
Biosimilars – současné poznatky o jejich zaměnitelnosti
Biosimilars – current knowledge on their interchangeability
The biosimilars cannot be regarded as chemical generics, because, unlike the latter ones, they are not and cannot be chemically identical to the original product. Even seemingly small differences in manufacturing of biosimilars may affect the immunogenic response. To date, there is little known about the mechanism of immunogenic responses. It has been pointed out that the acute therapy may be more immunogenic than the chronic one. Furthermore, experts incline to the belief that the intermittent administration is more likely to cause an immune response than continuous therapy, and that the theory of aggregation and detection of foreign epitopes as the main risk factor for immunogenicity is too simplistic. It is currently not possible to identify a direct safety risk associated with the substitution of biological drugs, because there is very little data dealing with this issue. It has been repeatedly confirmed that biological agents are not simply interchangeable even within the same ATC group due to their indications, side effects, and safety profiles that have been studied in the individual clinical trials designed specifically for every product. The current consensus in some EU countries is to treat the patient with the same agent until the urgent reasons force a change in the treatment.
Key words:
biosimilars, interchangeability of biosimilars, production of biosimilars, registration of biosimilars, immunogenicity
:
D. Vetchý 1; M. Vetchá 2
:
Ústav technologie léků, Farmaceutická fakulta, Veterinární a farmaceutická univerzita, Brno
1; Lékárna Na Mendlově náměstí, Brno
2
:
Čes. Revmatol., 21, 2013, No. 4, p. 200-205.
:
Comments
Na biosimilars nelze pohlížet stejně jako na chemické generické přípravky, protože na rozdíl od nich nejsou a nemohou být chemicky identické s původním originálním přípravkem. I zdánlivě malé rozdíly ve výrobě biosimilars mohou mít vliv na imunogenní odpověď. O mechanismu vzniku imunogenní odpovědi se stále ví jen velmi málo. Poukazuje se na to, že akutní terapie může být více imunogenní než chronická. Odborníci se dnes dále přiklání k názorům, že intermitentní podání vyvolá s větší pravděpodobností imunitní odpověď než kontinuální terapie, a že teorie agregace a odhalení cizích epitopů jako hlavního rizikového faktoru imunogenicity je příliš zjednodušená. V současné době není možné identifikovat přímé bezpečnostní riziko spojené se substitucí biologických léčivých přípravků, protože je velmi málo dat. Opakovaně bylo potvrzené, že biologické přípravky nejsou jednoduše zaměnitelné ani v rámci stejné ATC skupiny kvůli jejich indikacím, vedlejším účinkům a bezpečnostním profilům, které byly studované v individuálních klinických studiích navržených specificky pro jeden každý přípravek. Současný konsenzus v některých zemích EU je léčit pacienta stejným přípravkem, dokud naléhavé důvody nedonutí změnit léčbu.
