
Závažné lékové exantémy
Severe Drug Eruptions
The authors summarize the most severe drug eruptions, namely DRESS syndrome, acute generalized exanthematous pustulosis, Stevens-Johnson syndrome and toxic epidermal necrolysis. Both clinical aspects and current knowledge of etiopathogenesis of these disorders are discussed. Diagnostics, treatment and possible complications are also highlighted.
Key words:
drug eruption – DRESS syndrome – acute generalized exanthematous pustulosis – Stevens-Johnson syndrome – toxic epidermal necrolysis
:
H. Tomková 1; J. Štork 2
:
Kožní oddělení, Krajská nemocnice T. Bati, a. s., Zlín
přednosta prim. MUDr. Jan Šternberský, CSc.
1; Dermatovenerologická klinika, 1. LF UK a VFN Praha
přednosta prof. MUDr. Jiří Štork, CSc.
2
:
Čes-slov Derm, 88, 2013, No. 5, p. 203-212
:
Reviews (Continuing Medical Education)
Autoři podávají přehled nejzávažnějších lékových exantémů, kam patří DRESS syndrom, akutní generalizovaná exantemová pustulóza, Stevensův-Johnsonův syndrom a toxická epidermální nekrolýza. Diskutovány jsou jak klinické aspekty těchto onemocnění, tak současné poznatky etiopatogeneze. Důraz je kladen také na diagnostiku, léčbu a možné komplikace.
Klíčová slova:
lékový exzantém – DRESS syndrom – akutní generalizovaná exantemová pustulóza – Stevensův-Johnsonův syndrom – toxická epidermální nekrolýza
ÚVOD
Lékové exantémy jsou nezanedbatelnou součástí klinické praxe kožního lékaře. Jsou časté jak u pacientů hospitalizovaných (2–3 %), tak u pacientů v ambulantní péči (> 1 %). Většina reakcí je mírných, doprovázených pruritem a regredujících poměrně rychle po vysazení příslušného léku. Vyskytují se však i těžké, život ohrožující reakce, které jsou nepředvídatelné [53].
K těm nejtěžším patří DRESS syndrom, akutní generalizovaná exantemová pustulóza, Stevensův-Johnsonův syndrom a toxická epidermální nekrolýza. Právě těmito závažnými syndromy se v přehledu zabývá náš souhrnný článek.
DRESS SYNDROM
DRESS syndrom (léková reakce s eozinofilií a systémovými příznaky – „drug reaction with eosinophilia and systemic symptoms“), někdy označovaný také jako DHIS (léky indukovaný syndrom přecitlivělosti – „drug-induced hypersensitivity syndrome“) či DHS (syndrom lékové přecitlivělosti – „drug hypersensitivity syndrome“), je závažná potenciálně život ohrožující léková reakce, která zahrnuje febrilie, únavu, otoky obličeje a makulopapulózní exantém (obr. 1). K dalším známkám syndromu patří lymfadenopatie, hematologické abnormality (eozinofilie, lymfocytóza) a postižení vnitřních orgánů (nejčastěji ledviny, játra, plíce). Mortalita dostahuje 10 %, pokud není onemocnění poznáno a léčeno. Onemocnění se vyskytuje častěji u dospělých jedinců, stejně u mužů i žen [20, 53].


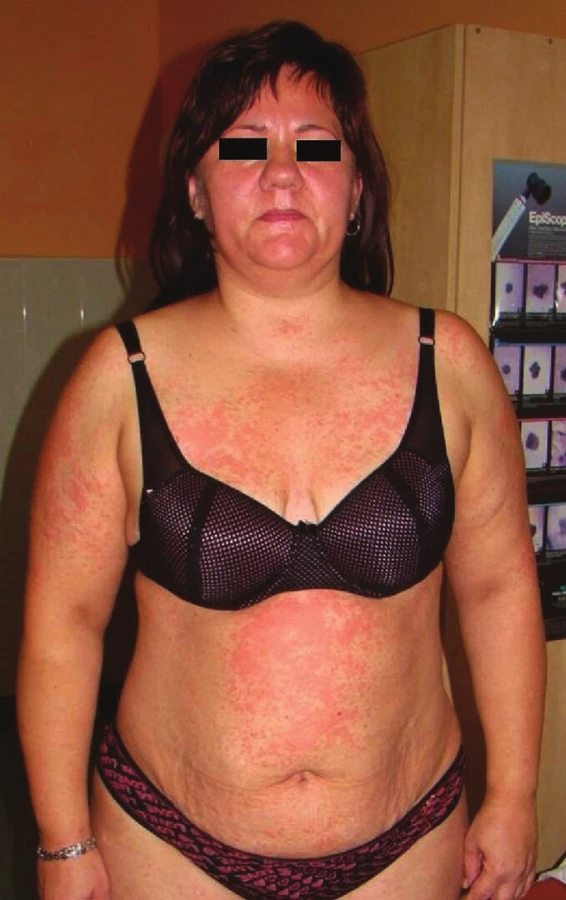
Léky způsobující DRESS syndrom jsou uvedeny v tabulce 1. Nejčastějšími z nich jsou antiepileptika (především fenytoin, karbamazepin, fenobarbital, primidon) a sulfonamidy (zejména dapson a sulfasalazin) [37, 46]. Uvádí se, že riziko DRESS syndromu po karbamazepinu a fenytoinu představuje 1–5 na 10 000 léčených pacientů, po lamotriginu je dokonce vyšší, jeden z 300 dospělých a jedno ze 100 léčených dětí [16, 47].
![Léky vyvolávající DRESS syndrom [20]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/121a63f0681e6d224a2192f0dd2318e9.png)
Přesné mechanismy patogeneze DRESS syndromu zatím nejsou známy. Podílí se zde abnormality detoxikačních enzymatických systémů s následnou kumulací reaktivních metabolitů léku, reaktivace latentních herpetických virů, jako je cytomegalovirus, EB virus, HHV-6 a HHV-7 [20]. Dalším faktorem je genetická predispozice asociovaná s určitými alelami HLA, jako je např. HLA-B*5801 u DRESS syndromu po allopurinolu [19].
DRESS syndrom vzniká akutně, nejčastěji během prvních dvou měsíců, obvykle za 2–6 týdnů, od nasazení léku a charakteristický je pro něj prodloužený průběh s častými exacerbacemi i přes vysazení příslušného léku [20, 53]. Průměrná doba od zahájení podávání léku do vzniku prvních příznaků této reakce je asi 25 dnů (rozmezí od 3 do 105 dnů) a v případě antiepileptik je latence nejdelší [49].
