
Lymfomatoidní papulóza
Lymphomatoid Papulosis
Lymphomatoid papulosis (LyP) is a rare skin disease of unknown etiology belonging to the indolent cutaneous T-cell lymphomas, affecting mostly persons between 30 and 40 years of age. Few isolated cases appear in childhood. We present 2 cases of a 38-year-old man and a 7-year-old girl with the diagnosis of LyP manifesting with waxing a waning papular eruptions on the trunk and limbs, histological background of lesions and clinical effect of local and systemic therapy.
Key words:
lymphomatoid papulosis – clinical manifestation – diagnosis – therapy
:
P. Horáčková 1; L. Drlík 1; L. Pock 2; E. Jašková 3
:
Dermatovenerologické oddělení, Šumperská nemocnice, a. s.
prim. MUDr. Lubomír Drlík
1; Dermatohistopatologická laboratoř, Praha
vedoucí lékař doc. MUDr. Lumír Pock, CSc.
2; Dermatovenerologické oddělení, Slezská nemocnice v Opavě
prim. MUDr. Eva Jašková
3
:
Čes-slov Derm, 88, 2013, No. 1, p. 17-20
:
Case Reports
Lymfomatoidní papulóza (LyP) je vzácné chronické onemocnění neznámé etiologie řadící se ke kožním T-buněčným lymfomům nízké malignity. Postihuje převážně osoby mezi 30. a 40. rokem života, sporadicky se vyskytuje v dětském věku. Uvádíme 2 případy 38letého muže a 7leté dívky s diagnózou LyP, které se manifestují recidivujícími papulózními projevy na trupu a končetinách, histologický podklad těchto projevů a vliv lokální a celkové terapie na jejich hojení.
Klíčová slova:
lymfomatoidní papulóza – klinická manifestace – diagnóza – terapie
ÚVOD
Lymfomatoidní papulóza (LyP) je definována jako chronické, recidivující, spontánně odeznívající papulonodulární kožní onemocnění s histologickými projevy napodobujícími maligní lymfomy. Jedná se o vzácné onemocnění (incidence u dospělých 1,2–1,9 na 1 milion, u dětí přesná incidence není známa). LyP popsal Macaulay v roce 1968 jako klinicky benigní onemocnění s histologickým nálezem maligního vzhledu. Vzhledem k tomu, že existují překryvné klinické a histologické obrazy, je LyP nejlépe řadit k lymfomům nízké malignity [1, 4, 5, 11, 15, 18, 22, 24]. Jedná se o poměrně vzácné onemocnění, proto uvádíme případy dvou našich nemocných.
POPIS PŘÍPADŮ
Případ 1
Třicetiosmiletý muž byl v roce 1998 léčen pro erythema chronicum migrans, v roce 2009 pro chlamydiovou uretritidu, jinak byl zdráv. Alergiemi netrpí, pracuje jako řidič. Dosud neužíval žádné léky, mimo průjmového onemocnění asi dva týdny před výsevem kožních projevů neměl zdravotní problémy.
Poprvé byl vyšetřen v kožní ambulanci v roce 2008, tehdy stav imponoval jako maloložisková parapsoriasis en plaques s histologickým nálezem chronická superficiální dermatitida bez patognomických rysů. Léčba lokálními steroidy s dobrým terapeutickým efektem a vymizením morf. Projevy však občasně recidivovaly. V květnu 2011 proběhla ataka výsevu drobných papul na bocích trupu, léčeno rovněž kortikosteroidním externem, ke zhojení došlo během několika týdnů.