Klíčová slova:
biosimilars, zaměnitelnost biosimilars, výroba biosimilars, registrace biosimilars, imunogenicita
Úvod
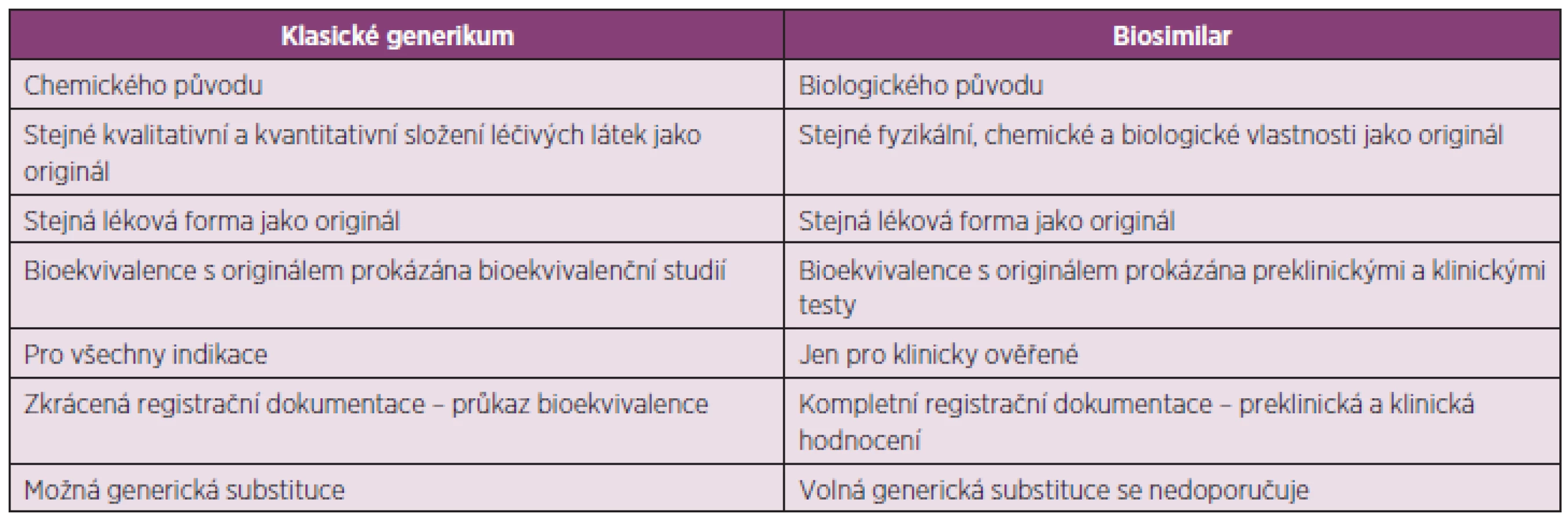
Biosimilars jsou podobnými biologickými přípravky, které se objevují na trhu po vypršení patentové ochrany originálních biologických přípravků. Nelze na ně pohlížet stejně jako na chemické generické přípravky, protože na rozdíl od nich nejsou a nemohou být chemicky identické s původním originálním přípravkem. Chemické generické léčivé látky jsou relativně malé molekuly, které se připravují chemickou syntézou a výsledkem jejich přípravy je látka s konkrétní, jasně definovanou strukturou, která je identická s originální léčivou látkou. Naproti tomu se biologické léčiva připravují biologicky, jsou produkovány živými systémy zahrnující bakterie, kvasinky a savčí buňky. Jsou to velké komplexní molekuly, obvykle proteiny nebo polypeptidy, které bývá prakticky nemožné jednoznačně charakterizovat. Jako u každého produktu získaného z živých systémů je i u biologických léčiv výsledná struktura variabilní. Jejich variabilita je dána prostorovým tvarem molekuly a typem a délkou každé navázané cukerné nebo uhlovodíkové skupiny. Sekvence aminokyselin musí být u biosimilars shodná s originálním biologickým přípravkem (1). Na rozdílnost výsledné struktury má vliv kromě vlastní variability biologických systémů také výrobní proces. Rozdíly mezi klasickými generiky a biosimilars jsou shrnuty v tabulce 1 (2).
Problematika biologických léčivých přípravků se dynamicky vyvíjí. Cílem příspěvku není detailně rozebírat problematiku biosimilars, ale předložit současný názor na problematiku zaměnitelnosti biologických přípravků a s ní souvisejících oblastí na základě aktuálních publikovaných validních informací za poslední rok.