Onemocnění obvykle začíná prodromálními symptomy, jako je svědění kůže a febrilie 38–40 °C, které předcházejí výsevu exantému obvykle o několik dnů. Nejčastější kožní manifestací DRESS syndromu je erytematózní morbiliformní svědivý exantém postihující nejdříve obličej a postupně přecházející na trup, horní končetiny, dolní končetiny. Může přejít v až erytrodermii, projevy mohou být později infiltrované a prosáklé. Na kůži však mohou být přítomna i atypická terčovitá ložiska, vezikuly, buly, purpura i sterilní pustuly, které mohou, ale nemusí, být folikulárně vázané. Při odeznívání exantému erytém nabývá likvidního zbarvení a dochází k lamelózní deskvamaci.
U rozsáhlého postižení kůže může být přítomna také cheilitida, eroze, zarudnutí hrdla a zvětšení tonzil. Častý je výrazný otok obličeje, především v periorbitální a centrofaciální oblasti, který může připomínat angioedém [20].
V histologickém vyšetření kůže může epidermis vykazovat spongiózu, jsou přítomny, zpravidla husté, perivaskulární lymfocytární infiltráty s příměsí eozinofilů a erytrocytárními extravazáty v prosáklém horním koriu. Zánětlivý infiltrát může obsahovat atypické lymfoidní buňky, může vykazovat lichenoidní uspořádání a epidermotropismus, takže může mít vzhled mycosis fungoides, vzácně jsou přítomny i granulomatózní infiltráty v koriu.
Systémové postižení zahrnuje nejčastěji lymfadenopatii a hematologické abnormality (leukocytóza, atypické lymfocyty, eozinofilie, eventuálně i trombocytopenie), nejčastěji postiženým vnitřním orgánem jsou játra (hepatitida). Následuje postižení ledvin (intersticiální nefritida), plic (intersticiální pneumonie, pleuritida, syndrom akutní respirační tísně) a srdce (myokarditida), v těžkých případech může být přítomna také manifestace neurologická (meningitida a encefalitida při reaktivaci HHV-6), gastrointestinální (gastroenteritida, krvácení) a endokrinní dysfunkce. Potenciálně fatální následky může mít především myokarditida a jaterní selhání. Endokrinní dysfunkce je vzácná v akutní fázi, spíše bývá pozdějším následkem DRESS syndromu, proto se doporučují endokrinologické kontroly do dvou let po prodělané reakci. Nejčastěji se jedná o rozvoj autoimunitní tyreoiditidy či diabetu I. typu při reaktivaci HHV-6 [20].
Dosud nebyla vytvořena obecně platná standardní diagnostická kritéria k potvrzení diagnózy DRESS syndromu, nejčastěji používaná kritéria jsou uvedena v tabulce 2.
![Diagnostická kritéria DRESS syndromu [5]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/7954af37c14f6e0c59965fec87c162e4.png)
Určení vyvolávajícího léku může být komplikované, především u hospitalizovaných pacientů léčených několika nově nasazenými léky. Pomoci nám může provedení epikutánního lékového testu (viz obr. 1) či lymfocytárního transformační testu, které mají dobrou specificitu, ale nízkou senzitivitu. Epikutánní lékový test je optimální provést za 2–6 měsíců po odeznění lékové reakce, bývá pozitivní zejména u antiepileptik (karbamazepin, fenytoin) a negativní u allopurinolu [11].
Diferenciálně diagnosticky odlišujeme DRESS syndrom od dalších těžkých lékových reakcí, jako je Stevensův-Johnsonův syndrom/toxická epidermální nekrolýza, akutní generalizovaná exantemová pustulóza a erytrodermie. Dále je důležité vyloučit kožní projevy asociované s akutními virovými infekcemi a vaskulitidy doprovázené eozinofilií [21], lymfomy či akutní lupus erythematosus.
Léčba DRESS syndromu po vysazení vyvolávajícího léku obvykle probíhá za hospitalizace. Lokálně aplikujeme lokální kortikosteroidy. Základem léčby pacienta se systémovým postižením je zahájení terapie systémovými kortikosteroidy, nejčastěji v dávce odpovídající 1 mg/kg denně prednisonu s pozvolným vysazováním během 3–6 měsíců pro potenciální riziko recidivy při rychlém ukončení léčby [21].
AKUTNÍ GENERALIZOVANÁ EXANTEMOVÁ PUSTULÓZA (AGEP)
AGEP, dříve označovaná jako toxická pustulodermie, je poměrně vzácný, spontánně regredující, akutní exantém, typicky provázený febriliemi a neutrofilní leukocytózou.