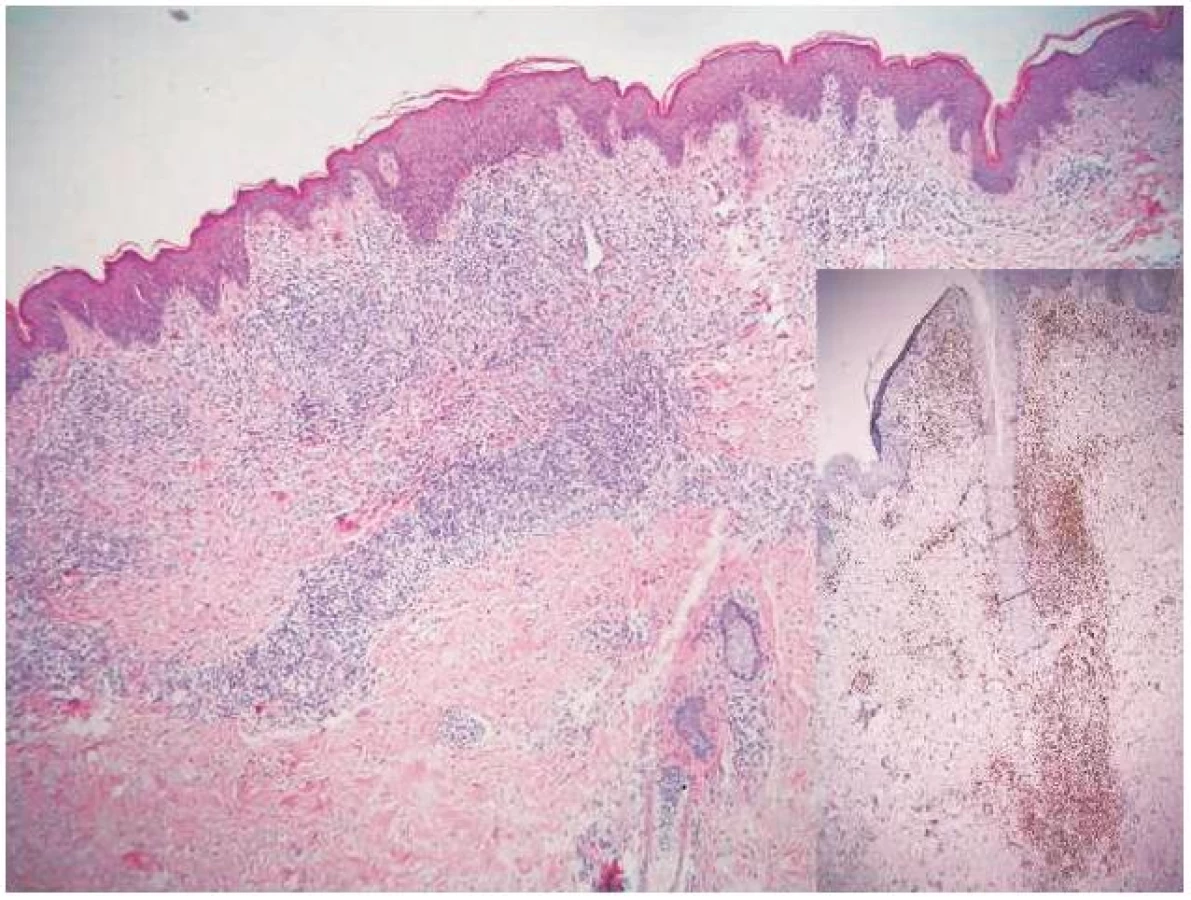
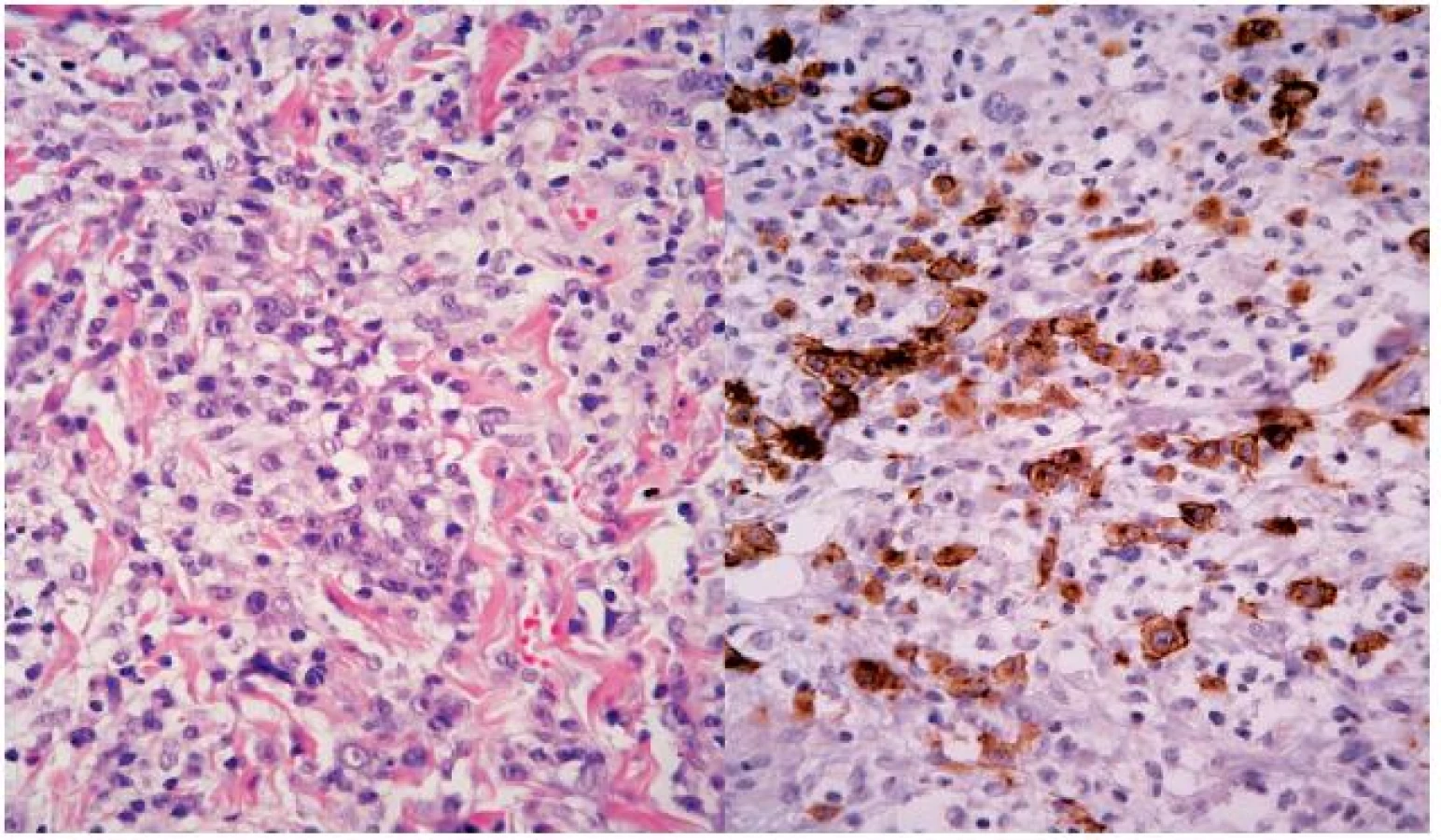
Koncem října 2011 se dostavil do kožní ambulance na vyšetření pro asi týden trvající výsev nesvědivých, červených papul až nodulů na trupu velikosti do 10 mm v průměru, z nichž některé v centru vykazovaly přítomnost krust a příškvarů (obr. 1). Aktuální projevy byly klinicky blízké obrazu pityriasis lichenoides et varioliformis acuta (PLEVA). V histologickém obraze byla popsána hyperkeratóza, parakeratóza, akantóza a papilomatóza, nečetné apoptotické keratinocyty ve stratum spinosum, ložiskovitě spongióza, ve středním koriu perivaskulárně středně husté infiltráty lymfocytů s příměsí histiocytů a nečetných fragmentů polynukleárů. Lymfocyty byly převážně CD3 pozitivní, velké buňky s velkým nepravidelným jádrem CD30 pozitivní, ojedinělé buňky přecházely do epidermis (obr. č. 2, 3). Na základě klinického a histologického vyšetření byla stanovena diagnóza LyP.
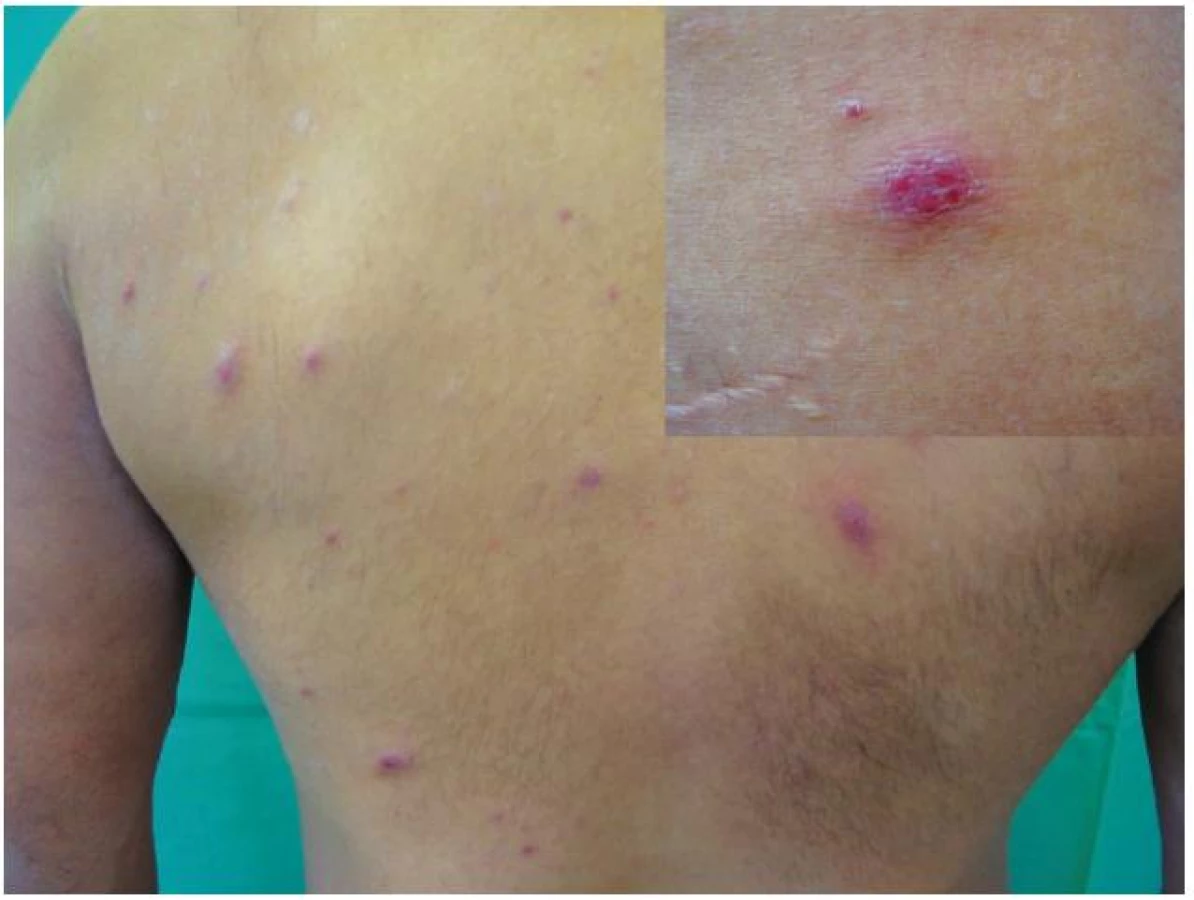
