
Pachydermoperiostosis
Pachydermoperiostosis
The present contribution describes a patient with pachydermoperiostosis, a rare disease associated with hyperperiostosis (occuring predominantly in long bones of forearms and shanks), and clubbing of the digits of the hands and feet. Patients frequently have arthralgia of both the small and large joints. The diagnosis is based on X-ray and clinical observations. Laboratory examination often shows unspecific changes such as elevated erythrocyte sedimentation and C-reactive protein level. Similar X-ray and clinical finding should be considered in the differential diagnosis, which include malignancies (most frequently lung and gastrointestinal cancer), but also, for instance, inherited heart defects. The disease can be diagnosed with the aid of X-ray and laboratory examination, however. We present a case report of our second patient with typical symptoms of this rare disease. Our first observation has been described as early as in 1960.
Key words:
pachydermoperiostosis, incidence, clinical picture
Authors:
J. Vachtenheim; A. Houzarová
Authors‘ workplace:
Revmatologická ordinace, interní oddělení, Krajská nemocnice, Jihlava
Published in:
Čes. Revmatol., 16, 2008, No. 3, p. 124-127.
Category:
Case Report
Overview
Příspěvek je věnován pachydermoperiostóze, která má vzácný výskyt a u které je v popředí výskyt hyperperiostózy, hlavně dlouhých kostí, zejména předloktí a bérců, dále paličkovité prsty a to nejen na horních ale i dolních končetinách. Současně bývá i artralgie velkých a malých kloubů. Rozhodujícími faktory v diagnóze jsou rentgenový důkaz změn kolem uvedených lokalizací a další klinické nálezy. Často zjišťujeme i vyšší hodnoty některých laboratorních výsledků, včetně vyšší sedimentace červených krvinek a C reaktivního proteinu. V diferenciální diagnóze nutno odlišit podobné rentgenové a klinické nálezy, hlavně při malignitách, zejména u karcinomu plic a trávicího traktu, ale i u vrozených srdečních vad. Většinou se ale pomocí laboratorních i rentgenových nálezů dá tato vzácná choroba spolehlivě diagnostikovat. V příspěvku je uvedena vlastní kazuistika již druhého pozorovaného pacienta s typickými projevy tohoto vzácného onemocnění. Naše první pozorování bylo popsáno již v roce 1960.
Klíčová slova:
pachydermoperiostosis, incidence, klinické projevy
Úvod
Pachydermoperiostóza (PDP) bývá popisována pod různými názvy a synonymy. Uvádíme alespoň nejčastěji citované v různé odborné a všeobecné literatuře: hyperplasia ossium et cutis, hyperostosis generalisata cum pachydermia, osteoarthropathia hypertrophiante, osteophytosis Friedreich-Erb-Arnold, la pachydermie plicatureé, syndrom Touraine-Sólente a další (1–3). Toto vzácné onemocnění bylo poprvé popsáno Friedreichem v roce 1868 (cit. v 1). Jak ukazují různé názvy téhož onemocnění, není zatím v nomenklatuře jednota. Nicméně, v posledních letech je již možno toto onemocnění podle průběhu, klinického a rentgenového (rtg) nálezu téměř bezpečně označovat jako PDP.
Onemocnění se v různé častosti vyskytuje familiárně a častěji u mužů než u žen. Nejčastěji bývají popisováni bratři, poprvé je popsal Erb v roce 1887 (3). Postižení bývají vysoké postavy. V naší literatuře je popsali poprvé Charvát s Mařátkou v roce 1946 (3). PDP se nejčastěji klinicky projeví mezi 10. až 30. rokem, ne vzácně i dříve anebo později. Hlavně periostotickými nánosy, nejvíce na dlouhých kostech, paličkovitými (Hippokratovými) prsty, zhrubělou až zřasnatou kůží obličeje a hlavně čela. Občas jsou popisovány i další viscerální a orgánové nálezy. Nicméně hlavní příznaky jak samotný název zní – se týkají pohybového aparátu a kůže. Incidence se uvádí 1 až 2 PDP na sto tisíc obyvatel (4, 5), jedná se tedy o vzácné onemocnění. Ve světové literatuře je doposud popsáno přibližně 350 až 400 případů (5). Víme, že jsou pacienti, jenž nejsou diagnostikováni, anebo vedeni pod jinou diagnózou (3). Většinou bývá klinický nález tak typický, že se dá, při pomocných vyšetřeních, snadno diagnostikovat. Osifikace a hyperperiostózy se týkají téměř vždy skeletu předloktí a bérců, někdy i lokalizací dalších. Pro samotnou diagnózu PDP rozhoduje téměř stoprocentní nález na popsaných lokalizacích a téměř nikdy nechybí paličkovité prsty a zhrubělá kůže. Některé viscerální příznaky mohou souviset i s jinými genetickými projevy na jiných orgánech.