K charakteristickým znakům AGEP patří:
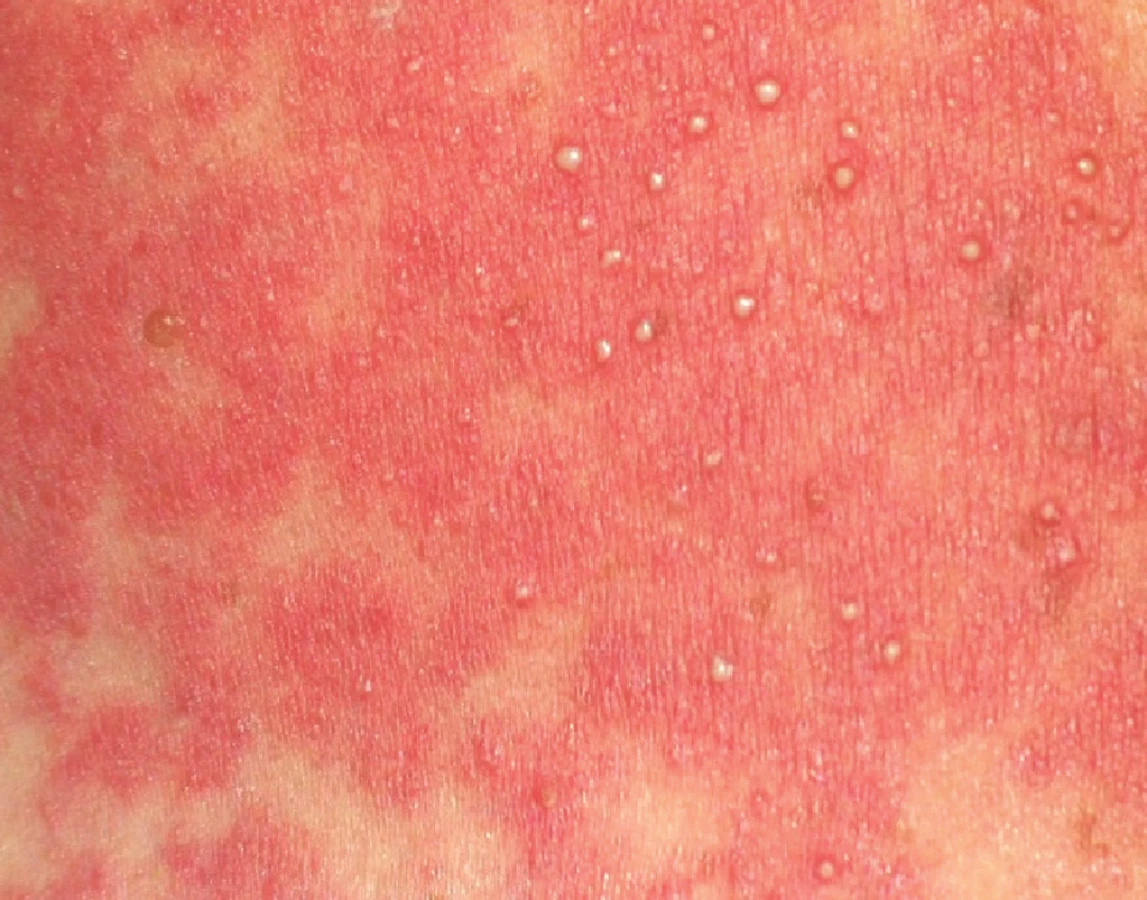
- četné malé intraepidermální nebo subkorneální pustuly o velikosti menší než 5 mm, které nejsou folikulárně vázané a vznikají na plochách prosáklého erytému (obr. 2);
- typické histologické změny;
- febrilie nad 38 °C;
- počet neutrofilních leukocytů nad 7 x 109/l;
- akutní rozvoj se spontánní regresí pustul během 15 dnů [38]. Tabulka 3 uvádí navržené bodovací skóre k potvrzení diagnózy AGEP [44].
![Diagnostické skóre k potvrzení AGEP [44]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d7bdb254f731126387e8031382fb0f79.png)
Incidence AGEP je odhadována na 1–5 případů na milion obyvatel ročně [53]. Onemocnění může vzniknout v kterémkoli věku, nejčastěji postihuje dospělé jedince. Ve studii 97 pacientů s AGEP byl průměrný věk pacientů 56 let, s mírnou převahou žen [43].
Asi 90 % případů je způsobeno léky, nejčastěji antibiotiky (aminopeniciliny a makrolidy), blokátory kalciového kanálu (diltiazem) a antimalariky (tab. 4). U dětí jsou za nejčastější spouštěcí faktory považovány virová infekce (např. coxsackie B4, parvovirus B19, Epstein-Barr virus, cytomegalovirus) a očkování. V ojedinělých případech nelze etiologii určit [4].
![Léky asociované s AGEP [38, 43, 45]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/41b4c50c49fa916eb9e563f968940ffa.png)
Genetická predispozice k rozvoji AGEP není známa. AGEP je neutrofilní zánětlivá reakce zprostředkovaná T lymfocyty. V patogenezi se iniciálně uplatňuje aktivace a migrace lékově specifických CD4+ a CD8+ T lymfocytů do kůže. Příliv cytotoxických CD8+ buněk vede k apoptóze keratinocytů a tvorbě subkorneálních vezikul, ke které přispívají i CD4+ buňky. Infiltrující CD4+ lymfocyty a keratinocyty uvolňují chemokin CXCL-8, který vede k chemoatrakci neutrofilů, a GM-CSF (granulocyte macrophage-colony stimulating factor), který brání apoptóze neutrofilů. Následně dochází k nahromadění neutrofilů v kůži a přeměně vezikul na pustuly [12]. K významným faktorům atrahujícím u AGEP do epidermis polymorfonukleáry patří IL-8 a IL-22. IL-22 je produkován populací Th17 společně s IL-17 a oba synergicky stimulují keratinocyty k tvorbě IL-8. V séru pacientů s AGEP byly detekovány zvýšené hladiny jak IL-8, tak IL-22 [24, 50].
K rozvoji kožního exantému může dojít během 1–3 týdnů po aplikaci léku, obvykle však do 2 dnů. Na prosáklých erytematózních plochách dochází k horečkou provázenému výsevu desítek až stovek drobných sterilních pustul, které nejsou folikulárně vázané, s predilekcí pro intertriga, trup a horní končetiny. Exantém většinou začíná na obličeji nebo v intertriginózní lokalizaci (zejména axily, třísla) odkud se rychle šíří, často i během hodin, na trup a končetiny[45]. Byla popsána i lokalizovaná varianta AGEP (ALEP) [3, 36].
Mírné, neerozivní postižení sliznic vzniká u 20 % případů. Postižení vnitřních orgánů je vzácné a obvykle je omezeno na mírné snížení clearance kreatininu a mírné zvýšení aminotransamináz [12].
Pustulózní fáze spontánně regreduje a během dvou týdnů přechází v generalizovanou deskvamaci projevů s límečkovitým olupováním kůže na periferii [53]. Splynutí pustul může vést k povrchovým erozím a v takovém případě může onemocnění připomínat až toxickou epidermální nekrolýzu.
V diagnostice AGEP je k vyloučení infekční etiologie doporučeno provedení stěru z pustuly. Laboratorně nacházíme neutrofílii a často i mírnou eozinofilii. Histopatologické vyšetření kůže ukazuje spongiformní nebo nespongiformní, subkorneální a/nebo intraepidermální pustuly, edém papilární dermis, infiltráty neutrofilů v dermis s perivaskulární akcentací, exocytózu eozinofilů a fokálně nekrotické keratinocyty [18]. K potvrzení asociace mezi AGEP a příslušným lékem může být užitečné provedení epikutánních testů. Tyto testy se provádějí nejdříve za měsíc po odeznění exantému a při pozitivním testu vzniká často pustulózní reakce. Jejich výpovědní schopnost je bohužel omezená, protože lékové epikutánní testy mají nízkou senzitivitu. Testy in vitro nejsou široce dostupné a jejich význam zatím nebyl ve větších studiích jednoznačně potvrzen [12].
Diferenciálně diagnostická rozvaha zahrnuje především generalizovanou akutní pustulózní psoriázu, dále pak subkorneální pustulózní dermatózu (Sneddon-Wilkinson), SJS, TEN, DRESS syndrom a bulózní impetigo [12]. Podle studie EuroSCAR svědčí v histologickém nálezu přítomnost eozinofilů, nekrotických keratinocytů, smíšeného intersticiálního a perivaskulárního infiltrátu ve střední dermis, a dále pak nepřítomnost stočených dilatovaných krevních cév pro diagnózu AGEP a proti diagnóze generalizované pustulózní psoriázy [19].