Byla ordinována 1krát denně krém-pasta obsahující 1 % clotrimazolu a 0,25 % hexamidindiisetionátu, dále 2krát denně mast s 0,05 % betamethazondipropionátu a 0,1 % gentamicinu, perorálně doxycyclinum 200 mg denně po dobu 14 dnů. V krevních odběrech byly zjištěny vysoké hodnoty antistreptolyzinu O – 778 IU/ml (ref. 0–200) a revmatoidního faktoru IgM 49,0 IU/ml (ref. 0–20) při normální hodnotě C-reaktivního proteinu. Další laboratorní vyšetření – krevní obraz s diferenciálním rozpočtem bílých krvinek, biochemické vyšetření včetně laktátdehydrogenázy a beta-2 mikroglobulinu bylo bez významných odchylek, stejně tak i antinukleární faktor a ENA-profil. Protilátky proti chlamydiím a yersiniím a vyšetření anti-HIV byly negativní. Prokázala se zvýšená hladina paměťových protilátek IgG proti viru Epstein-Barrové – index 6,70 (norma do 0,8), herpes simplex viru-1 – 8,65 IP (norma pod 0,90 IP), viru varicella-zoster – 1,20 IP (norma pod 0,90) a parvoviru B 19 – 9,65 IU/ml (norma pod 3,60), přičemž protilátky IgM ve všech uvedených vyšetřeních byly negativní. Vyšetření moči a močového sedimentu bylo bez patologického nálezu, RTG plic a ultrazvukové vyšetření lymfatických uzlin a břišních orgánů byly negativní. Pro nehojení kožních projevů byl v polovině listopadu přidán do terapie prednison 40 mg denně, který pacient užíval 1 měsíc a sám pro nelepšení stavu v polovině prosince 2011 vysadil. Na další kontrolu se dostavil 19. ledna 2012 a byl zcela zhojen (udával, že k úplnému zhojení ložisek došlo asi týden před tímto datem). Při vyšetření v dubnu 2012 byl pacient celkově i lokálně zcela asymptomatický, v dobré kondici. Kontrolní základní laboratorní odběry byly negativní. Na CT vyšetření plic a břicha byl nalezen v horním mediastinu retrosternálně útvar velikosti 20 x 17 x 17 mm, bylo vysloveno podezření na lymfom, dále změny na kolon charakteru kolitidy. Při PET CT vyšetření nedošlo k nasycení léze radiokontrastní látkou. Lymfom na základě tohoto výsledku byl vyloučen a byla doporučena dispenzarizace na plicní ambulanci.