Výroba biosimilars
Výroba biosimilars stejně jako originálních biologických přípravků ze savčích buněk začíná malým zmnožením základní kultury a následně kontrolou kvality získané startérové kultury. Startérová kultura je převedena do bioreaktoru pro přípravu zárodečné kultury k počátečnímu růstu buněk. Získaná zárodečná kultura je přemístěna do výrobního reaktoru, kde jsou optimální podmínky pro růst a produkci rekombinantních proteinů. Následuje ultracentrifugace a čištění získaného supernatantu. Rekombinantní protein se chromatograficky oddělí, odstraní se většina balastních kontaminantů, jako jsou ostatní proteiny, nukleové kyseliny, endotoxiny a viry a provede se finální čištění, ve kterém se eliminují zbývající stopové nečistoty nebo strukturálně blízce příbuzné látky (3). Buněčná kultura může být vyvinuta výrobcem, zakoupena z jiné společnosti nebo vyvinuta ve spolupráci s jinou společností. Tímto způsobem je možné zkrátit vývoj biosimilars až o 2 roky (4). Vlastnosti buněčné kultury a fermentační procesy jsou nejkritičtějšími kroky přípravy biologického léčiva vzhledem k jeho výsledné struktuře, čistotě a účinnosti. Většina rekombinantních proteinů vykazuje jednu nebo více post-translačních modifikací, které mají obvykle vliv na biochemické a terapeutické vlastnosti. Mezi nejčastěji se vyskytující post-translační modifikace patří glykosylace, karboxylace a hydroxylace, amidace a sulfatace, fosforylace, tvorba disulfidové vazby a štěpení bílkovin (5). I když je vztah mezi strukturou a terapeutickými vlastnostmi už pro mnoho post-translačních modifikací objasněn, zůstává stále mnoho neznámého, zejména v případě komplexu post-translačních modifikací, jakým je např. glykosylace. Výběr výrobního systému schopného generovat produkt s vhodnými post-translačními modifikacemi tak zůstává jedním z nejdůležitějších rozhodnutí, které musí výrobce udělat (5). Jakákoli změna v každém kroku výroby má vliv na produkci buněčné kultury vedoucí k rozdílnosti v kvalitě komerčních rekombinantních proteinů (6). Mezi faktory ovlivňující kvalitu vyráběného biologického přípravku, které mají vliv na účinnost a bezpečnost biologického léčiva, patří zdroj a typ použitého surového materiálu, chemické látky, které se mohou uvolnit při výrobě z kontejnerů a výrobních zařízení, podmínky výroby, zejména teplota, pH, míchání, počet čistících kroků, pomocné látky a další (6). Zjistilo se, že i zdánlivě malé rozdíly mohou mít vliv na imunogenní odpověď (1). Ze 6 přípravků, u kterých byla podána žádost o registraci v EU bez preklinického hodnocení, byla u 4 z nich registrace zamítnuta nebo byly staženy z důvodů neprokázané podobnosti v účinnosti a bezpečnosti s originálním přípravkem, u jednoho přípravku se musel modifikovat výrobní proces a musely se zopakovat klinické studie a jednomu přípravku byla odepřena indikace pro subkutánní podání, které se pokládá za nejrizikovější z hlediska imunogenicity (6). Proto se při vývoji biosimilars v první, analytické části soustředí na porovnávání nečistot a strukturálních a funkčních charakteristik s originálem. Ve druhé části pak přistupuje vlastní zkoušení v klinických fázích I.–III.