AGEP je spontánně regredující onemocnění s dobrou prognózou. Těžší případy jsou léčeny za hospitalizace. U starších a imunitně kompromitovaných pacientů může dojít ke komplikacím, jako je sekundární infekce kůže či hypokalcémie [45]. Mortalita u starších pacientů s komorbiditami se pohybuje mezi 1–2 % [39].
Léčebný přístup zahrnuje především vysazení vyvolávajícího léku, dále podpůrnou péči (antipyretika, případné doplnění tekutin a elektrolytů) a také symptomatickou terapii pruritu a zánětlivé kožní reakce. Lokální kortikosteroidní preparáty jsou preferovány před celkovou kortikoterapií [26], která se uplatňuje u refrakterních případů.
STEVENSŮV-JOHNSONŮV SYNDROM (SJS) A TOXICKÁ EPIDERMÁLNÍ NEKROLÝZA (TEN)
SJS a TEN jsou akutní, život ohrožující mukokutánní reakce charakterizované rozsáhlou nekrózou a cárovitým odlučováním epidermis a postižených sliznic.
SJS je méně těžká varianta onemocnění, u které odlučování epidermis (odloučenou či odlučující se – s pozitivním Nikolského fenoménem) postihuje méně než 10 % povrchu těla. Začíná prodromy, jako je únava a febrilie, které jsou následovány náhlým vznikem erytematózních nebo hemoragických makul či plochých splývajících ložisek s následným vznikem nekrózy a odlučování epidermis (obr. 3, 4). Sliznice jsou postiženy obvykle nejméně ve dvou lokalitách (oční, orální a genitální) – viz obrázek 4.



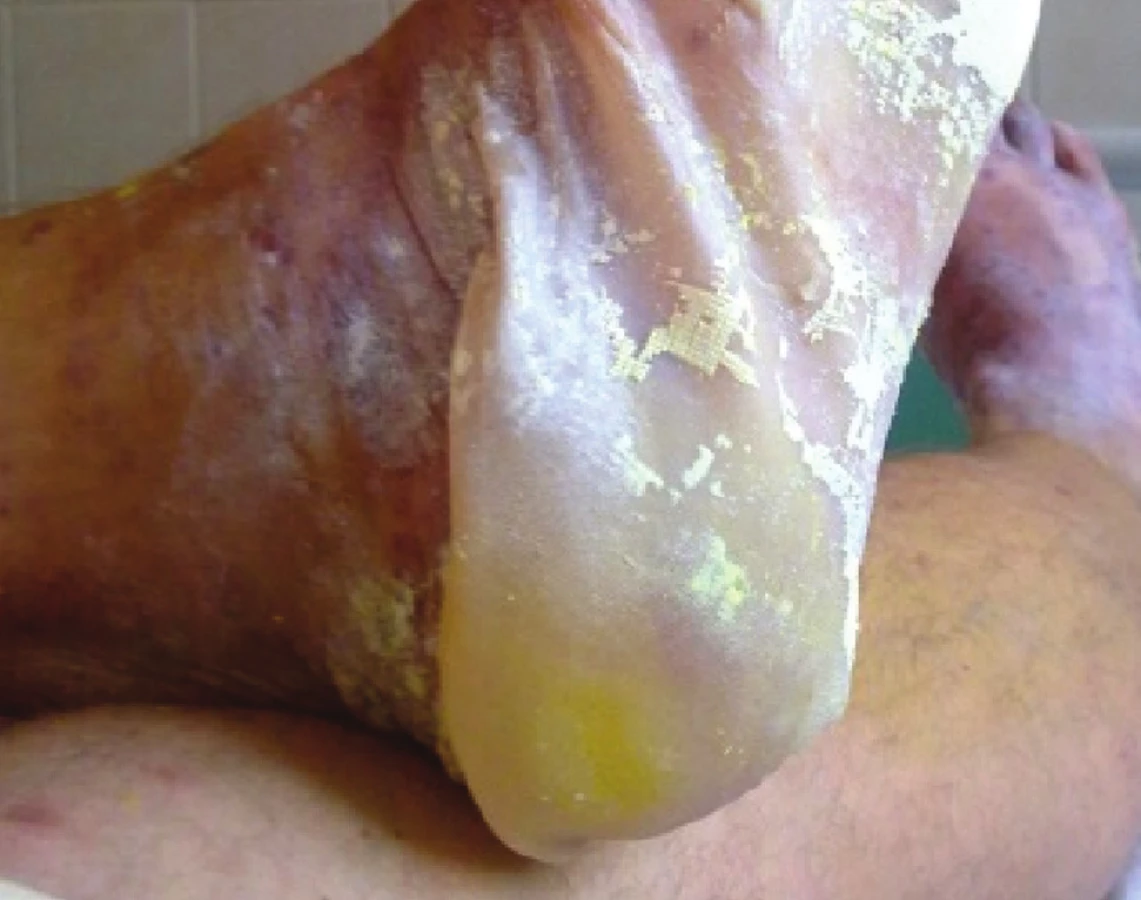



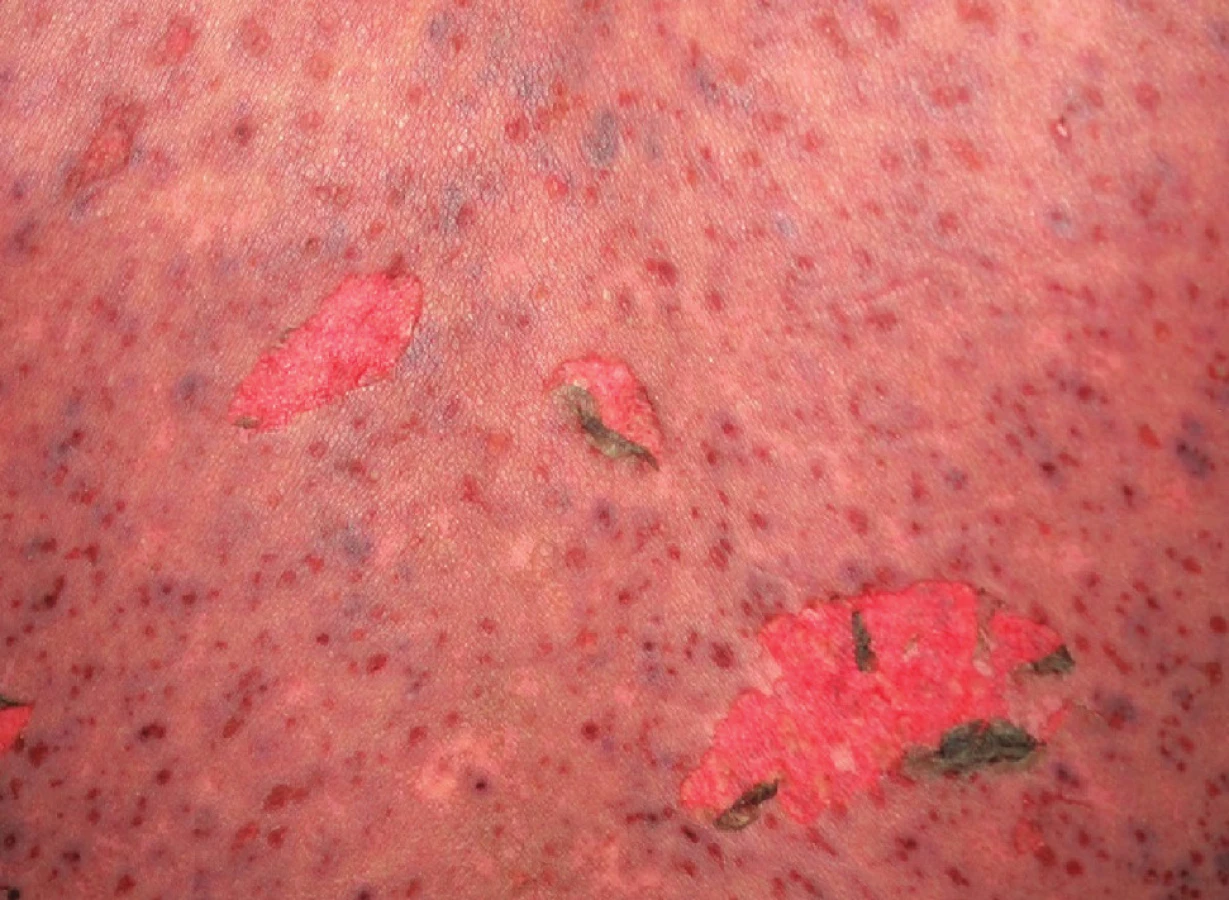
TEN (Lyellův syndrom) zahrnuje odlučování epidermis větší než 30 % povrchu těla. Začíná rovněž prodromálními symptomy, ale horečka bývá vyšší, často překračující 39 °C. Kožní léze začínají jako rozsáhlé erytematózní či hemoragické makuly nebo difuzní erytém, případně atypické terčovité splývající léze. Projevy zpravidla začínají na trupu, odkud se šíří na hlavu a paže, končetiny bývají postiženy méně, někdy mohou být přítomny na dlaních a ploskách.
Jako překryvný syndrom SJS/TEN označujeme pacienty s postižením kůže v rozsahu 10–30 % povrchu těla, SJS postihuje méně než 10 % [34, 41, 53].
K erupci kožních lézí dochází obvykle v průběhu 2–15 dnů. Kožní léze mohou progredovat v masivní nekrolýzu během 24 hodin, průměrné trvání progrese je však kratší než čtyři dny. Hojení po odeznění akutní fáze a při nepřítomnosti kožní infekce nastupuje již během několika dnů, v místech tlaku a v intertriginózních oblastech může trvat až dva týdny. Kůže bývá bolestivá na dotek, který vede k pozitivnímu Nikolského fenoménu [34, 41, 53].