Diagnóza je stanovena často náhodně, když pacient se dostaví k lékaři s jiným onemocněním anebo v souvislosti se samotnou PDP. K nejčastějším příznakům patří slabost, únava, malátnost, a pacienti mají sklon k infekčním onemocněním, zvláště k virózám horních cest dýchacích, vzácně ale i k závažnějším viscerálním afekcím. U našeho zde popisovaného nemocného, jsme např. zjistili polycystózu ledvin s následným vleklým selháním. Při celkovém vyšetření, včetně rtg nálezu na kloubech a skeletu při různé hyperostóze, je nutno při podobných nálezech vyloučit malignity (hlavně na plicích), vrozené vady srdce ale i jaterní cirhózu. Rtg nálezy se v pokročilém stadiu mohou objevit i na patách a osifikacích některých vazů a úponů šlach. Jak již bylo připomenuto, je často postižena kůže ve smyslu zhrubění a se zřasněním, hlavně na obličeji a nejvíce na čele. Toto je popisováno téměř v každé kazuistické práci.
Kazuistika
47letý muž byl poprvé hospitalizován na interním oddělení nemocnice v Jihlavě od 23. října do 2. listopadu 2006. Byl odeslán z revmatologické ordinace na další podrobné vyšetření na lůžkovém oddělení. Nikdy nebyl doposud zde anebo na jiném oddělení pro nějaké potíže hospitalizován.
V rodinné anamnéze udává, že otec zemřel v 39 letech na zhoubný nádor, ale neví nic o lokalizaci. Matka zemřela v 54 letech na selhání ledvin. Docházela nebo dojížděla sanitním autem na hemodialýzu. Pacient měl jednoho bratra, který zemřel v 45 letech. Měl mít „větší prsty“ jak na rukou, tak i nohou. Nevylučuje přítomnost paličkovitých prstů.
V osobní anamnéze neudává žádnou operaci anebo úrazy. Je nekuřák, pracuje jako automechanik. Je ženatý, má jedno zdravé dítě.
Nynější onemocnění: jeden týden před vyšetřením na revmatologické ambulanci začal pociťovat brnění a mravenčení ve všech prstech obou rukou. Otoky kolem kloubů doposud nepozoroval, ale měl je vždy „mohutnější“. Byl odeslán praktickým lékařem na neurologickou ambulanci a odtud na ambulanci revmatologickou. Při vyšetření udává trvalé brnění v posledních článcích obou rukou. Cit vnímá dobře, stejně tak teplo a chlad. Celkové projevy neudává.
Objektivní nález: výška 190 cm, hmotnost 95 kg, BMI 27,5. Při vyšetření pohybového aparátu na horních končetinách klouby funkční, bez periartikulárních otoků. Na posledních článcích prstů obou rukou typické paličkovité prsty (obr. 1 a 2). Na dolních končetinách klouby rovněž funkční. I zde na posledních článcích patrné paličkovité prsty (obr. 3). Na prstech sklíčkové nehty, zcela identické jako na prstech horních končetin. Kůže na obličeji a na čele zhrubělá, jinak beze změn. Při vyšetření páteře bolestivost neudává, hybnost je zachována. Je však nápadné zvětšení až hypertrofie dolních třetin předloktí a bérců. Po stránce interní bez patologických změn, TK 143/92 mmHg, puls pravidelný, 72/min.