Registrace biosimilars
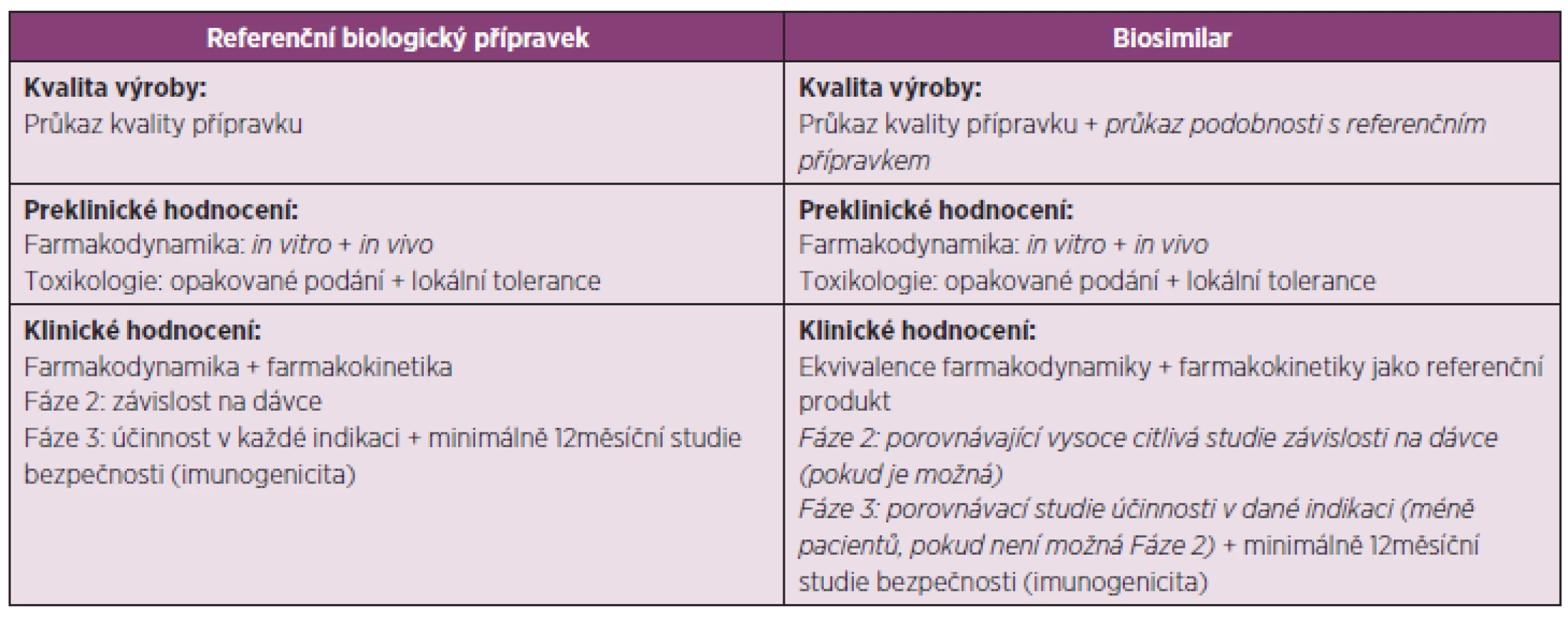
Registrace biosimilars je možná v Evropské unii od roku 2004 a pouze centralizovanou procedurou, která je povinná pro registraci léčivých přípravků vyrobených špičkovou technologií a zvláště pro přípravky, které jsou výsledkem biotechnologických procesů (např. rekombinantní DNA technologií, kontrolovanou expresí genů kódujících biologicky aktivní proteiny u prokaryont a eukaryont, včetně transformovaných savčích buněk, metodami hybridomu a monoklonálních protilátek). Tento způsob registrace byl zaveden z důvodu udržení vysoké úrovně vědeckého hodnocení uvedených léčivých přípravků v Evropské unii. Typ a množství požadovaných klinických zkoušení není stejné pro všechna biosimilars, ale liší se v závislosti na složitosti léčivé látky a jak úplně je charakterizovaná, na možnosti provedení vysoce citlivé porovnávající studie závislosti na dávce, na dostupnosti přijatelných cílových parametrů pro porovnání účinnosti, na typu a závažnosti známých bezpečnostních rizik pro danou terapeutickou skupinu a na možnosti extrapolace dat bezpečnosti a účinnosti na jiné indikace referenčního produktu, které nebyly v rámci vývoje biosimilar studovány (1, 7). Z hlediska bezpečnosti je zejména sledovaná imunogenicita. Jestliže je známé, že imunogenní odpověď je málo častá, bývá vyžadována navíc poregistrační studie (1). Příkladem přijatelných cílových parametrů pro porovnání účinnosti inzulinových přípravků je rychlost infuze glukózy, u přípravků obsahující faktor stimulující granulocytové kolonie pak absolutní počet neutrofilů, u přípravků obsahující folikuly stimulující hormony počet získaných oocytů v kontextu in vitro fertilizace, u přípravků se somatropinem šestiměsíční klinická studie účinnosti u dětí s deficitem růstového hormonu nebo u přípravků s erytropoetinem koncentrace hemoglobinu v krvi ± 0,5 g/dL (8). Pokud je klinická podobnost určité indikace s ověřenou hlavní indikací (např. stejný mechanismus účinku), data účinnosti a bezpečnosti na ni mohou být extrapolovány, jinak ne (1). Např. u faktoru stimulujícího granulocytové kolonie se účinnost porovnává absolutním počet neutrofilů. Při využití biosimilars k mobilizaci kmenových buněk z kostní dřeně u zdravých dárců je však vyžadována navíc studie porovnávající počet buněk CD34+ (8). Extrapolace na základě výše uvedených principů je však řadou klinických lékařů ostře kritizovaná, protože