Postižení sliznic s tvorbou erozí a krust obvykle o několik dnů předchází vzniku kožních lézí. Hojení slizničních projevů je pomalé, např. postižení v oblasti glans penis může perzistovat až dva měsíce [34].
Při laboratorním vyšetření bývá často zjištěna anémie a lymfopenie, přítomnost neutropenie však bývá známkou špatné prognózy. Těžký průběh může být komplikován závažnými metabolickými abnormalitami, sepsí, multiorgánovým selháním, plicní embolií a krvácením do gastrointestinálního traktu.
K určení tíže a předvídání mortality onemocnění byl zaveden bodovací index SCORTEN, který by měl být stanoven během prvních 24 hodin a znovu pak třetí den hospitalizace pacienta [2, 17]. Vzniká součtem 7 klinických proměnných, založených na klinických a laboratorních údajích pacienta, které jsou uvedeny v tabulce 5.
![Možné následky prodělaného SJS a TEN [42, 53]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/71cea7bce4d05dac342f562c7698075f.png)
Nejčastější příčinou TEN jsou léky, v ojedinělých případech je uváděna asociace s virovou infekcí (např. cytomegalovirus) či infekcí Mycoplasma pneumoniae, očkováním nebo expozicí chemickým látkám [41, 53]. Nejčastější léky jsou shrnuty v tabulce 6. Patří sem především allopurinol, antibiotika (sulfonamidy, peniciliny, cefalosporiny), antiepileptika (karbamazepin, lamotrigin, fenobarbital), analgetika a nesteroidní antiflogistika (piroxikam) [29].
![SCORTEN– prognostický bodovací systém u pacientů s TEN [2]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/373804e11c739ccc6c17d52e726c6c11.png)
Incidence je odhadována u SJS na 1,2–6 případů a u TEN na 0,4–1,2 případy na milion osob ročně. Onemocnění může vzniknout v kterémkoli věku. TEN je častější u dospělých starších 40 let [53]. Postihuje častěji osoby imunosuprimované (např. s HIV infekcí, lymfomy), u AIDS je až tisicínásobně vyšší riziko vzniku TEN než v normální populaci [41].
Genetické faktory asociované se zvýšeným rizikem SJS a TEN zahrnují především specifické alely HLA. Výrazně zvýšené riziko SJS a TEN po karbamazepinu bylo publikováno u pacientů s HLA-B*1502 [22], proto je před nasazením tohoto léku doporučen genetický screening pro tuto alelu u osob pocházejících z Asie [13]. U populace severní Evropy a Japonska je riziko SJS a TEN po karbamazepinu zvýšeno při přítomnosti alely HLA-A*3101 [28, 31]. Podobně bylo například zjištěno, že přítomnost alely HLA-B*5801 představuje riziko těžkých lékových reakcí, jako je SJS, TEN a DRESS syndrom po allopurinolu [19] a kombinace HLA-B*5701, HLA-DR7 a HLA--DQ3 je riziková pro podávání abakaviru [27].