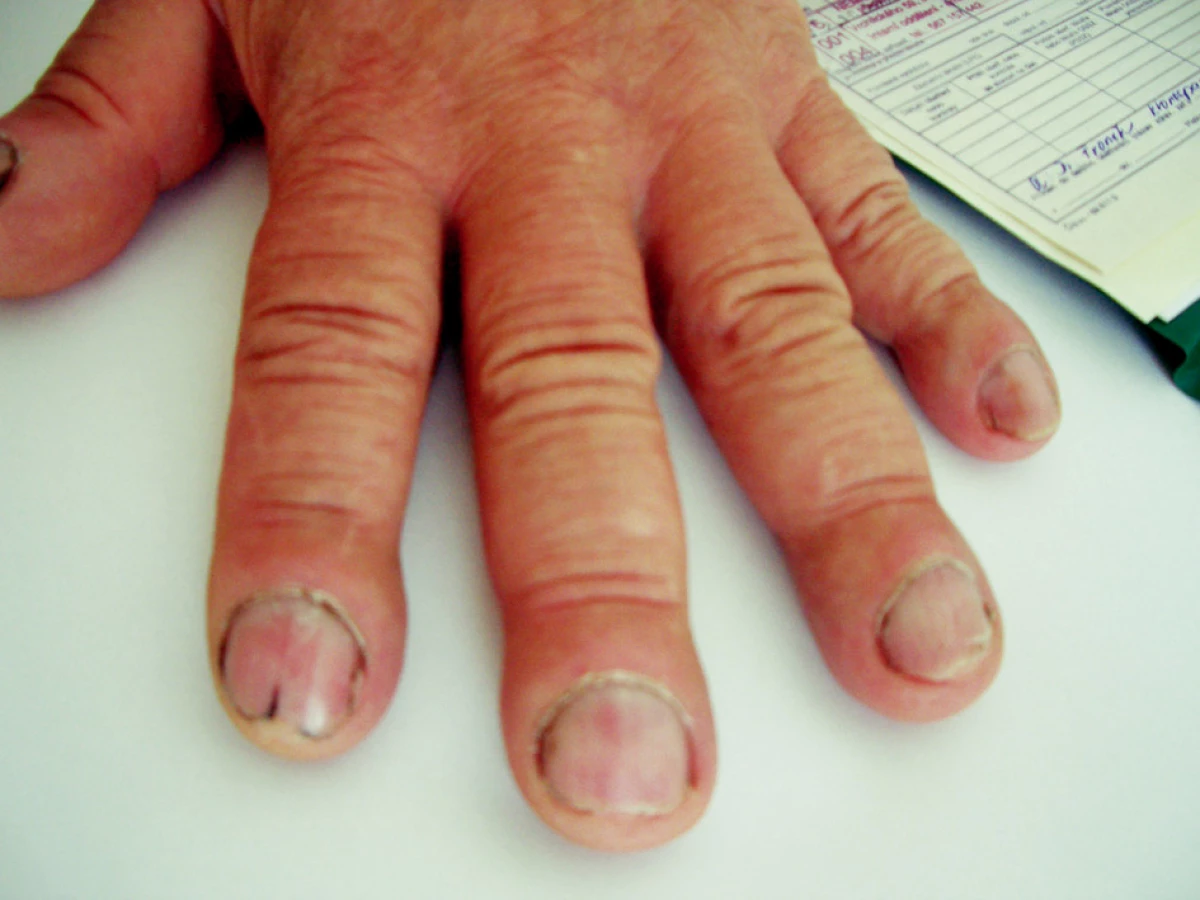
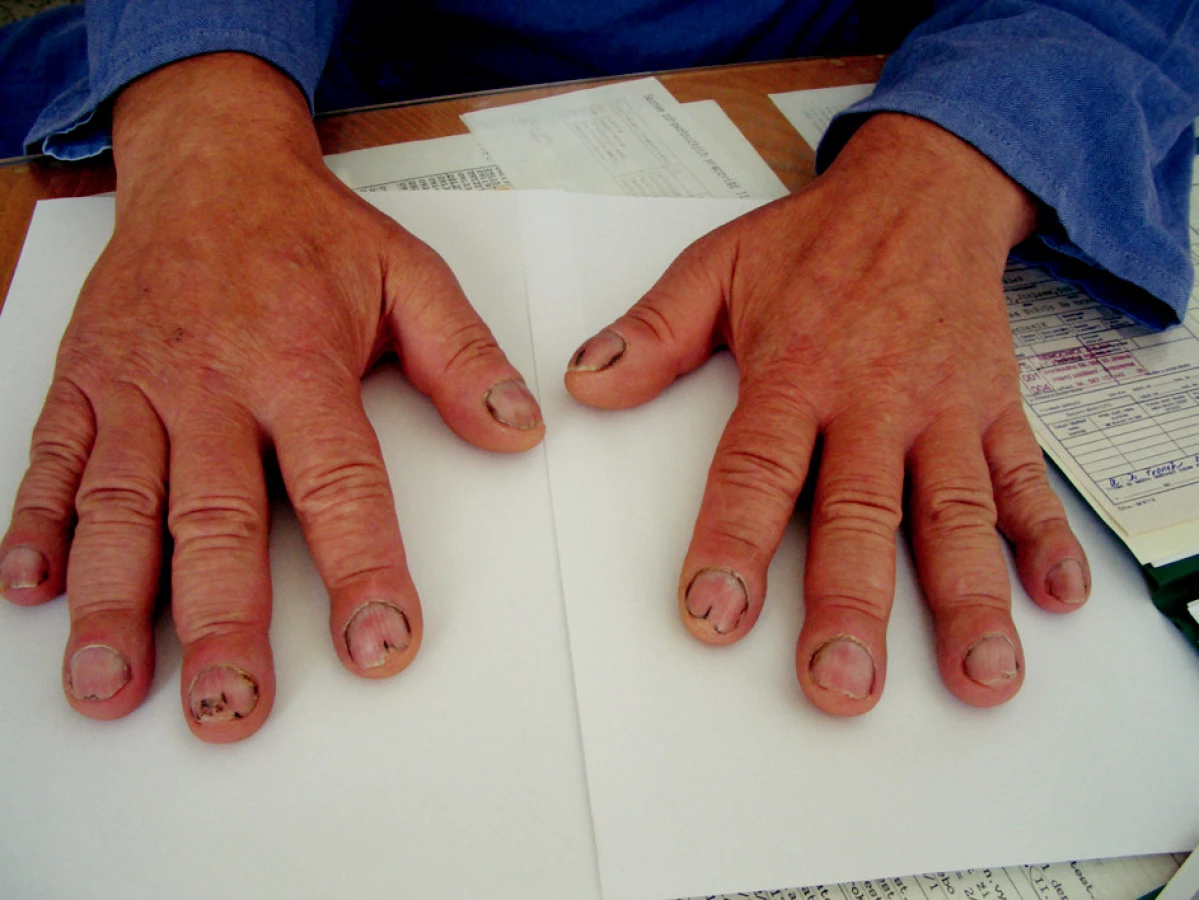
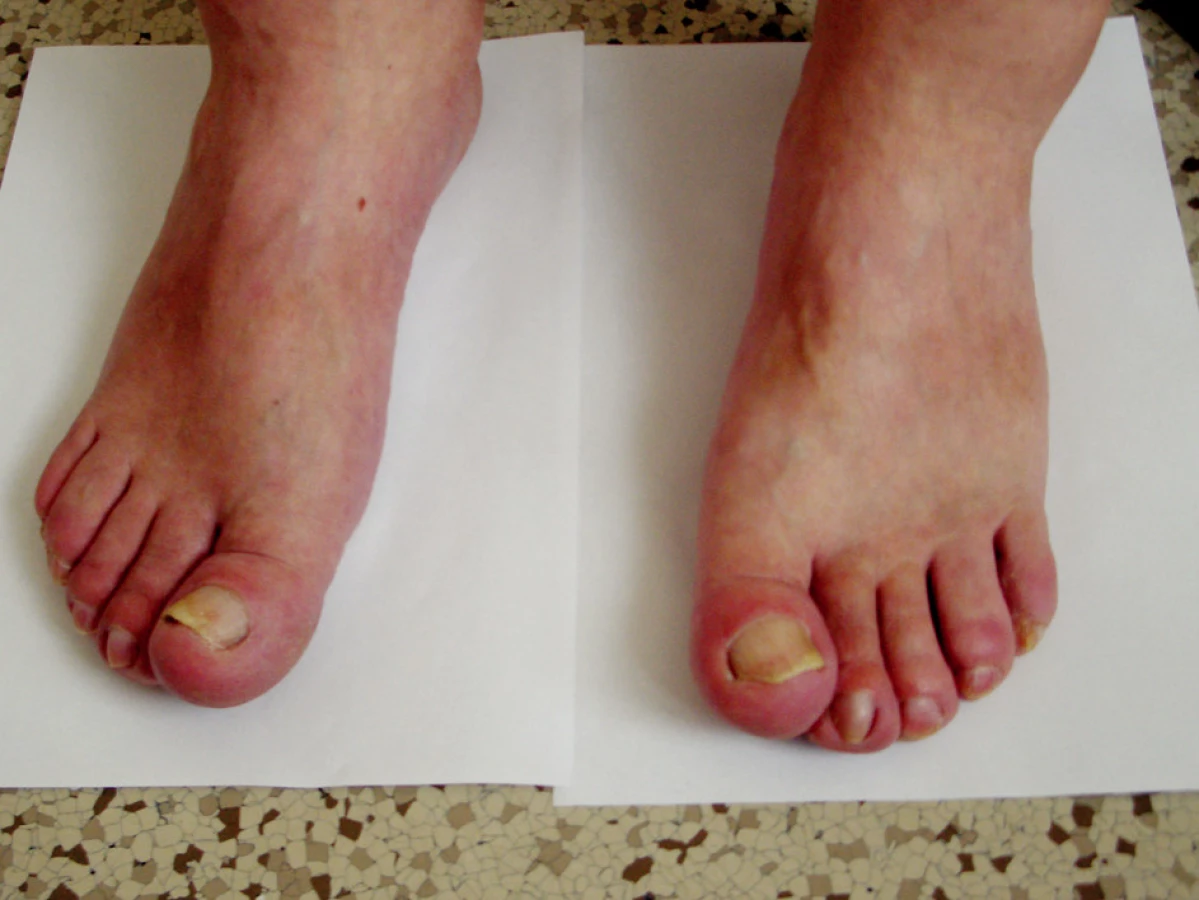
Pomocná vyšetření: sedimentace červených krvinek 21/45, C reaktivní protein 65 mg/l. Krevní obraz a koagulační faktory, včetně INR, bez patologických změn. V biochemickém spektru: močovina vyšší, 12.5 mmol/l, kreatinin 214 μmol/l, kyselina močová 526 μmol/l. Mineralogram, včetně kalcia a fosforu beze změn.V moči bílkovina ++, cukr a urobilinogen negativní, v sedimentu erytrocyty 0–20, leukocytů 0–20, epitelie 21–50. Glomerulární filtrace 0,463 ml/s, tubulární resorpce 0,970. Po stránce imunologické: RF-latexfixačně negativní, ANF, ENA, vyšetřeno Elisou negativní, anti ds-DNA, beta 2 glykoprotein a antikardiolipinové protilátky Elisou negativní. Stejně tak negativní TSH, fT3 a fT4 (na štítnou žlázu) negativní. Rovněž anti HbsAg a anti HCV protilátky negativní. Stolice 3krát na OK negativní.
Rtg nálezy: na plicích a na srdci bez patologických změn. Na skeletu rukou a nohou: v měkkých částech oboustranně drobná kovová tělíska, více vlevo (pravděpodobně poúrazového původu). Periostální apozice na obou radiích a ulnách (obr. 4). Obdobné apozice oboustranně i na metakarpech a na diafýzách. Na skeletu nohou v obou 5. metatarzech rovněž apozice. Na palcích nohou deformity typu hallux valgus a velké osteofyty. Na rtg obou bérců a předloktí patrná symetrická spikulovitá periostální reakce s maximem změn distálně (obr. 5) s necharakteristickým rozšířením kortikalis.