Případ 2
Sedmiletá dívka na jaře 2011 prodělala infekční mononukleózu, pro časté infekty horních cest dýchacích byla opakovaně léčena kúrami Ribomunyl tbl. (směs lyzátů Klebsiella pneumoniae, Streptococcus pneumoniae, Streptococcus pyogenes A a Haemophilus influenzae). Rodinná anamnéza byla nevýznamná. Od července 2011 se objevovaly výsevy nesvědivých tuhých papul až nodulů červenolividní barvy, na jejichž vrcholu se během několika týdnů vytvářely krusty a postupně se odhojovaly depigmentacemi (obr. 4). Výsev se zpočátku projevil jen na hrudníku, během půl roku se ojedinělé papuly začaly objevovat i na dolních končetinách.
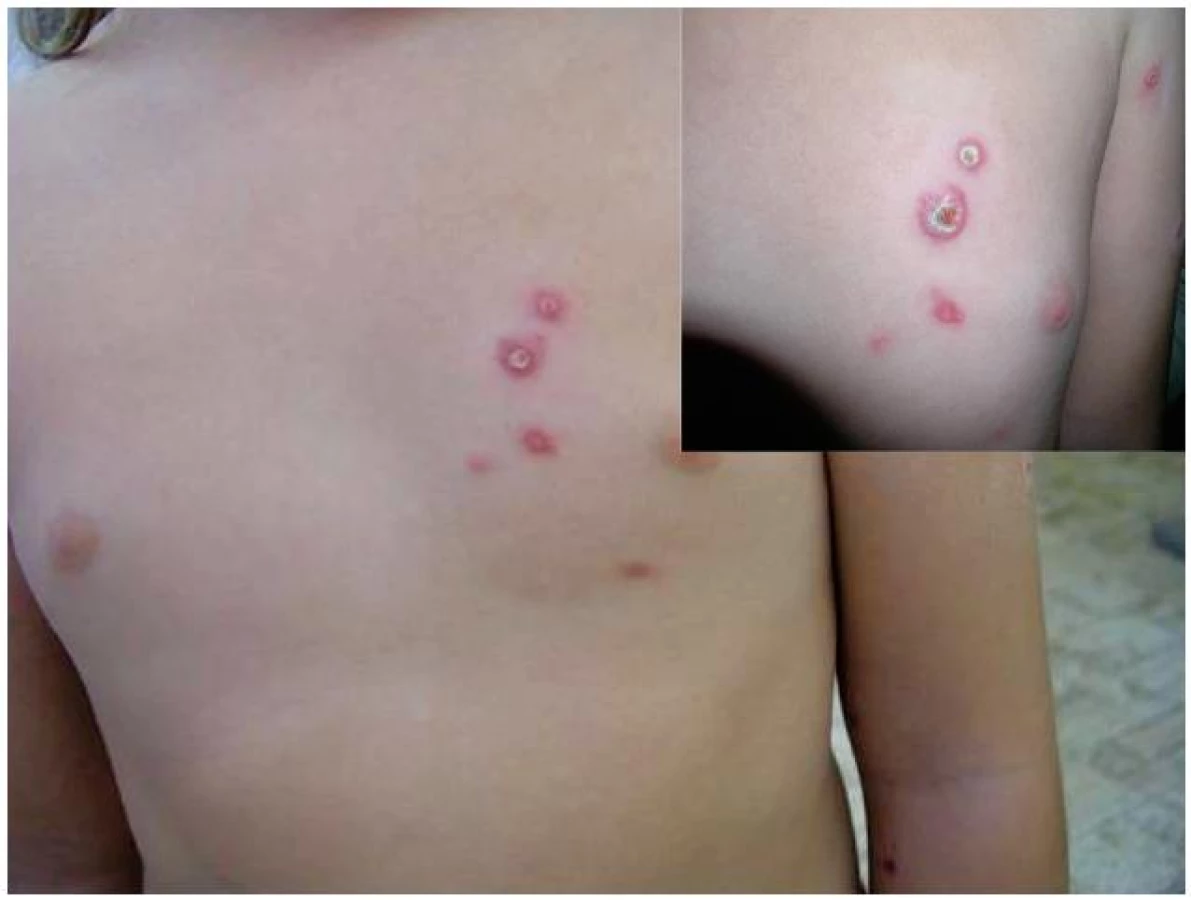
V srpnu 2011 provedená biopsie prokázala nespecifický granulomatózní zánět, v listopadu 2011 z dalšího bioptického vzorku bylo vysloveno podezření na lymfomatoidní papulózu. Byla popsána akantoticky rozšířená epidermis s hyperkeratózou, v koriu přítomný hustý infiltrát tvořený směsí středně velkých a velkých buněk, zachycen epidermotropismus, B lymfocyty se vyskytovaly jen ojediněle, převažovaly CD4 pozitivní T lymfocyty. CD8 pozitivní buňky byly v infiltrátu spíše disperzně. Četné velké buňky jsou CD30 pozitivní, ALK (Anaplastic lymphoma kinase) negativní.
