
Epidermolysis bullosa acquisita s očními komplikacemi
Epidermolysis Bullosa Acquisita with Ocular Complications
The authors present a case of a patient with epidermolysis bullosa acquisita and Crohn’s disease. The patient has been treated for 13 years with combined immunosuppressive and corticosteroid therapy with only limited morbostatic effect. Initially, classic form dominated in the clinical picture. Later, a bullous pemphigoid-like and a Brunsting-Perry pemphigoid-like lesions occurred, as well as a significant eye involvement.
Key words:
subepidermal blisters – epidermolysis bullosa acquisita – Crohn’s disease – anti-type VII collagen antibodies
Authors:
L. Drlík 1; L. Pock 2; J. Krtička 3
Authors‘ workplace:
Dermatovenerologické oddělení, Šumperská nemocnice, a. s.
přednosta prim. MUDr. Lubomír Drlík
1; Dermatohistopatologická laboratoř, Praha
vedoucí doc. MUDr. Lumír Pock, CSc.
2; Oční ambulance, Zábřeh na Moravě
vedoucí MUDr. Jakub Krtička
3
Published in:
Čes-slov Derm, 89, 2014, No. 1, p. 16-21
Category:
Case Reports
Overview
Autoři představují případ pacienta sledovaného a léčeného 13 roků pro epidermolysis bullosa acquisita a morbus Crohn. Kombinovaná léčba imunosupresivy a kortikosteroidy měla pouze omezený morbostatický efekt. Zpočátku v dermatologickém klinickém obraze dominovala klasická forma, v průběhu onemocnění došlo také k postižení napodobující bulózní pemfigoid a Brunstingův-Perryův pemfigoid. U pacienta se přidružilo významné oční postižení.
Klíčová slova:
subepidermální puchýře – epidermolysis bullosa acquisita – morbus Crohn – protilátky proti kolagenu VII
ÚVOD
Epidermolysis bullosa acquisita představuje jedno z nejvzácnějších imunobulózních onemocnění, které se vyskytuje v několika klinických formách. Léčba je málo efektivní, choroba má chronický průběh a pacienta významně omezuje. Častá je asociace se zánětlivým střevním onemocněním, oční postižení je literárně popsáno pouze sporadicky [1, 4, 7, 18, 19, 22, 25].
POPIS PŘÍPADU
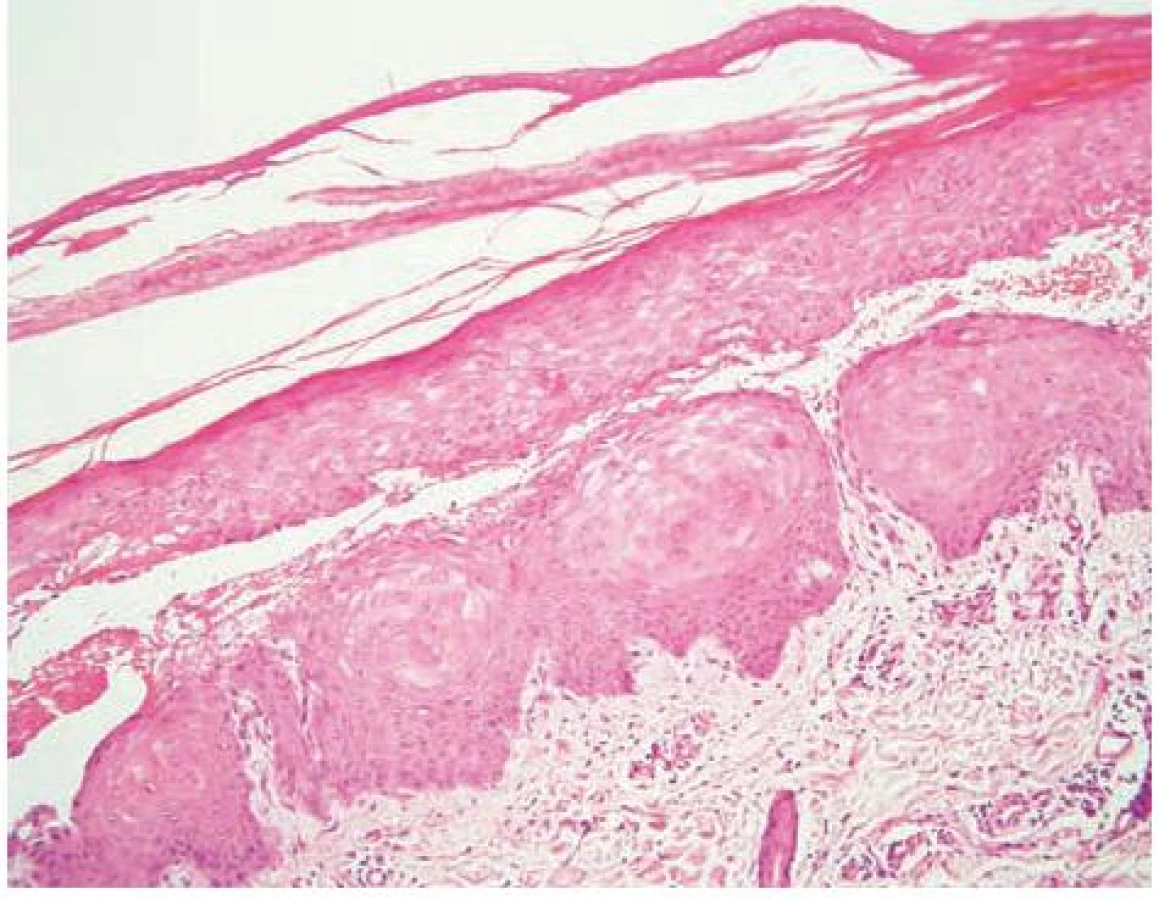
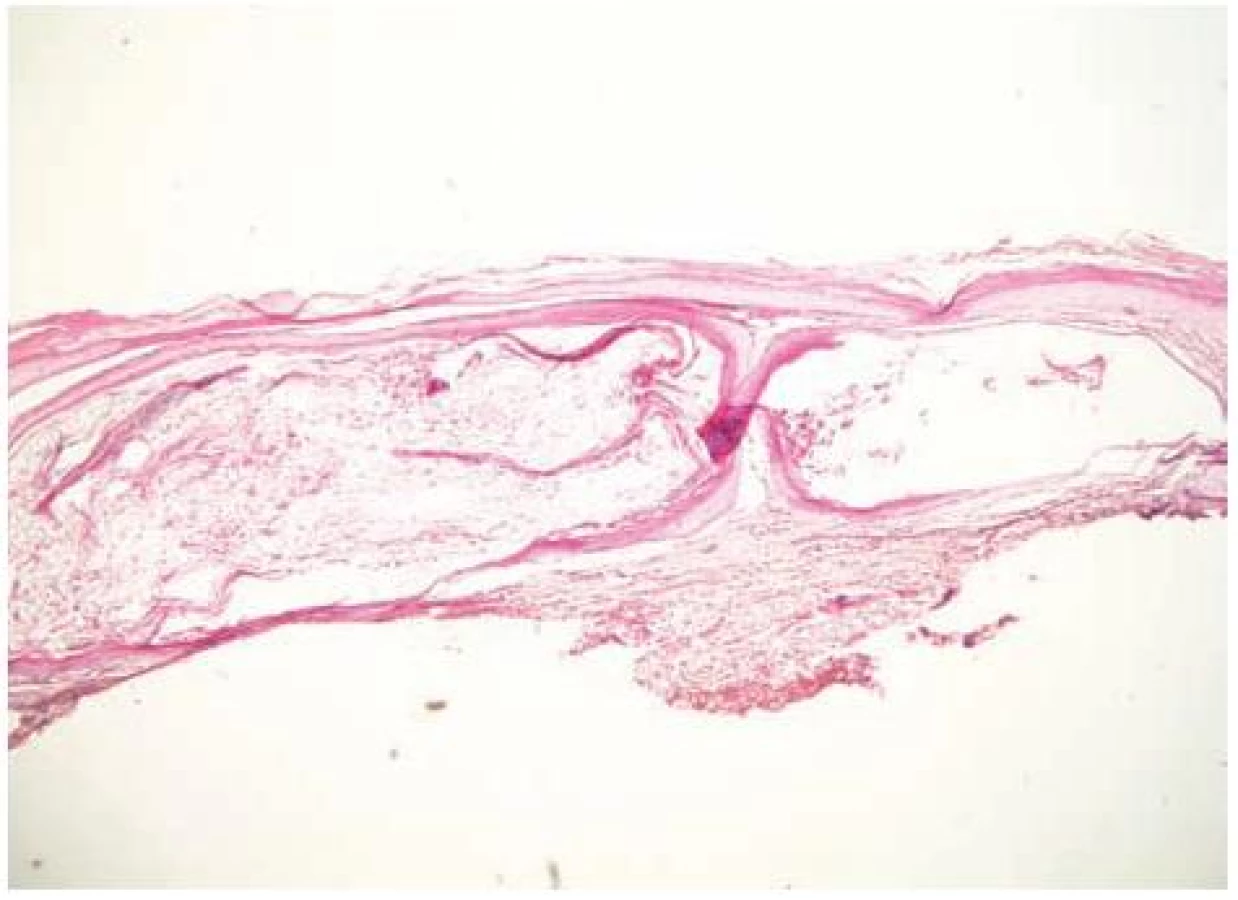
V březnu 2000 se 26letý pacient dostavil na vyšetření s několik měsíců trvající zvýšenou zranitelností kůže a tvorbou drobných puchýřů na hřbetech rukou (obr. 1). Jednalo se o kuřáka s příležitostným abúzem alkoholu, pracoval jako soustružník. Na dorzech a bočních stranách rukou a prstů se vyskytovaly vícečetné mělké erodované plošky a několik napjatých hemoragických puchýřů do velikosti 8–10 mm, okolní kůže nebyla zánětlivě postižena. V histologickém obraze z probatorní excize kůže dorza ruky byla prokázána subepidermální bula s pokročilou reepitelizací spodiny, mírná fibrotizace, několik tenkostěnných epidermoidních cyst charakteru milií. Uvedený obraz podporoval klinický předpoklad porphyria cutanea tarda (obr. 2, 3, 4). Laboratorní výsledky pro porfyrii nesvědčily: moč na porfyriny byla opakovaně negativní, biochemické vyšetření krve, krevní obraz v normě, nízká hladina sérového železa – 4,2 μmol/l (norma 10–28 μmol/l), sonografické