K dalším publikovaným genetickým faktorům patří pomalá acetylace vedoucí k prodloužené expozici lékům a jejich metabolitům [9] a polymorfismus genu pro receptor IL4 [48].
V patogenezi onemocnění se uplatňuje rozsáhlá apoptóza keratinocytů indukovaná buněčnou cytotoxickou reakcí. K objasnění patogeneze jsou studovány různé mechanismy vedoucí k rozsáhlé apoptóze epidermálních keratinocytů. Byly formulovány hypotézy založené jak na primární roli keratinocytů, zahrnující apoptózu v důsledku interakce molekul Fas a FasL (Fas ligand), tak hypotézy přisuzující hlavní roli buňkám imunitního systému.
Počet zánětlivých buněk infiltrujících postiženou kůži je však příliš malý na to, aby vysvětlil rozsáhlou apoptózu keratinocytů, proto bylo pátráno po solubilních mediátorech, které k apoptóze přispívají.
Nedávné studie podporují názor, že SJS a TEN jsou specifické lékové hypersenzitivní reakce iniciované CD8+ cytotoxickými T lymfocyty. Ukázalo se, že klíčovou molekulou zodpovědnou za diseminovanou apoptózu keratinocytů u SJS a TEN je granulysin. Lék může stimulovat CD8+ T buňky, CD56+ NK (natural killer) buňky a CD8+CD56+ NKT buňky, obsažené v puchýřích pacientů se SJS a TEN, k sekreci granulysinu. Důsledkem toho je apoptóza keratinocytů nevyžadující buněčný kontakt. Nejvyšší koncetrace z cytotoxických molekul dosahuje právě granulysin a pouze deplece granulysinu (ve srovnání s perforinem, granzymem B či solubilním Fas ligandem) redukuje cytotoxicitu. Studie ukázala také korelaci hladin granulysinu v tekutině puchýřů s klinickou tíží onemocnění. Injekce granulysinu do kůže myší pak vede k tvorbě projevů napodobujících SJS-TEN [23]. Další hypotéza předpokládá interakci léku s buňkami exprimujícími MHC I. třídy vedoucí k akumulaci lékově specifických cytotoxických T lymfocytů v puchýřích epidermis a uvolnění perforinu a granzymu B s následnou apoptózou keratinocytů. Tyto vědecké závěry podporují roli buněk imunitního systému v SJS a TEN, i když nevylučují roli Fas a FasL [30]. Nedávné studie potvrdily také roli oxidativního stresu keratinocytů v etiologii TEN [33], který přispívá k pro-apoptotickému procesu včetně stimulace exprese FasL a produkce zánětlivých cytosinů, jako je TNF alfa [1, 32].
Histologické vyšetření z kůže ukazuje v časné fázi vakuolizaci a nekrózu bazálních keratinocytů a také individuální nekrotické (přesněji apoptotické) keratinocyty v ostatních vrstvách epidermis. V pozdějším stadiu vidíme v mikroskopickém obraze nekrózu celé tloušťky epidermis, tvorbu subepidermální buly a odlučování epidermis nad bazální membránou a poměrně řídký lymfocytární infiltrát v dermis [53]. Tíže epidermální nekrózy ani hustota dermálního infiltrátu však není asociována s vyšší úmrtností na TEN [51].
Diferenciální diagnostika TEN zahrnuje erythema multiforme, exantémový lékový exantém, erytrodermii, generalizovaný bulózní fixní lékový exantém, fototoxickou reakci, popáleniny, polékový pemfigus, pemfigoid či lineární IgA bulózní dermatózu, syndrom toxického šoku, stafylokokový syndrom opařené kůže, AGEP, DRESS, paraneoplatický pemfigoid a akutní GVHD [34, 42, 53].
Léčba SJS a TEN zahrnuje včasné stanovení diagnózy a vysazení podezřelého léku. Studie 113 pacientů s TEN a SJS ukázala, že čím dříve je lék způsobující reakci vysazen, tím lepší je prognóza pacienta, přičemž léky s delším poločasem vykazovaly vyšší riziko úmrtí pacienta [15].
Podpůrná péče vyžaduje multioborovou spolupráci a zahrnuje ošetřování kůže a prevenci superinfekce, kontrolu febrilií a bolesti, doplnění tekutin a elektrolytů, nutriční podporu, dále pak spolupráci dalších specialistů při slizničním postižení, především kontroly oftalmologem.
V závislosti na rozsahu kožního a slizničního postižení je nutné zvážit překlad pacienta na jednotku intenzivní péče pro popáleniny [14].
Ošetřování kůže zahrnuje odstranění devitalizované epidermis, převazy neadherentním převazovým materiálem, prevenci a případnou léčbu bakteriální superinfekce. Nejčastější příčinou sepse u pacientů s TEN je Staphylococcus aureus a Pseudomonas aeruginosa [14, 34].
Celkově podávané kortikosteroidy byly řadu let doporučovány v celkové terapii i přes nedostatek důkazů podporujících jejich podávání [54]. Léčba celkovými kortikosteroidy ve vyšších dávkách v časném stadiu onemocnění může přispět ke snížení morbidity a mortality onemocnění, v pozdním stadiu je však kontraindikována [53]. Ve studii pacientů se SJS a TEN se doporučoval pulzní dexametazon v dávce 1,5 mg/kg/den intravenózně po dobu tří po sobě následujících dnů. Progrese onemocnění byla zastavena v průměru do tří dnů s následujícím hojením během dvou týdnů [25]. Nedávná retrospektivní kontrolovaná studie neprokázala zvýšení mortality způsobené léčbou kortikosteroidy [40].
Role intravenózního imunoglobulinu (IVIG), podávaného k blokaci interakce Fas-FasL a tím i apoptózy keratinocytů, v léčbě SJS a TEN je kontroverzní a zatím není dostatečně potvrzena většími studiemi. Zatímco mnohé studie nepotvrdily zlepšení přežití pacientů po léčbě IVIG [14, 40], v poslední době se v některých studiích testovalo podávání vyšších dávek IVIG, a to se slibnými výsledky: 1 g/kg/den po dobu tří dnů (celková dávka 3 g/kg) [35] či např. v jiné studii 400 mg/kg/den po dobu pěti po sobě jdoucích dnů [7].
K dalším zkoumaným terapeutickým možnostem patří cyklosporin A v dávce 3 mg/kg/den po dobu 10 dnů s vysazováním během jednoho měsíce [52] a plazmaferéza [10]. Prevencí recidivy této závažné lékové reakce je poučení pacienta, nutné je vyhnout se podání strukturálně podobného léku, který SJS či TEN způsobil.