bývá často pro jinou populaci pacientů, s jinými komorbiditami a s jinou konkomitantní medikací, než na které byla indikace klinicky ověřená. Extrapolace je podle klinických lékařů problematická i v rámci jednotlivých onemocnění, protože každé vykazuje odlišnou imunogenicitu (9). V Evropské unii se v rámci předregistračních studií biosimilars nevyžaduje průkaz zaměnitelnosti s originálním přípravkem. Naproti tomu v USA, kde je registrace biosimilars možná až od roku 2012, se průkaz zaměnitelnosti vyžaduje (10), resp. se bude vyžadovat prokázání, že opakované podání biosimilar jednomu pacientovi nepovede k většímu riziku v bezpečnosti a účinnosti, než jaké je po opakovaném podání referenčního biologického přípravku nebo po střídavém podání referenčního přípravku a biosimilar (11). Místo standardního zkříženého plánu studie probíhající ve dvou časových intervalech podávání a ve dvou sledech (two-period, two-sequence crossover design) se doporučuje modifikovaný Balaamův zkřížený plán studie (Biosimilar-Biosimilar, Originál-Originál, Biosimilar-Originál-Biosimilar, Originál-Biosimilar-Originál) (12). Požadavky na vývoj biosimilars a referenčního biologického přípravku jsou shrnuty v tabulce 2 (kurzívou jsou označené rozdíly).
Imunogenicita
Imunogenicita je schopnost látky po jejím určitém vpravení do organismu vyvolat imunitní odpověď (například tvorbu protilátek). Imunitní systém reaguje na proteinové molekuly v principu ze dvou důvodů. První a zcela nejběžnějším důvodem je vniknutí cizorodého proteinu (například z virů, bakterií aj.) do organismu. Druhým, méně běžným a nežádoucím důvodem je prolomení imunitní tolerance k vlastním tkáním, respektive proteinům, jimiž jsou tvořeny. Imunitní tolerance je u jednotlivých onemocnění obvykle narušena z doposud neznámých důvodů (genetické faktory aj.), ale může být iniciována i infekčně (např. bakterie z rodu Streptococcus nese protein silně podobný proteinům myokardu – tzv. molekulární mimikry). Faktory ovlivňující navození tvorby protilátek lze rozdělit do 4 kategorií na ty, které souvisejí s pacientem (stav, zejména stav imunitního systému), s léčbou (cesta podání, dávka, délka léčby, apod.), s výrobou a s povahou biologického léku (velikost, chemická struktura, respektive epitopy) (13). Jestli budou mít protilátky vytvořené proti biologickému přípravku vliv na léčbu, závisí na vazebném místě protilátky, na biologickém léčivu, na afinitě protilátky k