation of immunity. Annu Rev Immunol 1998; 6 : 545–568.
7. Sim RB, Reid KB. C1: molecular interactions with activating systems. Immunol Today 1991; 12 : 307–311.
8. Walport MJ, Ng YC, Lachmann PJ. Erythrocytes transfused into patients with SLE and haemolytic anaemia lose complement receptor type 1 from their cell surface. Clin Exp Immunol 1987; 69 : 501–507.
9. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology 198;199 : 265–285.
10. Herrmann M, Voll RE, Lolowos W, et al. Etiopathogenesis of systemic lupus erythematosus. Immunologist 2000; 8 : 345–350.
11. Walport MJ. Advances in immunology: Complement (first of two parts). NEJM 2001; 344 : 1058–1066.
12. Navratil JS, Korb LC, Ahearn JM. Systemic lupus erythematosus and complement deficiency: clues to a novel role for the classical complement pathway in the maintenance of immune tolerance. Immunopharmacol 1999; 42 : 47–52.
13. Law SK, Lichtenberg NA, Holcombe FH, et al. Interaction between the labile binding sites of the fourth and fifth human complement proteins and erythrocyte cell membranes. J Immunol 1980; 125 : 634–639.
14. Barilla-LaBarca ML, Atkinson JP. Rheumatic syndromes associated with complement deficiency. Curr Opin Rheumatol 2003; 15 : 55–60.
15. Mitchell DA, Pickering MC, Warren J, et al. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol 2002; 168 : 2538–2543.
16. Hauptmann G, Grosshans E, Heid E. Systemic lupus erythematosus and hereditary complement deficiency: A case with total C4 defect. Ann Dermatol Syphil Paris 1974; 101 : 479–496.
17. Hannema AJ, Kluin-Nelemans JC, Hack CE, et al. SLE like syndrome and functional deficiency of C1q in members of a large family. Clin Exp Immunol 1984; 55 : 106–114.
18. Pickering MC, Botto M, Taylor PR, et al. (2000) Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 76 : 227–324
19. Hauptmann G, Grosshans E, Heid E, et al. Lupus erythemateux aigu avec deficit complet de la fraction C4 du complement. Nouv Press Med 1974; 3 : 881–882.
20. Schur PH. Inherited complement component abnormalities. Annu Rev Med 1986; 37 : 333–346.
21. Ratnoff WD. Inherited deficiencies of complement in rheumatic diseases. Rheum Dis Clin N Am 1996; 22 : 75–94.
22. Glass D, Raum D, Gibson D, et al. Inherited deficiency of the second component of complement. Rheumatic disease associations. J Clin Invest 1976; 58 : 853–861.
23. Hauptmann G, Tappeiner G, Schifferli JA. Inherited deficiency of the fourth component of human complement. Immunodef Rev 1988; 1 : 3–22.
24. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiol 2002; 205 : 395–406.
25. Fielder AH, Walport MJ, Batchelor JR, et al. Family study of the major histocompatibility complex in patients with systemic lupus erythematosus: importance of null alleles of C4A and C4B in determining disease susceptibility. BMJ 1983;286 : 425–428.
26. Christiansen FT, Dawkins RL, Uko G, et al. Complement allotyping in SLE: association with C4A null. Aust NZ J Med 1983; 13 : 483–488.
27. Stone NM, Williams A, Wilkinson JD, et al. Systemic lupus erythematosus with C1q deficiency. Brit J Dermatol 2000; 142 : 521–524.
28. Berkel AL, Petry F, Sanal O, et al. Development of systemic lupus erythematosus in a patient with selective complement C1q deficiency. Eur J Ped 1997;156 : 113–115.
29. Botto M, Walport MJ. Hereditary deficiency of C3 in animals and humans. Int Rev Immunol 1993; 10 : 37–50.
30. Pickering MC, Botto M, Taylor PR, et al. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol 2000; 76 : 227–234.
31. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol 1997; 158 : 4525–4528.
32. Prodeus AP, Gorg S, Shen LM, et al. A critical role of complement in maintenance of self tolerance. Immunity 1998; 9 : 721–731.