vyšetření břicha a ledvin a RTG plic byly bez patologického nálezu. Pro sugestivní klinický obraz byla nasazena léčba Delagilem (chlorochin) 2krát týdně 125 mg a lokálními kortikosteroidy. Tato terapie podávaná několik týdnů kožní nález neovlivnila, vulnerabilita i tvorba puchýřů přetrvávala. Pro gastrointestinální obtíže – průjmy a bolesti břicha – byl pacient v červenci 2000 vyšetřen gastroenterologem, který stanovil diagnózu Crohnovy kolitidy, ordinoval Prednison (prednisonum) v úvodní dávce 60 mg/den, Salofalk (mesalazinum) 1500 mg/den, Sorbifer Durules (ferrosi sulfas) 200 mg/den, Imuran (azathioprinum) 100 mg/den, později i Pentazu (mesalamine). Léčbou došlo postupně ke stabilizaci stavu střevního onemocnění, které bylo hodnoceno jako středně těžké.
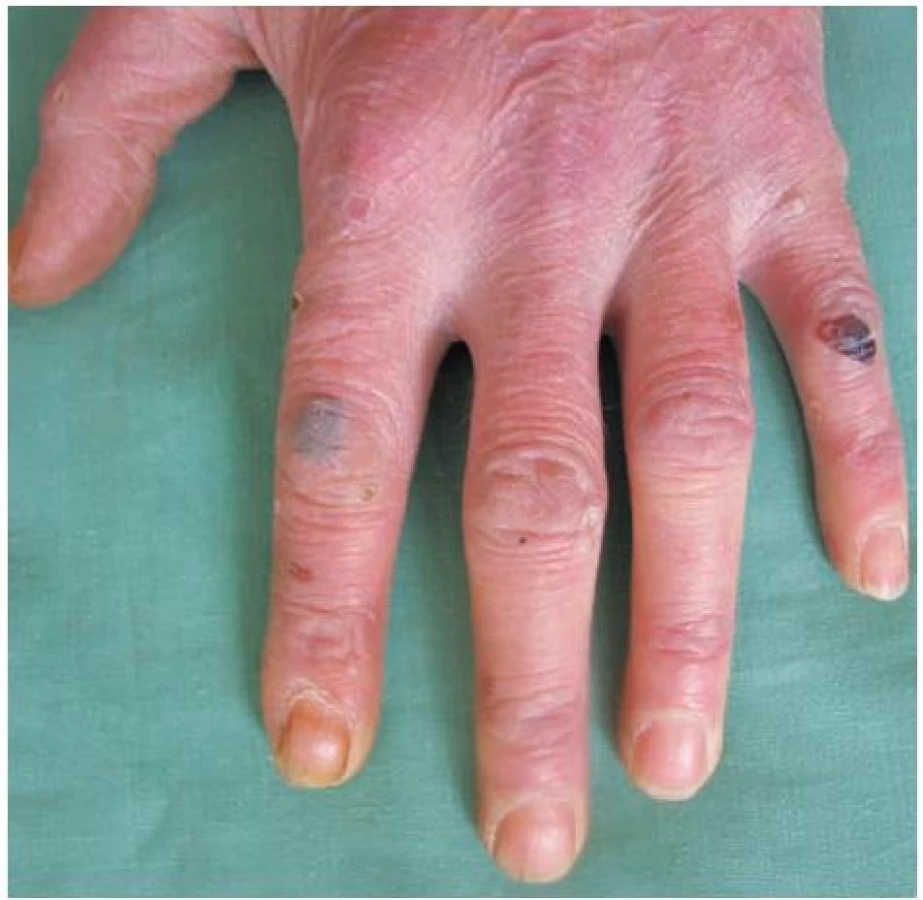
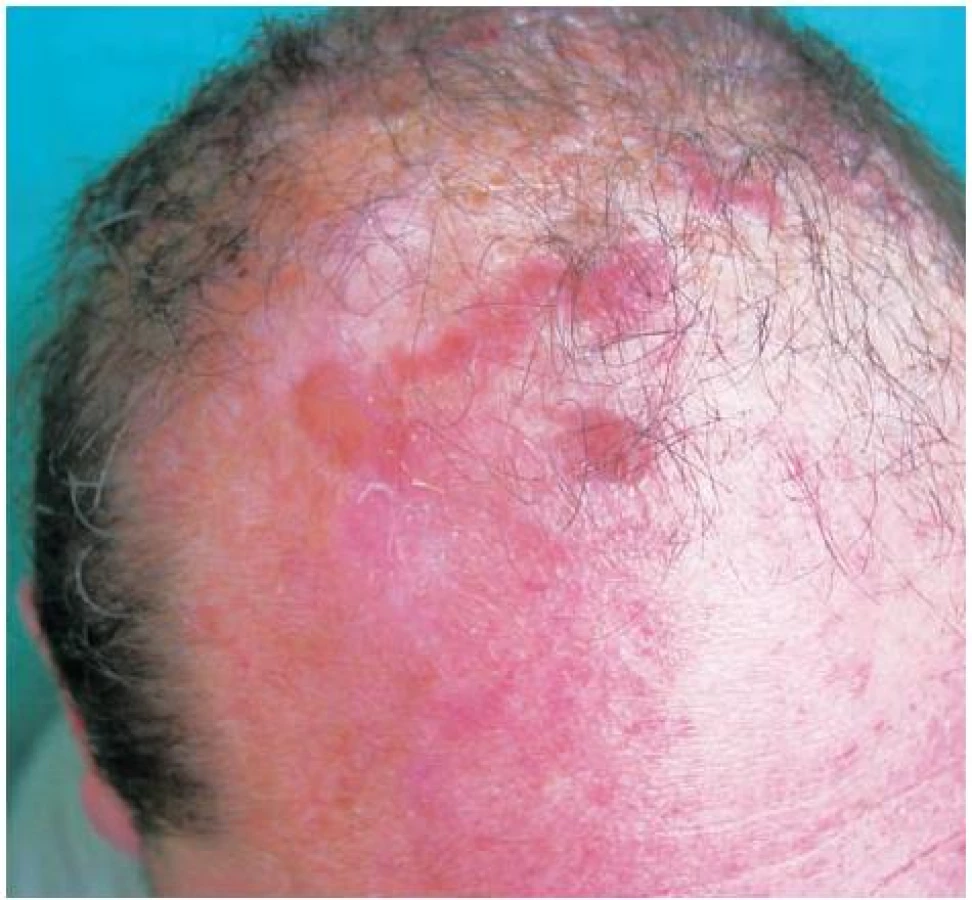
V červnu 2001 došlo mimo dosud postižených lokalit na dorzech rukou, předloktích a kolem loktů také k výsevům vícečetných puchýřů ventrálně i dorzálně na hrudníku a na temeni hlavy (obr. 5).
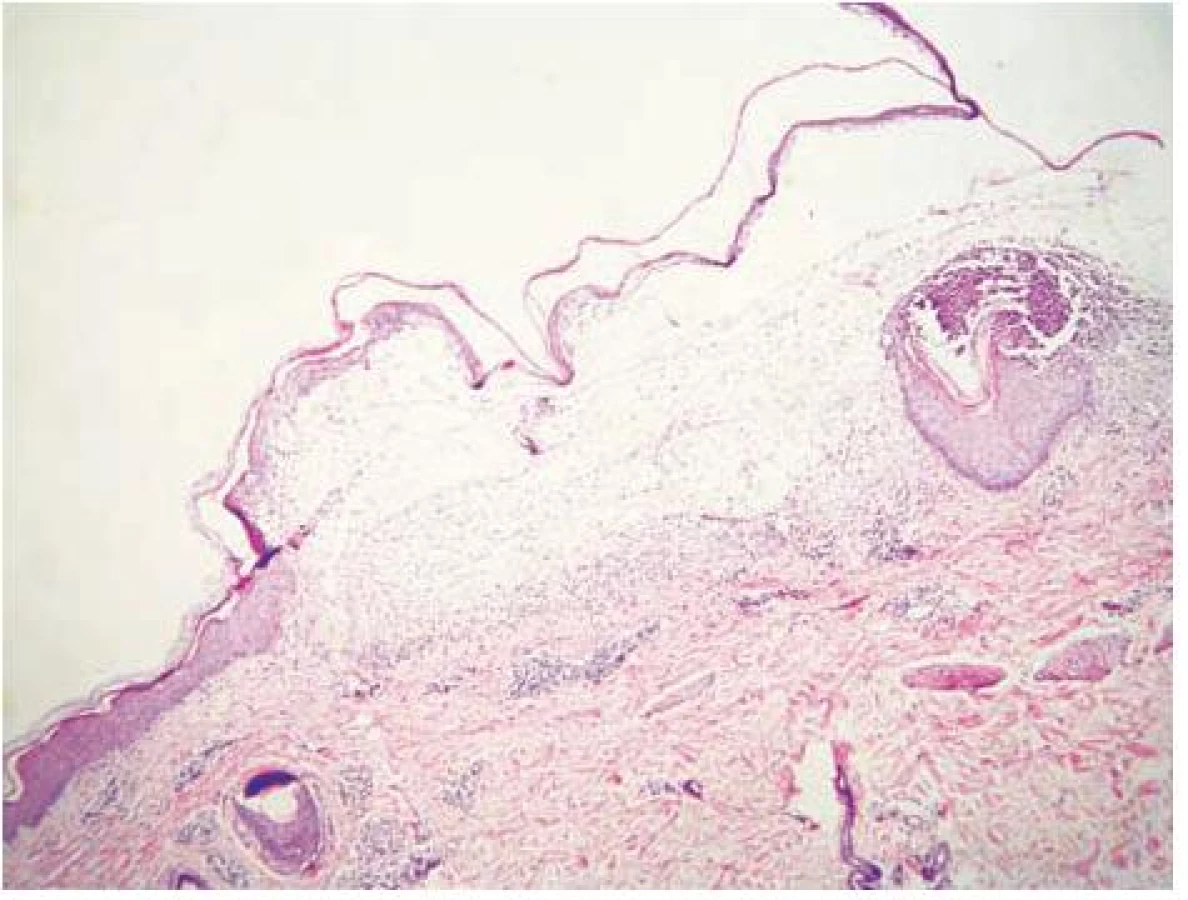
V histologických nálezech z probatorních excizí a vyšetření přímou imunofluorescencí (PIF) z oblasti sterna, ramene a hřbetu ruky byla nalezena subepidermální separace, subepidermální tenkostěnné epidermoidní cysty charakteru milií, lehké zmnožení cév pod štěrbinou, mírná subepidermální fibrotizace, velmi silná lineární pozitivita IgG a IgA, slabá pozitivita C3 na dermoepidermální junkci, fibrinogen negativní (obr. 6).