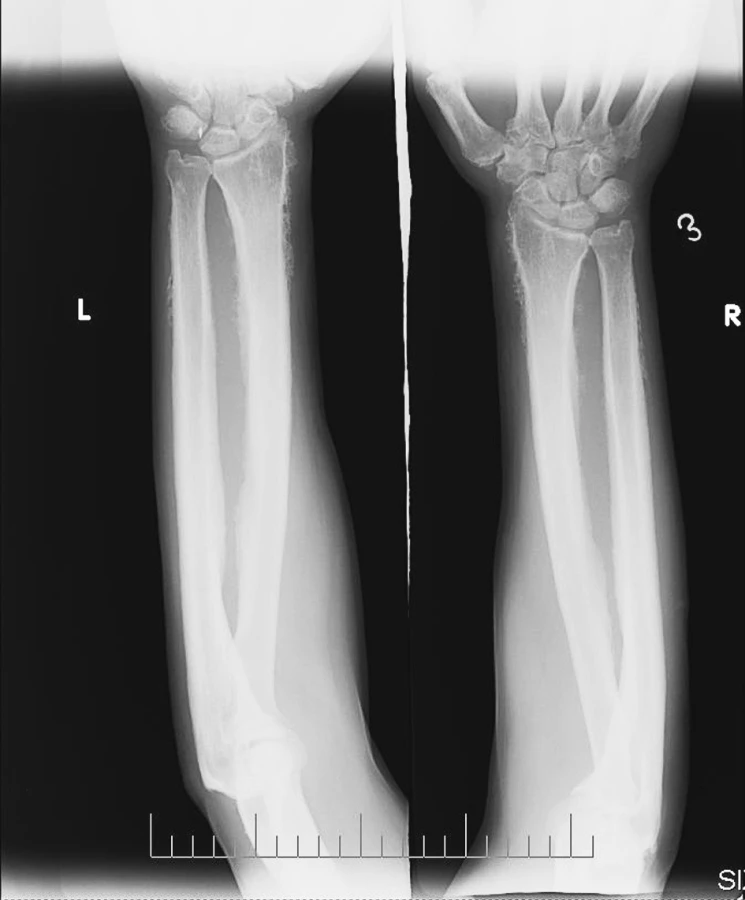
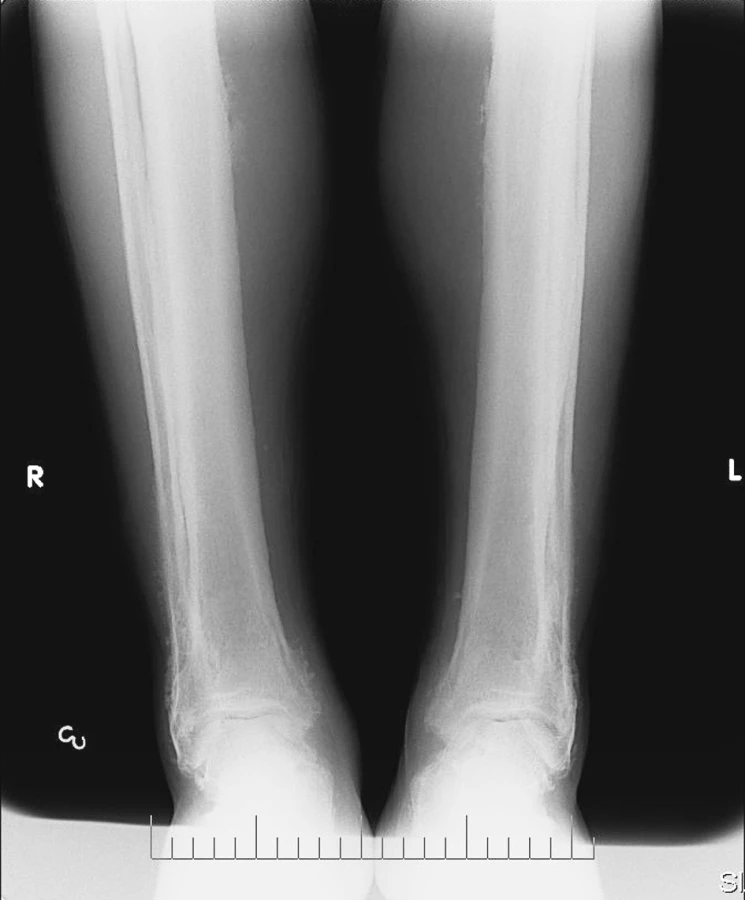
Závěr: nález nejspíše odpovídá klinické diagnóze PDP.