Byla ordinována kortikosteroidní externa, na ulcerovaná ložiska mast s 2 % mupirocinu a provedeno hematologické vyšetření, CT vyšetření plic, mediastina a CT břicha, vše s negativním nálezem. Krevní obraz, jaterní testy, elektroforéza bílkovin, imunoglobuliny, thymidinkináza, laktádehydrogenáza i beta-2 mikroglobulin v normě. Byla provedena imunofenotypizace periferní krve – vyšetření periferních lymfocytů neprokázalo patologickou populaci T lymfocytů, TCR (T-buněčný receptor) bylo negativní. Pacientka je v dispenzární péči dermatologické a hematologické ambulance, projevy se při uvedené léčbě jen velmi pomalu hojí.
DISKUSE
Typickými kožními projevy LyP jsou červené papuly a papuly až noduly s tvorbou hemoragických krust a nekróz, které mohou jevit tendenci ke spontánnímu hojení s tvorbou depigmentace, hyperpigmentace či jizev, nebo přetrvávají řadu měsíců i let [1, 3, 4]. Charakteristická je současná přítomnost projevů v různém stupni vývoje. Počet lézí kolísá od solitární až po několik set. Léze mohou být lokalizované na část těla nebo generalizované. Zvláště lokalizované léze u dětí bývají velmi torpidní a nereagují na léčbu. U těchto případů v literatuře popsaných pod názvem perzistující agminované LyP jsou doporučována pečlivá a opakovaná staging vyšetření [8, 19]. Onemocnění postihuje nejčastěji trup a proximální partie končetin, ojediněle též ruce včetně dlaní, obličej a genitál. Výsev je zpravidla asymptomatický, může však být provázen svěděním [4, 9, 15, 16, 18, 25]. Etiologie LyP není známa, zvažovaný původ působením HTLV-1, EB viru, nebo jiných herpetických virů – herpes simplex virus 1 nebo 2, herpesvirus 6 – nebyl prokázán [5, 20]. LyP představuje přibližně 15 % kožních T-buněčných lymfomů. Může se objevit v jakémkoli věku, převážně mezi 30. – 40. rokem, popsané věkové rozpětí pacientů bylo 8 měsíců až 84 let. Postihuje muže 2krát častěji nežli ženy [2]. Doba trvání nemoci je velmi variabilní – od několika měsíců přes více než čtyřicet let [4, 5].
Histologické obrazy LyP imitují maligní lymfomy. Jsou popsány 3 histopatologické typy LyP, které se mohou vyskytovat u jednoho pacienta současně a korelují se stářím jednotlivých kožních lézí. Typ A je charakterizován přítomností velkých atypických blastů, včetně jedno - a vícejaderných buněk připomínajících Reedové-Sternbergovy buňky (typické pro morbus Hodgkin), rozptýlených v zánětlivém infiltrátu sestávajícím z histiocytů, malých lymfocytů, neutrofilů a/nebo eozinofilů. Velké atypické buňky se nacházejí v nevelkém počtu v čerstvých lézích, četné jsou v případě plně vyvinuté léze a mohou kompletně chybět v regredujících lézích. Typ B je méně častý, charakterizovaný superficiálním perivaskulárním infiltrátem složeným z malých nebo středně velkých atypických T-buněk s cerebriformními jádry, CD3+, CD4+, CD8-, ale CD30-, vykazujícími epidermotropismus a napodobujícími tak mycosis fungoides. Typ C představuje shluky velkých CD30+ T-buněk, histologicky připomínající anaplastický velkobuněčný lymfom. Nově byl popsán typ připomínající primární agresivní epidermotropní CD8+ cytotoxický T-lymfom [2]. Koexistovat mohou zejména typ A a B [4, 5, 23, 24]. Klinické projevy LyP histologického typu A se odhojují v průběhu 4–6 týdnů v závislosti na velikosti lézí, na rozdíl od klinických projevů LyP histologického typu B, které se odhojují spíše v průběhu měsíců [23]. V případě LyP histologického typu C je zvýšené riziko vzniku maligního lymfomu [3].