Prognóza z hlediska mortality je udávána do 5 % u SJS a 25–30 % u TEN [24, 42]. Přes 50 % nemocných má trvalé následky z poškození: srůsty víček, spojivek, sliznice jícnu a vulvovaginální, entropion, zarůstání řas, jizvení kůže, poruchy pigmentace, eruptivní névy, perzistující eroze sliznic, fimózu, dystrofie až ztrátu nehtů vlasů, hypohidrózu, xerostomii aj. (tab. 7).
![Léky způsobující TEN [6, 29, 41]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/b6802abb369470c60cf4088b59784913.png)
ZÁVĚR
Závažné lékové exantémy se vyznačují výraznou morbiditou a mortalitou. Mohou napodobovat některá jiná kožní onemocnění a mít závažný průběh komplikovaný orgánovým postižením. Vždy je proto důležité správné vyhodnocení klinického obrazu a pečlivá anamnéza, vedoucí k vysazení příslušného léku a zahájení podpůrné léčby [8].
Do redakce došlo dne 16. 9. 2013.
Adresa pro korespondenci:
MUDr. Hana Tomková Ph.D.
Kožní oddělení
Krajská nemocnice T. Bati, a. s.
Havlíčkovo nábřeží 600
762 75 Zlín
e-mail: tomkova@bnzlin.cz
Sources
1. ARNOLD, R., SEIFERT, M., ASADULLAH, K., VOLK, H. D. Crosstalk between keratinocytes and T lymphocytes via Fas/Fas ligand interaction: modulation by cytokines. J. Immunol., 1999, 162, p. 7140–714.
2. BASTUJI-GARIN, S., FOUCHARD, N., BERTOCCHI, M. et al. SCORTEN: A severity-of-illness score for toxic epidermal necrolysis. J. Invest. Dermatol., 2000, 115, p. 149–153.
3. BETTO, P., GERMI, L., BONOLDI, E., BERTAZZONI, M. Acute localized exanthematous pustulosis (ALEP) caused by amoxicillin-clavulanic acid. Int. J. Dermatol., 2008, 47, p. 295–296.
4. BIRNIE, A. J., LITLEWOOD, S. M. Acute generalized exanthematous pustulosis does not always have a drug-related cause. Br. J. Dermatol., 2008, 159, p. 492–493.
5. BOCQUET, H., BAGOT, M., ROUJEAU, J. C. Drug-induced pseudolymphoma and drug hypersensitivity syndrome (Drug Rash with Eosinophilia and Systemic Symptoms: DRESS). Semin. Cutan. Med. Surg., 1996, 15, p. 250–257.
6. BREATHNACH, S. M. Erythema multiforme, Stevens - -Johnson syndrome and toxic epidermal necrolysis. In Burns, T., Breathnach, S., Cox, N., Griffiths, C. Rook’s Textbook of Dermatology. 8th ed., Wiley-Blackwell: Hoboken 2010.
7. CAMPIONE, E., MARULLI, G. C., CARROZZO, A. M. et al. High-dose intravenous immunoglobulin for severe drug reactions: efficacy in toxic epidermal necrolysis. Acta Derm. Veneol., 2003, 83, p. 430–432.
8. CETKOVSKÁ, P., PIZINGER, K., ŠTORK, J. Kožní změny u interních onemocnění. Grada: Praha 2011, 1. vydání, s. 213–222.
9. DIETRICH, A., KAWAKUBO, Y., RZANY, B. et al. Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp. Dermatol., 1995, 4, p. 313–316.
10. EGAN, C. A., GRANT, W. J., MORRIS, S. E. et al. Plasmapheresis as an adjunct treatment in toxic epidermal necrolysis. J. Am. Acad. Dermatol., 1999, 40, 458–461.
11. ELZAGALLAAI, A. A., KNOWLES, S. R., RIEDER, M. J. et al. Patch testing for the diagnosis of anticonvulsant hypersensitivity syndrome: a systematic review. Drug Safety, 2009, 32, p. 391–408.
12. FERNANDO S. L. Acute generalized exanthematous pustulosis. Australas J Dermatol 2012, 55, p. 87-92.
13. FERRELL, P. B. Jr., McLEOD, H. L. Carbamazepine, HLA-B*1502 and risk of Stevens-Johnson syndrome and toxic epidermal necrolysis: US FDA recommentations. Pharmacogenomics, 2008, 9, p. 1543–1546.
14. FIROZ, B. F., HENNING, J. S., ZARZABAL, L. A., POLLOCK, B. H. Toxic epidermal necrolysis: five years of treatment experience from a burn unit. J. Am. Acad. Dermatol., 2012, 67, p. 630–635.
15. GARCIA-DOVAL, I., LeCLEACH, L., BOCQUET, H. et al. Toxic epidermal necrolysis and Stevens-Johnson syndrome: does early withdrawal of causative drugs decrease the risk of death? Arch. Dermatol., 2000, 136, p. 323–327.
16. GUBERMAN, A. H., BESAG, F. M., BRODIE, M. J. et al. Lamotrigine-associated rash: risk/benefit considerations in adults and children. Epilepsia, 1999, 40, p. 985–991.
17. GUEGAN, S., BASTUJI-GARIN, S., POSZEPCZYNSKA--GUIGNE, E. et al. Performance of the SCORTEN during the first five days of hospitalization to predict the prognosis of epidermal necrolysis. J. Invest. Dermatol., 2006, 126, p. 272–276.
18. HALEVY, S., KARDAUN, S. H., DAVIDOVICI, B., WECHSLER, J. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br. J. Dermatol., 2010, 163, p. 1245–1252.
19. HUNG, S. I., CHUNG, W. H., LIOU, L. B. et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci., USA 2005, 102, p. 4134–4139.
20. HUSAIN, Z., REDDY, B. Y., SCHWARTZ, R. A. DRESS syndrome. Part I. Clinical perspectives. J. Am. Acad. Dermatol., 2013, 68, p. e1–14.
21. HUSAIN, Z., REDDY, B. Y., SCHWARTZ, R. A. DRESS syndrome. Part II. Management and therapeutics. J. Am. Acad. Dermatol., 2013, 68, p. 709e1-9.
22. CHUNG, W. H., HUNG, S. I., HONG, H. S. et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature, 2004, 428, p. 486.
23. CHUNG, W. H., HUNG, S. I., YANG, J. Y. et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nature Medicine, 2008, 14, p. 1343–1350.