biologickému léčivu a na titru protilátek (14). Vytvořené protilátky tak nemusí mít žádné zjevné klinické důsledky, mohou snižovat léčebnou účinnost nebo vyvolat nežádoucí účinky (např. přecitlivělost nebo aplazii červené krevní řady) (14). V současnosti se přehodnocuje dosud přijímaný názor, že pravděpodobnost imunitní odpovědi je nejvyšší po subkutánním podání, menší po intramuskulárním a nejmenší po intravenózním podání. Poukazuje se také na to, že akutní terapie může být více imunogenní než chronická (8). Odborníci se dnes dále přiklání k názorům, že intermitentní podání vyvolá s větší pravděpodobností imunitní odpověď než kontinuální terapie, že teorie agregace a odhalení cizích epitopů jako hlavního rizikového faktoru imunogenicity je příliš zjednodušená, a že imunitní odpověď je méně pravděpodobná u pacientů se slabým nebo oslabeným imunitním systémem nebo u těch, kteří užívali imunosupresiva (13). Např. filgastrim je tedy považován za relativně bezpečný z hlediska imunogenicity, protože se používá u imunokomprimovaných osob. Vně této populace je však schopen vyvolat imunitní reakce obdobně jako jiný biologický přípravek (15). Naproti tomu může být imunogenicita významná např. u starších mužů a dalších podskupinách se zhoršenou odpovědí na erytropoetickou stimulaci (16). Obecně u přípravků s rekombinantním lidským erytropoetinem Binocrit, Retacrit a Eporatio se porovnáním se zjistil obdobný bezpečnostní profil (16). Nežádoucí aplazie červené krevní řady po erytropoetinu byla přisouzena agregaci vyvolané wolframem uvolněným z primárního obalu přípravku (17). U generického somatotropinu byla zase zvýšená imunogenicita výsledkem vyššího množství proteinu z hostitelské buňky (18). O mechanismu vzniku imunogenní odpovědi se stále ví jen velmi málo a je stále velkým problémem. Proto se v současné době vyvíjí tzv. biobetters, modifikované biologické přípravky (biologické přípravky vyšších generací), které vykazují nižší imunogenicitu a agregaci, mají prodloužený účinek, jsou stabilnější anebo více účinné (13, 19). Aktuální pokyny ke způsobu testování imunogenicity jsou ve směrnici Evropské lékové agentury EMA/CHMP/BMWP/86289/2010, která řeší problematiku výběru vhodných metodik pro hodnocení autoprotilátek, standardů skrínovacích a konfirmačních technik, neutralizační kapacity protilátek i problematiku identifikace, hodnocení a monitorování rizika imunogenicity (20).
Substituce biologických léčivých přípravků