Sonografie břicha: cholecystolitiáza, oboustranně polycystické ledviny, levá dystopická a v levém hypogastriu. Spirometrie: normální ventilační hodnoty. Echokaradiografie: koncentrická hypertrofie levé komory s poruchou relaxace, lehká difúzní hypokinéza levé komory s mírnou depresí funkce, EF 52 %, toky na chlopních bez patologie. Bez perikardiální tekutiny. Doporučeno opakovat echokardiografii za půl roku s ohledem na lehkou dysfunkci levé komory.
Počítačová tomografie ledvin s kontrastem: dystopie levé ledviny, částečně zkřížená a pánevní. Jedna z cyst pravé ledviny má vysokou denzitu, bez sycení po kontrastní látce, patrně druhotně změněný obsah, snad po krvácení. Útvar maligního charakteru nesledovatelný.
Neurologické vyšetření: projevy nejpravděpodobněji symetrické distální polyneuropatie senzo-motorické s větším postihem dolních končetin. Závěr: pachydermoperiostóza, abusus alkoholu, persona simplex. Urologické vyšetření potvrzuje chronickou ledvinovou nedostatečnost při oboustranné polycystóze.
Koncem listopadu 2006 stav komplikován femoropopliteální flebotrombózou pravé dolní končetiny s plicní embolií středního rozsahu a následnou půlroční antikoagulační léčbou. Zjištěn kombinovaný trombofilní stav. Hematologem stav uzavřen jako homozygot FVL (Factor 5 Leiden), mírná hyperhomocysteinemie, nadále sledován hematologem.
Po všech uvedených vyšetřeních a dalších ambulantních kontrolách stav veden jako PDP se senzo-motorickou polyneuropatií horních a dolních končetin, dále jako chronická ledvinová nedostatečnost na podkladě polycystózy ledvin se sekundární lehkou anémií a hyperparathyreózou, asymptomatickou hyperurikemií, stav po femoropopliteální flebo trombóze pravé dolní končetiny s plicní embolií středního rozsahu, při kombinovaném trombofilním stavu – homozygot FVL (faktor V. Leiden), mírná hyperhomocysteinemie. Pacient kontrolován revmatologem, nefrologem, dle potřeby urologem a hematologem. Koncem roku 2007 stav stabilizovaný i po stránce ledvinové nedostatečnosti, bez progrese dusíkatých látek. Léčba cílená a přizpůsobená uvedeným diagnózám, později již bez antikoagulancií. Dostává hypolipidemika, Allopurinol, jen nárazově analgetika anebo i nesteroidní antirevmatika (Iboprufen několik dnů s přestávkami), bez projevů krvácení a s dobrou tolerancí.
Diskuse
Příčina tohoto vzácného onemocnění sice není známá, ale většina autorů se přiklání k názoru o často genetickém původu. Téměř vždy se jedná o kazuistická sdělení. V naší literatuře poprvé – a dosti detailně – PDP popsali Charvát s Mařátkou v roce 1946 u 21letého muže. Od té doby se v naší literatuře objevilo několik dalších, rovněž kazuistických sdělení, a všichni se přiklánějí ke genetickému původu (1, 2, 6–10). Jeden z nás s Pánkem (3) popsal PDP u 29letého muže, který měl bratra se stejným onemocněním a průběhem. I u nyní popsaného našeho pacienta se jedná o rodinné postižení. Navíc u pacienta jsou další afekce genetického původu, hlavně mnohočetná ledvinová polycystóza s chronickým selháním jen pomalu progredujícím. Toto je charakteristické u pacientů jen s polycystózou. Trvá různě dlouho až do nutnosti zahájit hemodialýzu. Navíc u pacienta je hematologem diagnostikován kombinovaný trombofilní stav, mírná hyperhomocysteinemie. Je shoda s některými autory (9–11) i vzhledem ke kombinaci s jinými genetickými projevy, hlavně na kůži a sliznici (2, 9–11). V literatuře je polycystóza ledvin u PDP popsána mimořádně vzácně (5). O to je naše nynější kazuistika cennější. Dosud jen Castori a spol. (5) našli v literatuře 68 publikovaných rodin s 204 pacienty. Mutaci našli u 37 rodin s autosomální recesivní formou. Bachmeyer a spol. (12) popisují kožní postižení, hlavně zhrubělou kůži a zejména na čele, s typickými projevy na skeletu, jako autosomální dominantní formu přenosu. O tomto genetickém původu se ale zmínili již uvedení autoři Tourain a Solente v roce 1935 (2). I proto francouzská literatura cituje PDP jako onemocnění Tourain-Solente (3, 13).