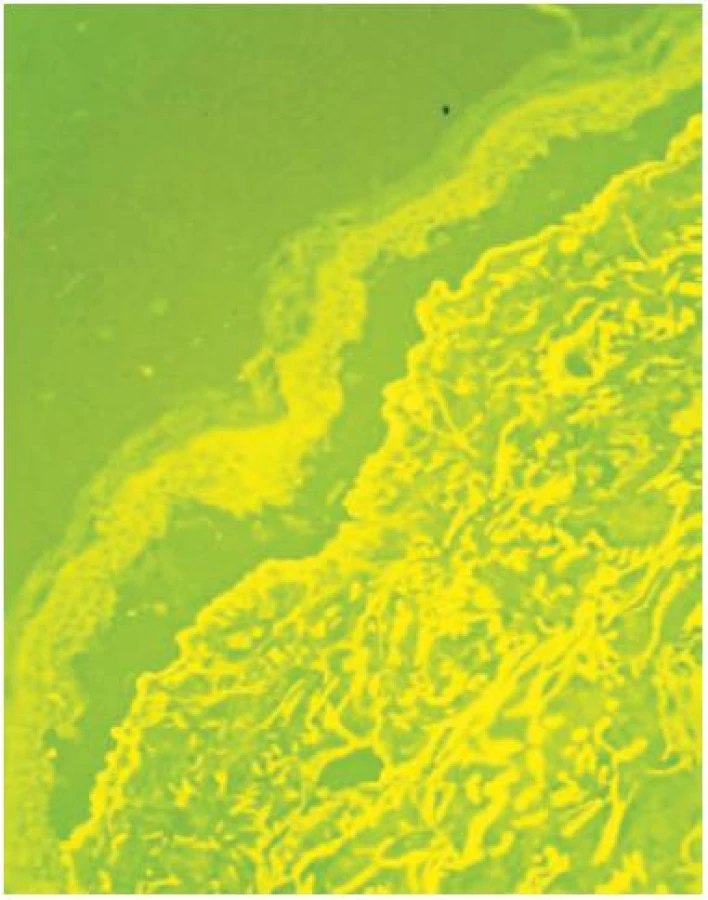
Salt split skin test prokázal IgG lineární pozitivitu na dermální straně dermoepidermální separace, zčásti také na epidermální straně. Uvedený nález odpovídal epidermolysis bullosa acquisita. Do medikace byl přidán Sandimmun Neoral (cyclosporinum) 300 mg/den (4 mg/kg hmotnosti) a vysazen Imuran. Při této léčbě došlo k rychlé regresi kožních projevů, po snížení dávky cyklosporinu ale docházelo k občasným exacerbacím, zejména na hřbetech rukou, temeni hlavy a seboroické predilekci na hrudníku. Projevy zanechávaly vcelku nenápadné atrofické jizvy, na dorzech rukou se tvořila milia. Pacient udával zranitelnost ústní sliznice s tvorbou drobných erozí, genitální sliznice nebyly v průběhu onemocnění postiženy, růst a charakter nehtů nebyl negativně ovlivněn. V dalším průběhu přetrvávaly kožní projevy v různé míře intenzity, střídala se období relapsů a remisí.
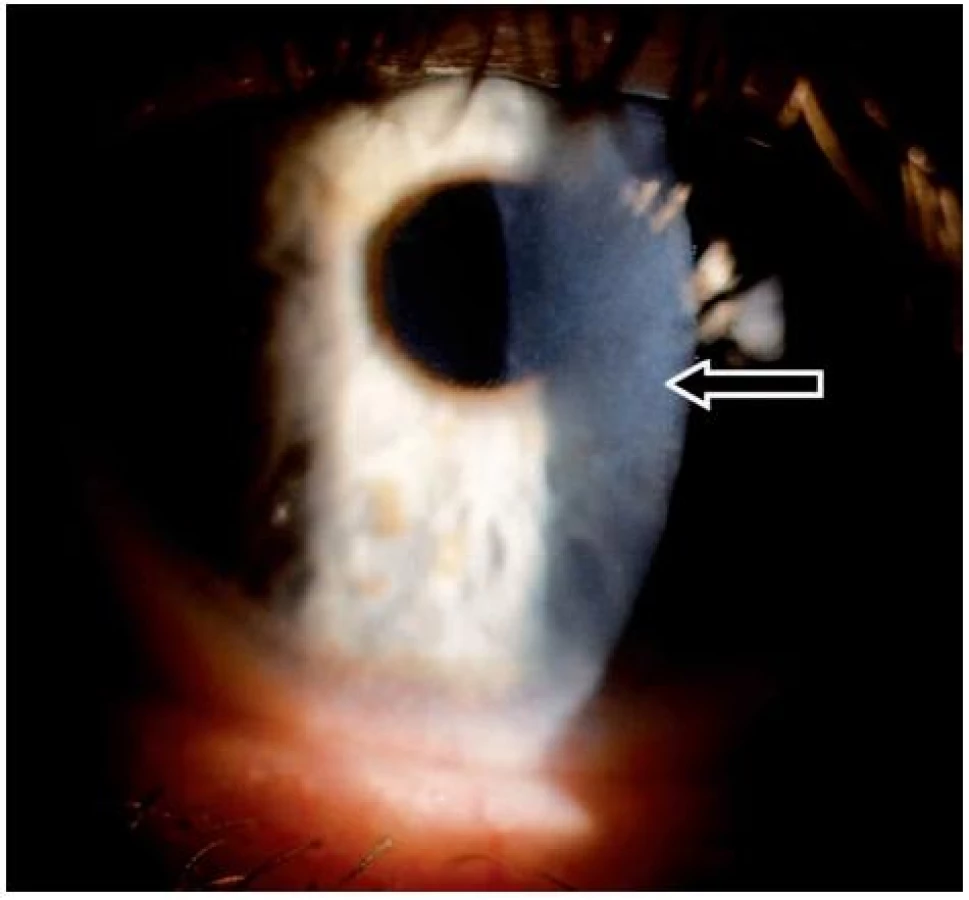
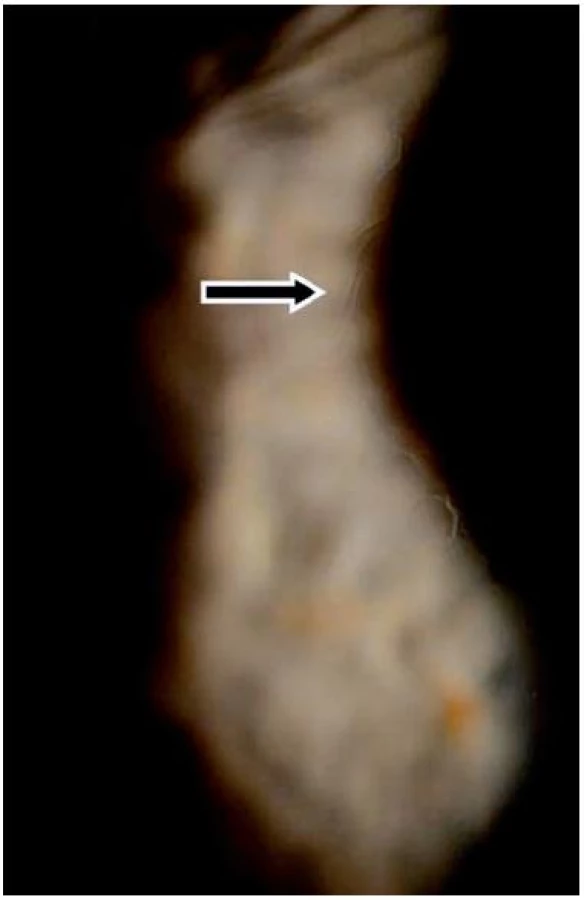
V listopadu 2006 při oftalmologické kontrole byly nalezeny subepiteliální fibrózní pruhy na tarzální spojivce, symblefara v zevním koutku obou očí, byla doporučena aplikace umělých slz ke snížení tření a náchylnosti k tvorbě erozí. V březnu roku 2007 byl zjištěn chronický zánět okrajů víček s jizvením vývodů mazových žláz, drobná symblefara v zevních koutcích obou očí a subepiteliální fibróza na horních tarzálních spojivkách. Stav imponoval jako začínající jizevnaté