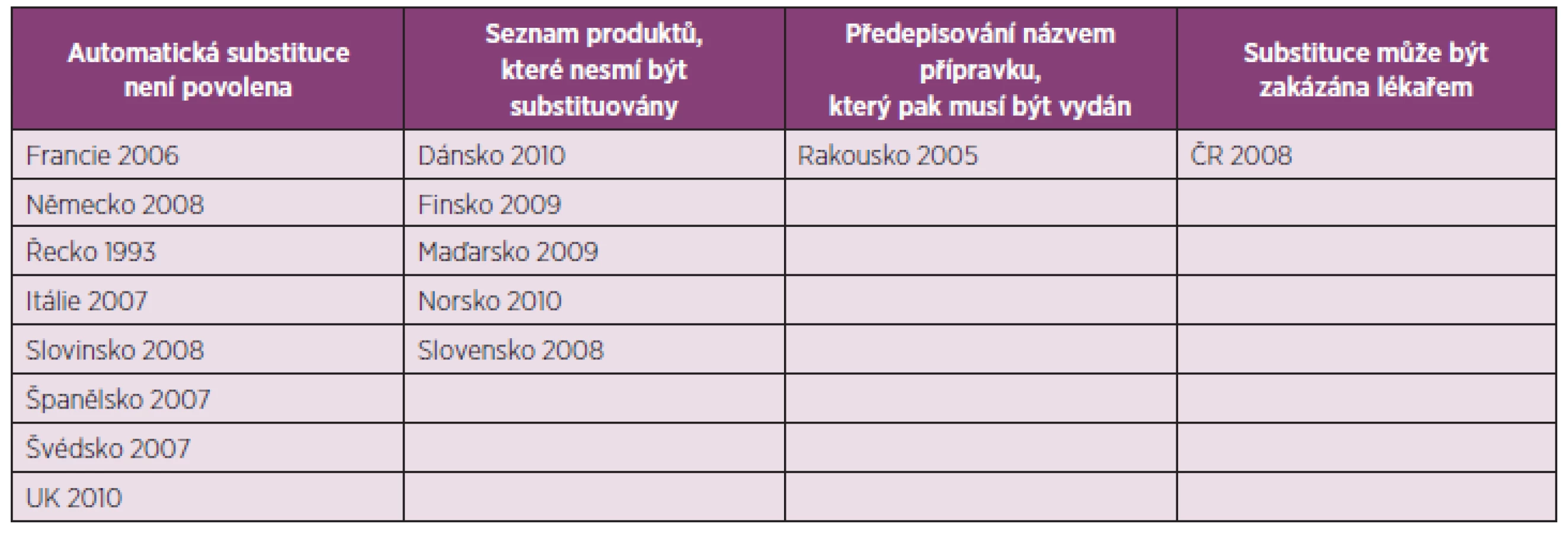
V současnosti není možné identifikovat přímé bezpečnostní riziko spojené se substitucí biologických léčivých přípravků, protože je velmi málo dat. Klinická zkoušení nebývají zaměřené na nežádoucí účinky související se substitucí. Navíc, většina popsaných substitucí se týká substituce mezi originálními přípravky a ne mezi originálním přípravkem a biosimilar (21). Za bezpečnou a efektivní se považuje substituce mezi různými inzulinovými přípravky, protože je evidováno jen málo hypersenzitivních reakcí (21). I kvůli velmi omezenému množství dat týkajících se záměn podobných biologických přípravků se v současné době přijímá v EU legislativa, která výrazně změní farmakovigilanci biologických přípravků. V každé klinické studii bude muset být uveden název a číslo šarže biologického léčiva. Takto bude v budoucnu možné záměny lépe vyhodnotit (21). Obecně Evropská léková agentura nemůže regulovat substituci podobných biologických přípravků, to je v kompetenci každého státu EU (1). V tabulce 3 jsou uvedeny státy EU, které regulují automatickou substituci biologických přípravků podobnými biologickými přípravky, datum přijetí a princip zavedených regulací (22).
Závěr
Je doložené, že opakovaná záměna podobných biologických léčivých přípravků s originálním přípravkem může zvýšit imunogenicitu (1). Automatická substituce není vhodná i z praktického hlediska, zejména z pohledu aplikace přípravku pacientem (např. různé typy injekcí). Rozhodně se nedoporučuje předepisovat biosimilar INN názvem, protože může mít jiný INN název než referenční přípravek, se kterým byl porovnáván (př. Retacrit – epoetin-zeta versus Eprex – epoetin-alfa) (21). Opakovaně bylo potvrzeno, že biologické přípravky nejsou jednoduše zaměnitelné ani v rámci stejné ATC skupiny kvůli jejich indikacím, vedlejším účinkům a bezpečnostním profilům, které byly studovány v individuálních klinických studiích navržených specificky pro jeden každý přípravek (23). Jedním z posledních případů, který potvrzuje dané tvrzení, byl výskyt anafylaktických reakcí po substituci za biosimilární rituximab v Mexiku (24). I když se odhaduje, že 20% snížení ceny 5 biologických přípravků ušetří EU více než 1,6 miliard eur, je současný konsenzus v některých zemích EU léčit pacienta stejným přípravkem, dokud naléhavé důvody nedonutí změnit léčbu (10, 21).
Adresa pro korespondenci:
Doc. PharmDr. Mgr. David Vetchý, Ph.D.