24. KABASHIMA, R., SUGITA, K., SAWADA, Y. et al. Increased circulating Th17 frequencies and serum IL-22 levels in patients with acute generalized exanthematous pustulosis. J. Eur. Acad. Dermatol. Venereol., 2011, 25, p. 485–488.
25. KARDAUN, S. H., JONKMAN, M. F. Dexamethasone pulse therapy for Stevens-Johnson syndrome/toxic epidermal necrolysis. Acta Derm. Venereol., 2007, 87, p. 144–148.
26. LEE, H. Y., CHOU, D., PANG, S. M. et al. Acute generalized exanthematous pustulosis: analysis of cases managed in a tertiary hospital in Singapore. Int. J. Dermatol., 2010, 49, p. 507–512.
27. MALLAL, S., NOLAN, D., WITT, C. et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet, 2002, 359, p. 727–732.
28. McCORMACK, M., ALFIREVIC, A., BOURGEOIS, S. et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N. Engl. J. Med., 2011, 364, p. 1134–1143.
29. MOCKENHAUPT, M., VIBOUD, C., DUNANT, A. et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. The EuroSCAR study. J. Invest. Dermatol., 2008, 128, p. 35–44.
30. NICKOLOFF, B. J. Saving the skin from drug-induced detachment. Nature Medicine, 2008, 14, p. 1311–1313.
31. OZEKI, T., MUSHIRODA, T., YOWANG, A. et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepin-induced cutaneous adverse drug reactions in Japanese population. Hum. Mol. Genet., 2011, 20, p. 1034–1041.
32. PAQUET, P., NIKKELS, A., ARRESE, J. E. et al. Macrophages and tumor necrosis factor alpha in toxic epidermal necrolysis. Arch. Dermatol., 1994, 130, p. 605–607.
33. PAQUET, P., PIERARD, G. E. Glutathione-S-transferase pi expression in toxic epidermal necrolysis: a marker of putative oxidative stress in keratinocytes. Skin Pharmacol. Physiol., 2007, 20, p. 66–70.
34. PEREIRA, F. A., MUDGIL, A. V., ROSMARIN, D. M. Toxic epidermal necrolysis. J. Am. Acad. Dermatol., 2007, 56, p. 181–200.
35. PRINS, C., KERDEL, F. A., PADILLA, R. S. et al. Treatment of toxic epidermal necrolysis with high-dose intravenous immunoglobulins: multicenter retrospective analysis of 48 consecutive cases. Arch. Dermatol., 2003, 139, p. 26–32.
36. RASTOGI, S., MODI, M., DHAWAN, V. Acute localized exanthematous pustulosis (ALEP) caused by ibuprofen. A case report. Br. J. Oral. Maxillofac. Surg., 2009, 49, p. 132–134.
37. ROUJEAU, J. C. Clinical heterogeneity of drug hypersensitivity. Toxicology, 2005, 209, p. 123–129.
38. ROUJEAU, J. C., BIOULAC-SAGE, P., BOURSEAU, C. et al. Acute generalized exanthematous pustulosis. Analysis of 63 cases. Arch. Dermatol., 1991, 127, p. 1333–1338.
39. ROUJEAU, J. C. Neutrophilic drug eruption. Clin. Dermatol., 2000, 18, p. 331–337.
40. SCHNECK, J., FAGOT, J. P., SEKULA, P. et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR study. J. Am. Acad. Dermatol., 2008, 58, p. 33–40.
41. SCHWARTZ, R. A., McDONOUGH, P. H., LEE, B. W. Toxic epidermal necrolysis. Part I. Introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J. Am. Acad. Dermatol., 2013, 69, p. 173e1-13.
42. SCHWARTZ, R. A., McDONOUGH, P. H., LEE, B. W. Toxic epidermal necrolysis. Part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J. Am. Acad. Dermatol., 2013, 69, p. 187e1-16.
43. SIDOROFF, A., DUNANT, A., VIBOUD, C. et al. Risk factors for acute generalized exanthematous pustulosis (AGEP) - results of a multinational case-control study (EuroSCAR). Br. J. Dermatol., 2007, 157, p. 989–996.
44. SIDOROFF, A., HALEVY, S., BAVINCK, J. N. et al. Acute generalized exanthematous pustulosis (AGEP) - a clinical reaction pattern. J. Cutan. Pathol., 2001, 28, p. 113–119.
45. SPEECKAERT, M. M., SPEECKAERT, R., LAMBERT, J., BROCHEZ, L. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts. Eur. J. Dermatol., 2010, 20, p. 425–433.
46. TAS, S., SIMONART, T. Drug rash with eosinophilia and systemic symptoms (DRESS). Acta Clin. Belg., 1999, 54, p. 197–200.
47. TENNIS, P., STERN, R. S. Risk of serious cutaneous disorders after initiation of use of phenytoin, carbamazepine, or sodium valproate: a record linkage study. Neurology, 1997, 49, p. 542–546.
48. UETA, M., SOTOZONO, C., INATOMI, T. et al. Association of IL4R polymorphisms with Stevens-Johnson syndrome. J. Allergy Clin. Immunol., 2007, 120, p. 1457–1459.
49. UM, S. J., LEE, S. K., KIM, Y. H. et al. Clinical features of drug-induced hypersensitivity syndrome in 38 patients. J. Investig. Allergol. Clin. Immunol., 2010, 20 : 556–562.
50. UMAYAHARA, T., SHIMAUCHI, T., FUJIYAMA, T. et al. Paediatric acute generalized exanthematous pustulosis induced by paracetamol with high serum levels of interleukin-8 and -22: A case report. Acta Derm. Venereol., 2013, 93, p. 362–363.
51. VALEYRIE-ALLANORE, L., BASTUJI-GARIN, S., GUÉGAN, S. et al. Prognostic value of histologic features of toxic epidermal necrolysis. J. Am. Acad. Dermatol., 2013, 68, p. e29-35.
52. VALEYRIE-ALLANORE, L., WOLKENSTEIN, P., BROCHARD, L. et al. Open trial of ciclosporin treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis. Br. J. Dermatol., 2012, 163, p. 847–853.
53. WOLFF, K., JOHNSON, R. A. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed., McGraw - -Hill: Columbus 2009, p. 552–581.
54. WORSWICH, S., COTLIAR, J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol. Ther., 2011, 24, p. 207–218.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2013 Issue 5
Most read in this issue
- Severe Drug Eruptions
- Evaluation of Frequency of the Food Allergy to wheat Flour, Cow’s Milk, Eggs, Soya and Peanuts in 240 Patients with Atopic Eczema Older than 14 Years