změny I. stadia očního jizevnatého pemfigoidu. Jizevnaté změny působily snížení počtu drobných slzných žláz a pohárkových hlenotvorných buněk ve spojivkové tkáni a tím sníženou tvorbu vodné složky slzného filmu a poruchu jeho stability narušením lipidové složky. Byla doporučena aplikace umělých slz. Od začátku roku 2008 se zhoršila zraková ostrost levého oka, na rohovce byly nalezeny první projevy poruchy funkce bazální membrány epitelu rohovky – spontánní odlučování vrchních vrstev epitelu s bolestivými projevy erozí a mikrocystické prostory ve tvaru otisků prstů v úrovni bazální membrány epitelu rohovky.
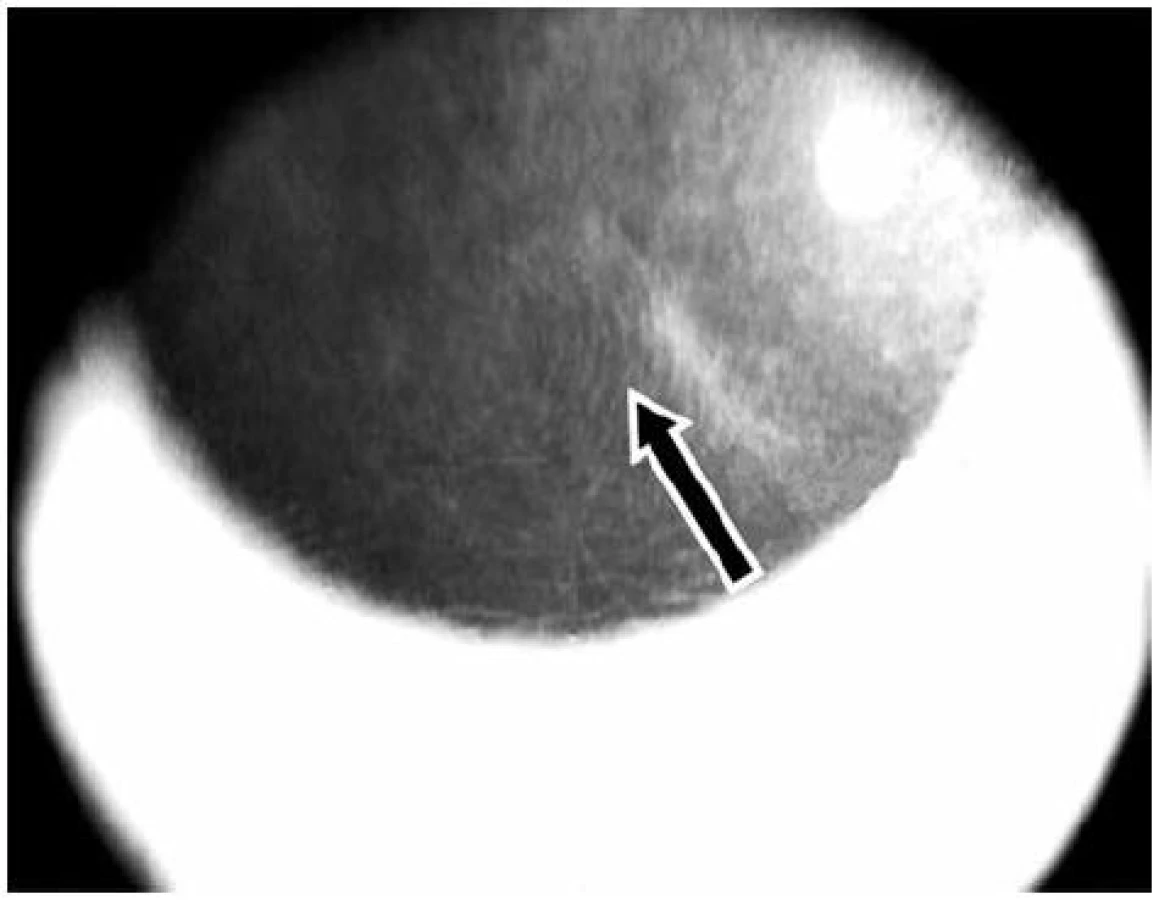
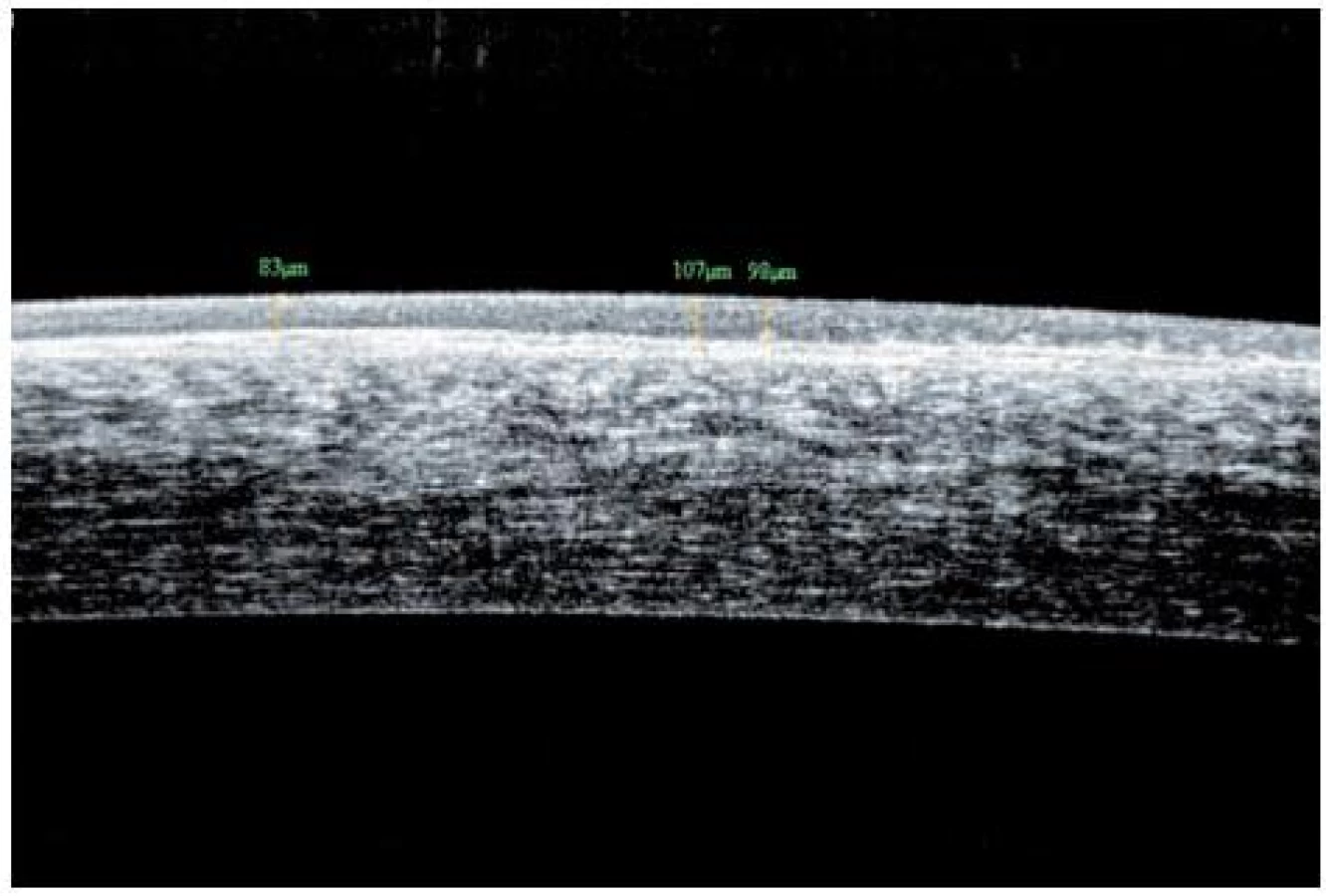
V roce 2009 došlo ke zhoršení stavu – další pokles zrakové ostrosti v důsledku zvětšování rozsahu mikrocystických změn bazální membrány a rozvoje subepiteliální fibrózy rohovky. V reakci na tuto změnu byla zvýšena dávka Sandimmunu z 200 na 300 mg denně a Medrolu (methylprednisolonum) na 16 mg/den. Úprava celkové imunosupresní léčby nevedla k příznivé klinické odpovědi v očním nálezu, naopak bylo nutno začít léčit arteriální hypertenzi, na jejímž vzniku se mohly podávané léky podílet. V průběhu dalšího sledování očního nálezu stále dochází k pozvolné progresi postižení. Projevy očního diskomfortu se nestupňují, ale přidává se pokles zrakové ostrosti i jizevnaté změny na rohovce dříve méně postiženého pravého oka (oftalmologické postižení dokumentováno na obr. 7, 8, 9, 10, 11). Aktuální léčba pacienta zahrnuje Medrol 8 mg/den, Sandimmun 200 mg/den, Salofalk 1500 mg/den, umělé slzy a doplňující antiporotickou, gastroprotektivní a antihypertenzní léčbu.