Ústav technologie léků Farmaceutická fakulta, Veterinární a farmaceutická univerzita
Palackého třída 1/3
612 42 Brno
telefon: 541562860
e-mail: vetchy@email.cz
Sources
1. Weise M, Bielsky M-C, De Smet K, et al. Biosimilars: what clinicians should know. Blood 2012; 120 : 5111–7.
2. Scotté F, Launay-Vacher V, Rey JB. Colony stimulating factors (CSF) biosimilars. Progress? Target Oncol 2012;7 Suppl 1: S17–24.
3. Ahmed I, Kaspar B, Sharma U. Biosimilars: impact of biologic product life cycle and European experience on the regulatory trajectory in the United States. Clin Ther 2012; 34 : 400–19.
4. Vetchý D, Vetchá M. Biosimilars – podobné biologické léčivé přípravky. Remedia 2010; 20 : 74–7.
5. Walsh G. Post-translational modifications of protein biopharmaceuticals. Drug Discov Today 2010; 15 : 773–80.
6. Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin 2012; 28 : 1053–8.
7. European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. EMA/CHMP/BMWP/403543/2010. Available from: URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
8. European Medicines Agency, Committee for Medicinal Products for Human Use. Biosimilar Guidelines. Available from: URL: www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000408.jsp&mid=WC0b01ac058002958c.
9. IV. zimní konference nemocničních lékárníků, Available from: URL: www.zdravky.cz/kongresovy-list/aktualne/iv-zimni-konference-nemocnicnich-lekarniku.
10. Calvo B, Zuñiga L. The US approach to biosimilars: The long-awaited FDA approval pathway. BioDrugs 2012; 26 : 357-61.
11. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Available from: URL: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf.
12. Chow SC, Yang LY, Starr A, Chiu ST. Statistical methods for assessing interchangeability of biosimilars. Stat Med 2013; 32 : 442–8.
13. Barbosa MD, Kumar S, Loughrey H, Singh SK. Biosimilars and biobetters as tools for understanding and mitigating the immunogenicity of biotherapeutics. Drug Discov Today 2012; 17 : 1282–8.
14. Cohen BA, Oger J, Gagnon A, Giovannoni G. The implications of immunogenicity for protein-based multiple sclerosis therapies. J Neurol Sci 2008; 275 : 7–17.
15. Vulto AG, Crow SA. Risk management of biosimilars in oncology: each medicine is a work in progress. Target Oncol 2012; 7 Suppl 1: S43–9.
16. Abraham I, MacDonald K. Clinical safety of biosimilar recombinant human erythropoietins. Expert Opin Drug Saf 2012; 11 : 819–40.
17. Seidl A, Hainzl O, Richter M, et al. Tungsten-induced denaturation and aggregation of epoetin alfa during primary packaging as a cause of immunogenicity. Pharm Res 2012; 29 : 1454–67.
18. European Medicines Agency. European Assessment Report on Omnitrope. Available from: URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000607/WC500043692.pdf.
19. Simoens S, Verbeken G, Huys I. Biosimilars and market access: a question of comparability and costs? Target Oncol 2012; 7 : 227–31.
20. European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. EMA/CHMP/BMWP/86289/2010. Available from: URL: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf.
21. Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther 2012; 12 : 1473–85.
22. Niederwieser D, Schmitz S. Biosimilar agents in oncology/haematology: from approval to practice. Eur J Haematol 2011; 86 : 277–88.
23. Kalodiki E, Fareed J. New and generic anticoagulants and biosimilars: safety considerations. Clin Appl Thromb Hemost 2011; 17 : 136–9.
24. Lapadula G, Ferraccioli GF. Biosimilars in rheumatology: pharmacological and pharmacoeconomic issues. Clin Exp Rheumatol 2012; 30 (Suppl 73): S102–6.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2013 Issue 4
Most read in this issue
- Polymyalgia rheumatica – is the biological therapy effective?
- Regulatory T cells as a tool in modulation of immune system
- Biosimilars – current knowledge on their interchangeability
- Circulating Heat Shock Protein 90 (HSP90) in patients with rheumatoid arthritis and axial spondylarthritis