Drobné recidivující léze klinicky napodobují folikulitidu nebo reakci po bodnutí hmyzem. V úvahu také přicházejí lymfomatoidní lékové reakce [11]. Dále je nutné odlišit maligní kožní lymfomy, zejména při nálezu blastů CD30+, sekundární syfilis a syphilis maligna, papulonekrotický tuberkulid. Klinická podobnost existuje zejména s pityriasis lichenoides et varioliformis acuta (PLEVA). Toto onemocnění ale mívá spíše kratší trvání, postihuje mladší jedince, nevyvíjejí se u ní noduly a nevyskytují se CD30+ blasty. Nově byly literárně zaznamenány překryvné případy PLEVA a LyP, popsána možná souvislost s infekcí parvoviry B 19 [12]. Při histologickém nálezu, který je velmi sugestivní pro maligní lymfom, je nezbytná klinickopatologická korelace s klinickým obrazem a průběhem onemocnění [4, 5]. Rovněž lze k diferenciální diagnostice maligních kožních lymfomů a LyP využít metodu PCR analýzy klonální přestavby TCR-γR (T-buněčný receptor γ) [2].
Léčba pacientů s LyP je neuspokojivá, onemocnění je do určité míry limitované, je tedy nutno zvážit riziko a benefit eventuální aktivní terapie. Agresivní terapeutické postupy jako systémová chemoterapie nebo celotělové ozáření jsou neadekvátní. U pacientů s relativně málo početnými spontánně regredujícími lézemi je zvykem dlouhodobá observace bez aktivní terapie. V případě četných papulonodulů či jizvících lézí je nejefektivnější nízkodávkovaný metotrexát (5–20 mg/týden). Dobrý terapeutický efekt má též PUVA, ale při jejím přerušení dochází často v krátké době k relapsům choroby [1, 4, 5, 9]. Mezi další možnosti patří topická, intralezionální či systémová aplikace kortikosteroidů, tetracyklinová antibiotika, imiquimod, interferon alfa, fotodynamická léčba a lokální radioterapie [10, 16, 17]. Průběh onemocnění je chronický, ve vlnách, často trvající celá desetiletí, s epizodami akutní exacerbace [3]. Obecně LyP vykazuje benigní chování s 10letým přežitím ve 100 % případů. Nicméně v 10–20 % případů (u dětí méně než v 10 %) je LyP předcházena, koexistuje, nebo je následována maligním lymfomem, zejména MF, Hodgkinským lymfomem nebo nodálním anaplastickým velkobuněčným lymfomem [16, 24]. Vzhledem ke zvýšenému riziku vzniku maligních onemocnění, které se dále zvyšuje s věkem a délkou trvání LyP, je nutná dlouhodobá observace. Vhodné je také histologické monitorování, hematologické kontroly, sledování stavu lymfatických uzlin a sérové hladiny laktátdehydrogenázy a beta-2 mikroglobulinu [3, 7, 14, 15, 16, 21, 22, 24, 25].
ZÁVĚR
Podle World Health Organization – European Organization for Research and Treatment of Cancer (WHO-EORTC) klasifikace je LyP řazena mezi kožní lymfomy nízké malignity [6, 7]. Naše případy byly pro LyP charakteristické, odpovídaly klinickým a histopatologickým nálezem a také průběhem literárně popsaným případům. Diagnóza byla stanovena na základě klinicko-histopatologické korelace. Léčba celkovými a lokálními kortikosteroidy byla částečně úspěšná, pacienti jsou zavzati do dlouhodobé dispenzární dermatologické a hematologické péče.
Do redakce došlo dne 4. 9. 2012.
Adresa pro korespondenci:
MUDr. Pavlína Horáčková
Dermatovenerologické oddělení
Šumperská nemocnice, a. s.
Nerudova 640/41
78701 Šumperk
e-mail: pavlinahorackova@email.cz
Sources
1. ADAM, Z., KREJČÍ, M., VORLÍČEK, J. et. al. Hematologie – Přehled maligních hematologických nemocí, 2.vydání, Grada: Praha, 2008, s. 159. ISBN 978-80-247-2502-4.
2. AKILOV, O. E., PILLAI, R. K., GRANDINETTI, L. M. et al. Clonal T-cell receptor γ-chain gene rearrangements in differential diagnosis of lymfomatoid papulosis from skin metastasis of nodal anaplastic large-cell lymphoma. Arch. Dermatol., 2011, 147, 8, p. 943–947.
3. BELJAARDS, R., WILLEMZE, R. The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas. Br. J. Dermatol., 1992, 126, 6, p. 596–602.
4. BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. Dermatology. 3rd edition, Mosby: London, 2008, p. 1880–1882. ISBN 978-1-4160-3269-4.
5. BURGDORF, W. H. C., PLEWIG, G., WOLFF, H. H., LANDTHALER, M. Braun-Falco’s Dermatology. 3rd edition, Springer: Heidelberg, 2009, p.1295–1296. ISBN 978-3-540-29312-5.