DISKUSE
Epidermolysis bullosa acquisita (EBA) byla popsána G. T. Elliottem v roce 1895 [9]. Jedná se o velmi zřídka se vyskytující klinicky heterogenní imunobulózní onemocnění s prevalencí v Evropě 0,2/milion obyvatel. Častější výskyt je popisován na Dálném východě a u Afroameričanů, manifestace může být od dětského do pozdně dospělého věku, většinou začíná v dospělosti. Onemocnění je charakterizované zvýšenou kožní fragilitou, tvorbou subepidermálních puchýřů na místech vystavených tlaku nebo traumatům, následným vznikem milií a jizev, ztrátou nehtů [2, 3, 11, 13, 14, 20, 26, 31]. Etiopatogenetickým podkladem je tvorba IgG (v menšině případů IgA) protilátek proti 145 kDa nekolagenní NC1 doméně kolagenu VII, který představuje základní stavební prvek kotvících fibril. Poškozením kotvících fibril je tak onemocnění příbuzné s dystrofickou formou epidermolysis bullosa congenita. Predispozičním faktorem vzniku onemocnění je výskyt HLA-DR2. V důsledku působení protilátek proti kolagenu VII dochází k odloučení epidermis včetně bazální membrány od koria a vzniku subepidermálních puchýřů (protilátky váží komplement, následně dochází k atrakci neutrofilů, jejich degranulaci s uvolněním elastázy a gelatinázy B [10, 11, 13, 15, 22, 23, 28, 30].