33. Paul E, Carrol MC. SAP less chromatin triggers systemic lupus erythematosus. Nature Medicine 1999; 5 : 607–608.
34. Gommerman JL, Carrol MC. Negative selection of B lymphocytes: a novel role for innate immunity. Immunol Rev 2000; 173 : 120–130.
35. Trouw LA, Roos A, Daha MR. Autoantibodies to complement components. Mol Immunol 2001; 38 : 199–206.
36. Seelen MA, Trouw LA, Daha MR. Diagnostic and prognostic significance of anti-C1q antibodies in systemic lupus erythematosus. Curr Opin Nephrol Hypertens 2003; 12 : 619–624.
37. Agnello V, Koffler D, Eisenberg JW, et al. C1q precipitins in the sera of patients with systemic lupus erythematosus and other hypocomplementemic states: characterization of high and low molecular weight types. J Exp Med 1971; 134 : 228–241.
38. Trouw LA, Daha MR. Role of anti-C1q autoantibodies in the pathogenesis of lupus nephritis. Expert Opin Biol Ther 2005; 5 : 243–251.
39. Trendelenburg M. Antibodies against C1q in patients with systemic lupus erythematosus. Springer Semin Immunopathol 2005; 27 : 276–285.
40. Antes U, Heinz HP, Loos M. Evidence for the presence of autoantibodies to the collagen-like portion of C1q in systemic lupus erythematosus. Arthritis Rheum 1988; 31 : 457–464.
41. Uwatoko S, Aotsuka S, Okawa, M, et al. C1q solid-phase radioimmunoassay: evidence for detection of antibody directed against the collagen-like region of C1q in sera from patients with systemic lupus erythematosus. Clin Exp Immunol 1987; 69 : 98–106.
42. Uwatoko S, Mannik M. Low-molecular weight C1q-binding immunoglobulin G in patients with systemic lupus erythematosus consists of autoantibodies to the collagen-like region of C1q. J Clin Invest 1988; 82 : 816–824.
43. Trouw LA, Groeneveld TW, Seelen MA, et al. Anti-C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J. Clin Invest 2004; 114 : 679–688.
44. Holers VM. Anti-C1q autoantibodies amplify pathogenic complement activation in systemic lupus erythematosus. J Clin Invest 2004; 114 : 616–619.
45. Flierman R, Daha R. Pathogenic role of anti C1q autoantibodies in the development of lupus nephritis – a hypotesis. Molecular Immunol 2007; 44 : 133–138.
46. Horvath L, Czirjak L, Fekete B. High levels of antibodies against Clq are associated with disease activity and nephritis but not with other organ manifestations in SLE patients. Clin Exp Rheumatol 2001; 19 : 667–672.
47. Siegert CE, Daha MR, Halma C, et al. IgG and IgA autoantibodies to C1q in systemic and renal diseases. Clin Exp Rheumatol 1992; 10 : 19–23.
48. Siegert CE, Daha MR, Swaak AJ, et al. The relationship between serum titers of autoantibodies to C1q and age in the general population and in patients with systemic lupus erythematosus. Clin Immunol Immunopathol 1993; 67 : 204–209.
49. Marto N, Bertolaccini ML, Calabuig E, et al. Anti-C1q antibodies in nephritis: correlation between titers and renal disease activity and positive predictive value in systemic lupus erythematosus. Ann Rheum Dis 2005; 64 : 444–448.
50. Moroni G, Trendelenburg M , Del Papa N, et al. Anti-C1q antibodies may help in diagnosing a renal flare in lupus nephritis. Am J Kidney Dis 2001; 37 : 490–498.
51. Coremans IE, Spronk PE, Bootsma H, et al. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am J Kidney Dis 1995; 26 : 595–601.
52. Wisnieski JJ, Naff GB. Serum IgG antibodies to C1q in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum 1989; 32 : 1119–1127.
53. Trendelenburg M, Schifferli JA. Apoptosis and C1q: possible explanations for the pathogenesis of systemic lupus erythematosus. J Rheumatol 2000; 59 : 172–175.