Nelze uvést přesný počet publikovaných pacientů. Nicméně nutno opět připomenout, že téměř každá práce popisuje 1 až 3 pacienty. Jinak se jedná jen o souhrnné hledání co nejvíce prací s uváděním počtu nemocných (5). Jisté ale je, že ne každý pacient je publikován. Stejně tak je ale pravděpodobné, že ne každý pacient s primární hyperostózou a popsaným nálezem je jako PDP diagnostikován. Dle některých autorů je možno v nynější době diagnosticky využít i magnetickou rezonanci (4). Autoři této práce chtějí připomenout pečlivé diagnostické zaměření u nálezů paličkovitých prstů s hyperostotickými nánosy na již popsaných lokalizacích v tomto příspěvku.
MUDr. Julius Vachtenheim, CSc.
Revmatologická a interní ordinace
Vrchlického 57
586 01 Jihlava
e-mail: vachtenheim@ji.cz
Sources
1. Charvát J, Mařátka Z. Hyperostosis generalisata cum pachydermia. Čas Lék čes 1946; 15 : 508–516.
2. Miková D. Pachydermoperiostóza–vývoj kostních změn po 25 letech od počátku nemoci. Vnitř Lék 1989; 35 : 68–73.
3. Vachtenheim J, Pánek Z. Die Pachydermoperiostose. Radiol Diagn 1960; 1 : 473–479.
4. Anandacoomarasamy A, Bak HS, Peduto A, et al. Magnetic resonance imaging in pachy-dermodactyly. J Rheumatol 2005; 32 : 2239–41.
5. Castori M, Sinibaldi L, Mingarelli L, et al. Pachydermoperiostosis: an update. Clin Genet 2005; 68 : 477–486.
6. Dudová N. Hyperostosis generalisata cum pachydermia. Vnitř Lék 1958; 4 : 345–349.
7. Grepl J. Pachydermoperiostitis. Srovnání dvou pozorování v různém evolučním stadiu. Čs Radiol 1979; 33 : 183–190.
8. Janečka V, Bruna J. Pachydermoperiostitis. Čs Radiol 1974; 28 : 353–359.
9. Štěpánek V, Metelka J. Generalizovaná hyperostóza s pachydermií a s hypertrofií sliznice trávicí trubice. Čs Rentgenol 1958; 12 : 246–249.
10. Točík J. Leukoplakia a pachydermia sliznice hlasivek. Čs Otolaryng 1953;32 : 312–315.
11. Schroder K, Goerdt S, Sieper J, et al. Psoriatic onycho-periostitis (POOP). Hautarzt 1997; 48 : 500–505.
12. Bachmeyer C, Blum L, Cadranel JF, Delfraissy JF. Myelofibrosis in a patient with pachy-dermoperiostosis. Clin Exp dermatol 2005; 30 : 646–648.
13. Toepfer M, Rieger J, Pfluger T, et al. Primary hypertrophic osteoarthropathy (Touraine-So lente-Gole Syndrom). Dtsch Med wschr 2002; 127 : 1013–16.
14. Rezgui-Marhout L, Edouira-Khomsi W, Bouslama K, et al. Pachydermoperiostosis, a report of two cases. J Radiol 2005; 86 : 340–343.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2008 Issue 3
Most read in this issue
- Rizartróza karpometakarpálního (CMC) kloubu palce ruky
- Difuzní idiopatická skeletální hyperostóza (DISH) ve stáří, výjimečné deformující postižení
- Nové poznatky v patogenezi postchlamydiové reaktivní artritidy
- Krikofaryngeální achalasie u myozitidy