Onemocnění se vyskytuje v následujících klinických formách – klasická (napodobující porphyria cutanea tarda a hereditární epidermolysis bullosa), formy napodobující bulózní pemfigoid, jizvící pemfigoid (s převažujícím postižením sliznic), Brunstingův-Perryův pemfigoid a IgA bulózní dermatózu. Uvedené klinické formy se mohou u konkrétního pacienta měnit v průběhu doby, případně koexistují [2, 13, 14, 17, 20, 31]. V histologickém nálezu dominuje subepidermální puchýř jako výraz alterace v oblasti bazální membrány, dermální neutrofilní infiltrát s příměsí eozinofilů a monocytů. U starších lézí je v popředí fibróza. Histologické odlišení od dalších autoimunitních onemocnění charakterizovaných subepidermálními puchýři může být obtížné. Přímá imunofluorescence z perilezionální kůže prokazuje depozita IgG, komplementu a IgM v zóně bazální membrány, respektive sublamina densa. V případech IgA bulózní dermatózy-like jsou to uloženiny IgA. Salt split skin test odliší bulózní pemfigoid (u bulózního pemfigoidu jsou depozita uložena na epidermální straně puchýře, zatímco u EBA na dermální straně). Dalšími diagnostickými možnostmi jsou nepřímá imunofluorescence a zejména elektronová mikroskopie, která prokazuje snížený počet kotvících fibril a uloženiny denzního materiálu. Zlatým standardem moderní diagnostiky je imunoelektronová mikroskopie. Metodou Western-blot je možno prokázat v krvi pacienta protilátky proti kolagenu VII. EBA je asociována se zánětlivými střevními onemocněními (v 25–50 % případů) – zejména s morbus Crohn. Naopak u Crohnovy choroby jsou v séru v desítkách procent případů detekovatelné protilátky proti kolagenu VII [12, 21, 22]. Další možné asociace – revmatoidní artritida, plicní fibróza, diabetes mellitus, thyreoiditida, lupus erythematosus, myelom, lymfomy. Jsou popsány také léky indukované případy [8]. Komplikacemi mohou být bakteriální superinfekce, fibrózy, které způsobují omezenou hybnost prstů rukou či omezují chůzi při postižení plantárních oblastí, dále to jsou striktury ezofagu a laryngeální stenózy, oslepnutí. V diferenciální diagnóze je nutné odlišit bulózní a jizvící pemfigoid, porphyria cutanea tarda, epidermolysis bullosa congenita dystrophica, bulózní lupus erythematosus, dermatitis herpetiformis Duhring a IgA lineární dermatózu [2, 3, 26]. Léčebné možnosti jsou omezené, praktické terapeutické zkušenosti jsou pro nízkou incidenci malé, neexistují větší soubory léčených pacientů. Torpidní jsou zejména mechanobulózní klasická forma a forma napodobující jizvící pemfigoid. Efekty léčby jsou udávány při podávání kolchicinu, rituximabu (monoklonální chimerická protilátka proti CD 20 B lymfocytům), infliximabu (anti TNF alfa). Kortikosteroidy mají omezený efekt zejména u klasické formy, prospěšnější jsou u bulózní pemfigoid- -like formy. Dalšími možnostmi terapie jsou sulfony a imunosupresiva, případně intravenózní imunoglobuliny. Cyklosporin bývá úspěšný ve vyšších dávkách (5 až 6 mg/kg/den). Popsány jsou také efekty léčby mykofenolát mofetilem [3, 5, 6, 11, 14, 16, 24, 27, 29, 30]. Nutná je edukace pacienta s cílem omezit traumatizaci kůže.
ZÁVĚR
Klinické dermatologické projevy u našeho pacienta odpovídaly v počátku onemocnění klasické mechanobulózní formě. V dalším průběhu se současně objevovaly puchýře na temeni hlavy (charakteru Brunstingova-Perryho pemfigoidu) a na trupu, které byly blízké bulóznímu pemfigoidu – projevy ve všech lokalitách se vyskytovaly v určitých obdobích nemoci současně. Oční postižení nebylo zpočátku zjevné, oftalmologické vyšetření bylo negativní. Projevy spojené s jizevnatým postižením spojivky a rohovky se manifestovaly v roce 2006 – tedy šest roků od vzniku kožních obtíží a tedy také po šesti letech kontinuální kortikosteroidní a imunosupresivní léčby. Zpočátku se jednalo o poruchy slzného filmu s očním diskomfortem a prchavými poruchami zrakové ostrosti. Později se rozvinuly zřetelné jizevnaté změny na spojivce a rohovce, s progresí docházelo k poklesu zrakové ostrosti. Prognóza očního postižení je nejistá, jizevnaté změny rohovky zvolna progredují. Základem oční léčby zůstává substituce nedostatku slzného filmu. Možnost úspěšného chirurgického řešení neprůhledné rohovky alogenní transplantací je významně snížena právě poruchou slzného filmu, který je nezbytný pro úspěšné přihojení transplantátu. Charakter choroby navíc s vysokou pravděpodobností povede k recidivě poruchy na epitelu rohovkového štěpu. Literárně jsou popsány možnosti léčby – resekce spojivky s kryoterapií, autologní štěpy, transplantace amniotické membrány – s neurčitými vyhlídkami na úspěch [7, 25]. Léčba pacienta probíhá v koordinaci s terapií zánětlivého střevního onemocnění a oftalmologického postižení kombinací celkově podávaných kortikosteroidů a cyklosporinu. Podávání kolchicinu nebylo zvažováno (významné nežádoucí vedlejší gastrointestinální účinky léku), diagnóza EBA byla stanovena až po zjištění Crohnovy nemoci. V průběhu 13letého sledování a léčby pacienta docházelo k atakám vzplanutí choroby. Dermatologické projevy většinou nebyly nijak dramatické, reagovaly na zvýšené dávky kortikosteroidů a cyklosporinu, nicméně pacienta omezovaly zejména při manuální práci, přispěly k přiznání plného invalidního důchodu. V současné době je zvažováno nasazení biologické léčby (anti TNF alfa), která by mohla příznivě ovlivnit patologické procesy ve všech postižených orgánech a dovolila omezit dlouhodobě podávanou imunosupresivní a kortikosteroidní léčbu.
Do redakce došlo dne 25. 6. 2013.
Adresa pro korespondenci:
Prim. MUDr. Lubomír Drlík
Dermatovenerologické oddělení
Nemocnice Šumperk, a.s.
Nerudova 41
787 52 Šumperk
e-mail: drlik@nemspk.cz
Sources
1. BAUER, J. W., SCHAEPPI, H., METZE, D. et al. Ocular involvement in IgA-epidermolysis bullosa acqusita. Br. J. Dermatol., 1999, 141, p. 887–892.
2. BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. Dermatology. 2th Ed., Mosby Elsevier, 2008, p. 441–445, ISBN 9781416029991.
3. BURNS, T., BREATHNACH, S., COX, N., GRIFFITHS, C. Rook’s Textbook of Dermatology. 8th Ed., Wiley -Blackwell Publishing Ltd., 2012, Vol. 2, p. 40.51–40.57, ISBN 978-1-4051-6169-5.