54. Lienesch DW, Sherman KE, Metzger A, et al. Anti-Clq antibodies in patients with chronic hepatitis C infection. Clin Exp Rheumatol 2006; 24 : 183–185.
55. Saadoun D, Sadallah S, Trendelenburg M, et al. Anti-1q antibodies in hepatitis C virus infection. Clin Exp Immunol 2006; 145 : 308–312.
56. Fremeaux-Bacchi V, Noel LH, Schifferli JA. No lupus nephritis in the absence of anti-C1q autoantibodies? Nephrol Dial Transplant 2002;17 : 2041–2043.
57. Kumar A, Gupta R, Varghese T, et al. Anti-C1q antibody as a marker of disease activity in systemic lupus erythematosus. Indian J Med Res 1999; 10 : 190–193.
58. Monova D, Monov S, Rosenova K, et al. Autoantibodies against C1q: view on association between systemic lupus erythematosus disease manifestation and C1q autoantibodies. Ann Rheum Dis 2002; 61 : 563–564.
59. Trendelenburg M, Marfurt J, Gerber SA, et al. Lack of occurrence of severe lupus nephritis among anti-C1q autoantibody negative patients. Arthritis Rheum 1999; 42 : 187–188.
60. Trendelenburg M, Lopez-Trascasa M, Potlukova E, et al. High prevalence of anti C1q antibodies in biopsy–proven active lupus nephritis, Nephrol Dial Transplant 2006; 21 : 3115–3121.
61. Mosca M, Chimenti D, Pratesi F, et al. Prevalence and clinico-serological correlations of anti enolase, anti C1q, and dsDNA antibodies in patients with systemic lupus erythematosus J Rheumatol 2006; 33 : 695–697.
62. Horák P, Heřmanová Z, Zadražil J, et al. C1q complement component and antibodies reflect SLE activity and kidney involvement. Clin Rheumatol 2006; 25 : 532–536.
63. Kazatchkine MD, Fearon DT. Deficiencies of human complement receptors type 1 (CR1, CD35) and type 2 (CR2, CD21). Immunodef Rev 1990; 2 : 17–41.
64. Iida K, Mornaghi R, Nussenzweig V. Complement receptor (CR) deficiency in erythrocytes from the patients with systemic lupus erythematosus. J Exp Med 1981; 155 : 1427–38.
65. Tausk FA, McCutchan A, Spechko P, et al. Altered erythrocyte C3b receptor expression, immune complexes, and complement activation in homosexual men in varying risk groups for acquired immune deficiency syndrome. J Clin Invest 1986; 78 : 977–82.
66. Kanto T, Hayashi N, Takehara T, et al. Low expression of erythrocyte complement receptor type 1 in chronic hepatitis C patients. J Med Virol 1996; 50 : 126–34.
67. Sadallah S, Hess C, Trendelenburg M, et al. Autoantibodies against complement receptor 1 (CD35) in SLE, liver cirrhosis and HIV-infected patients Clin Exp Immunol 2003;131 : 174–181.
68. Davies KA, Hird V, Stewart S, et al. A study of in vivo immune complex formation and clearance in man. J Immunol 1990; 144 : 4613–4620.
69. Trouw LA, Roos A, Daha MR. Autoantibodies to complement components. Molecular Immunol 2001; 38 : 199–206.
70. West CD, Witte DP, McAdams A. Composition of nephritic factor-generated glomerular deposits in membranoproliferative glomerulonephritis type 2. Am J Kidney Dis 2001; 37 : 1120–1130.
71. D’Incan M, Tridon A, Ponard D, et al. Acquired angioedema with C1 inhibitor deficiency: Is the distinction between type I and type II still relevant? Dermatology 1999; 199 : 227–230.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2008 Issue 1
Most read in this issue
- Antibodies directed against complement components and systemic lupus erythematosus
- Idiopathic retroperitoneal fibrosis: Less common cause of low back pain. Use of tamoxifen in the treatment of the diesease
- Influence of supervised physical therapy on spinal mobility and pain in patients with ankylosing spondylitis
- Cytokines BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand) and their role in autoimmune diseases