6. CETKOVSKÁ, P. Nová WHO-EORTC klasifikace kožních lymfomů. Čes-slov. Derm., 2006, 81, 2, p. 69–76.
7. GRUBER, R., SEPP, N. T., FRITSCH, P. O., SCHMUTH, M. Prognosis of lymphomatoid papulosis. Oncologist, 2006, 11, 8, p. 955–957.
8. HEALD, P., SUBTIL, A., BRENEMAN, D. et al. Persistent agmination of lymphomatoid papulosis: an equivalent of limited plaque mycosis fungoides type of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol., 2007, 57, 6, p. 1005–1011.
9. HSU, Y.-J., SU, L.-H., HSU, Y.-L. et al. Localized lymphomatoid papulosis. J. Am. Acad. Dermatol., 2010, 62, 2, p. 353–356.
10. HUGHES, P. S. H. Treatment of lymphomatoid papulosis with imiquimod 5% cream. J. Am. Acad. Dermatol., 2006, 54, 3, p. 546–547.
11. JUNG, J., LEVIN, E. C., JARRETT, R. et al. Lymphomatoid Drug Reaction to Ustekinumab. Arch. Dermatol., 2011, 147, 8, p. 992–993.
12. KEMPF, W., KAZAKOV, D.V., PALMEDO, G. et al. Pityriasis lichenoides et varioliformis acuta with numerous CD30+ cells: a variant mimicking lymphomatoid papulosis and other cutaneous lymphomas. A clinicopathologic, imunohistochemical, and molecular biological study of 13 cases. Am. J. Surg. Pathol., 2012, 36, 7, p. 1021–1029.
13. MACAULAY, W. L. Lymphomatoid papulosis. Arch. Dermatol., 1968, 97, 1, p. 23–30.
14. MARTORELL-CALATAYUD, A., HERNANDÉZ-MARTÍN, A., COLMENERO, I. et al. Lymphomatoid papulosis in children: report of 9 cases and review of the literature. Actas Dermosifiliogr., 2010, 101, 8, p. 693–701.
15. ORTIZ-ROMERO, P. L., LOPEZ-ESTEBARANZ, J. L., GIL-MARTIN, R. et al. Lymphomatoid papulosis: a study of 18 cases. JEADV, 2006, 1, 3, p. 205–216.
16. PALLER, A. S, MANCINI, A. J. Hurwitz clinical pediatric dermatology. 3rd edition, Elsevier Saunders: Philadelphia, 2006, p. 99. ISBN 978-0-7216-0498-5.
17. RODRIGUES, M., McCORMACK, C., YAP, L. M. et al. Successful treatment of lymphomatoid papulosis with photodynamic therapy. Australas. J. Dermatol., 2009, 50, 2, p. 129–132.
18. ŠTORK, J. et al. Dermatovenerologie. 1. Vydání. Galén: Praha, 2008, s. 405. ISBN 978-80-7262-371-6.
19. TORRELO, A., COLMENERO, I., GOIRIZ, R. Persistent agmination of lymphomatoid papulosis. Pediatric dermatology, 2009, 26, 6, p. 762–764.
20. WANG, H. H., LACH, L., KADIN, M. E. Epidemiology of lymphomatoid papulosis. Cancer, 1992, 70, 12, p. 2951–2957.
21. WANG, H. H., MYERS, T., LACH, L. J. et al. Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis. Cancer, 1999, 86, 7, p. 1240–1245.
22. WESTON, W. L., LANE, A. T., MORELLI, J. G. Color Textbook of Pediatric Dermatology. 4th edition, Mosby Elsevier: Philadelphia, 2007, p. 162. ISBN 978-0-323-04909-2.
23. WILLEMZE, R., MEYER, C., VAN VLOTEN, W., SCHEFFER, E. The clinical and histological spectrum of lymphomatoid papulosis. Br. J. Dermatol., 1982, 107, 2, p. 131–144.
24. WOLFF, K., GOLDSMITH, L. A., KATZ, S. I. et al. Fitzpatrik’s dermatology in general medicine. 7th ed., The McGraw-Hill Companies: New York, 2008, p. 1392–1394. ISBN 978-0-07-146690-5.
25. YANCOVITZ, M., WALTERS, R. F., KAMINO, H., BROWN, L. H. Acral lymphomatoid papulosis. J. Am. Acad. Dermatol., 2010, 62, 3, p. 530–531.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2013 Issue 1
Most read in this issue
- Drug Reactions Affecting the Nail Unit
- Lymphomatoid Papulosis
- Erythema Gyratum Repens
- Food Reactions in Adolescent and Adult Patients with Atopic Eczema