4. CAUX, F., KIRTSCHIG, G., LEMARCHAND-VENENCIE, F. et al. IgA-epidermolysis bullosa acquisita in a child resulting to blindness. Br. J. Dermatol., 1997, 137, p. 270–275.
5. CETKOVSKÁ, P., PIZINGER, K., RESL, V. Mycofenolát mofetil v léčbě vybraných kožních nemocí. Čes.-slov. Derm., 80, 2005, 1, p. 11–14.
6. CETKOVSKÁ, P., PIZINGER, K., SKÁLOVÁ, A. Klinický případ: Svědivé puchýře na končetinách a na trupu. Čes.-slov. Derm.,76, 2001, 4, s. 215–216.
7. DANTAS, P. E., NISHIWAKI-DANTAS, M. C., SEQUIM, M. H., CURSINO, J. W. Bilateral corneal involvement in epidermolysis bullosa acquisita. Cornea, 2001, 20 (6), p. 664–667.
8. DELBALDO, C., CHEN, M., FRIEDLI, A. et al. Drug-induced epidermolysis bullosa acquisita antibodies to type VII collagen. J. Am. Acad. Dermatol., 2002, 46, p. 161–164.
9. ELLIOTT, G. T., Two cases of epidermolysis bullosa. J. Cutan. Genitourinar. Dis., 1895, 13, p. 10.
10. GAMMON, W., HEISE, E., BURKE, W. et al. Increased frequency of HLA-DR2 in patients with auto-antibodies to epidermolysis bullosa acquisita antigen: evidence that expression of autoimmunity to type VII collagen is HLA class II allele assotiated. J. Invest. Dermatol., 1998, 91, p. 228–232.
11. GUPTA, R., WOODLEY, D. T., CHEN, M. Epidermolysis bullosa acquisita. Clin. Dermatol., 2012, 30 (1), p. 60–69.
12. HUNDORFEAN, G., NEURATH, M. F., SITARU, C. Autoimmunity against type VII kolagen in inflammatory bowel disease. J. Cell. Mol. Med., 2010 (10), p. 2393–2403.
13. CHEN, M., KIM, G. H., PAKASH, L. et al. Epidermolysis Bullosa Acquisita: Autoimmunity to Anchording Fibril. Autoimmunity, 2012, 45 (1), p. 91–101.
14. ISHII, N., HAMADA, T., DAINICHI, T. et al. Epidermolysis bullosa acquisita: what’s new? J. Dermatol., 2010, 37 (3), p. 220–230.
15. ISHII, N., YOSHIDA, M., HISAMATSU, Y. et al. Epidermolysis bullosa acquista sera react with distinct epitopes on the NC1 and NC2 domains of type VII collagen: study using immunobloting of domain-specific recombinant proteins and postembeding immunoelectron microscopy. Br. J. Dermatol., 2004, 150, p. 843–851.
16. KIRTSCHIG, G., MURRELL, D., WOJNAROWSKA, F. et al. Interventions for mucous membrane pemphigoid/cicatricial pemphigoid and epidermolysis bullosa acquisita: a systematic literature review. Arch. Dermatol., 2002 (138), p. 380–384.
17. KURZHALS, G., STOLZ, W., MEURER, M. et al. Acquired epidermolysis bullosa with the clinical feature of Brunsting-Perry cicatricial bullous pemphigoid. Arch. Dermatol., 1991, 127, p. 391–395.
18. LAFOREST, C., HUILGOL, S. C., CASSON, R. et al. Autoimmune bullous diseases: ocular manifestations and management. Drugs, 2005, 65 (13), p. 1767–1779.
19. LANG, P. G., TAPERT, M. J. Severe ocular involvement in a patient with epidermolysis bullosa acquisita. J. Am. Acad. Dermatol., 1987 (16), p. 439–443.
20. PLEWIG, G., WOLFF, H. H., LANDTHALER, M. Braun ĐFalco’s Dermatology. 3th Ed. Springer Medicin Verlag Heidelberg, 2009, p. 662–663, ISBN 278-3-540-29312-5.
21. RAAY, T. L., LEVINE, J. B., WEISS, W. et al. Epidermolysis bullosa acquisita and inflammatory bowel disease. J. Am. Acad. Dermatol., 1982 (6), p. 242–252.
22. REDDY, H., SHIPMAN, A. R., WOJNAROWSKA, F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin. Exp. Dermatol., 2013, 38 (3), p. 225–230.
23. SCHIMANOVICH, I., MIHAI, S., OOSTINGH, G. J. et al. Granulocyte-derivered elastase and gelatinase B are recquired for dermal-epidermal separation induced by autoantibodies from pacients with epidermolysis bullosa acquisita and bullous pemphigoid. J. Pathol., 2004, 204, p. 519–527.
24. SCHMIDT, E., HUNZELMANN, N., ZILLIKENS, D. et al. Rituximab in refractory autoimmune bullous diseases. Clin. Exp. Dermatol., 2006, 31, p. 503–508.
25. SPRAUL, C. W., BUCHWALD, H. J., LANG, G. K., LANG, G. E. Reccurent corneal ulcer in a patient with Crohn’s disease assotiated with epidermolysis bullosa acquisita. Klin. Monbl. Augenheilkd., 2003, 220 (6), p. 423–426.
26. ŠTORK, J. et al. Dermatovenerologie. Galén: Praha, 2008, p. 207–208, ISBN 978-80-7262-371-6.
27. TREBING, D., ZIEMER, A. Acquired epidermolysis bullosa acquisita with a highly varied clinical picture and successful treatment with mycophenolate mofetil. Hautarzt, 2001, 52, p. 717–721.
28. UMEMOTO, N., DEMITSU, T., TODA, S. et al. A case of nonscarring subepidermal blistering disease associated with autoantibodies reactive with both type VII collagen and laminin 5. Dermatology, 2003, 207, p. 61–64.
29. WALLET-FABER, N., FRANCK, N., BATTEUX, F. et al. Epidermolysis bullosa acquisita following bullous pemphigoid, successfuly treated with the anti-CD20 monoclonal antibody rituximab. Dermatology, 2007, 215, p. 252–255.
30. WOODLEY, D. T., CHANG, C., SADAT, P. et al. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological and immunological features of epidermolysis bullosa acquisita. J. Invest. Dermatol., 2005, 124, p. 958–964.
31. WOODLEY, D. T., CHEN, M. Epidermolysis bullosa acquisita. In Wolff, K., Goldsmith, L. A., Katz, S. I. Fitzpatrik’s Dermatology in General Medicine. 7th Ed. McGraw-Hill Medical, 2008, p. 494–500, ISBN 978-0-07-146690-5.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2014 Issue 1
Most read in this issue
- Dermatoskopická diagnostika alopecií
- Léčba periungválních virových bradavic pulzním barvivovým laserem
- Epidermolysis bullosa acquisita s očními komplikacemi