
Keratodermia punctata palmaris et plantaris typ 1:
popis případu matky a dcery
Keratosis Punctata Palmaris et Plantaris Type 1: Case Study of a Mother and Daughter
Palmoplantar keratodermas are an extensive group of heterogenous acquired and inherited diseases. Modern molecular genetic methods have helped to clarify the etiopathogenetic relationships among the specific forms. Diagnosis is currently based on the clinical and histopathologic pictures and molecular genetics. Treatment is symptomatic, aiming to decrease palmoplantar hyperkeratosis. In our presentation we describe the case of mother and daughter with punctate palmoplantar keratosis type I.
Key words:
Keratosis Punctata Palmaris et Plantaris – Type 1
:
L. Smižanský-Bari 1; L. Drlík 1; L. Pock 2
:
Dermatovenerologická ambulance Mohelnice
vedoucí lékař prim. MUDr. Lubomír Drlík
1; Dermatohistopatologická laboratoř s. r. o., Praha
vedoucí doc. MUDr. Lumír Pock, CSc.
2
:
Čes-slov Derm, 90, 2015, No. 6, p. 243-247
:
Case interpretation
Palmoplantární keratodermie jsou rozsáhlou heterogenní skupinou získaných a dědičných kožních poruch. Etiopatogenetické vztahy mezi jednotlivými formami byly částečně objasněny pomocí moderních molekulárních genetických metod. Základem stanovení diagnózy je v současnosti klinický, histopatologický obraz a molekulární diagnostika. Terapie je symptomatická a cílem je zmenšit rozsah postižení, snížit palmoplantární hyperkeratózu. V naši práci představujeme případ matky a dcery s keratodermia punctata palmaris et plantaris typ I.
Klíčová slova:
keratodermia punctata palmaris et plantaris – typ 1
ÚVOD
Palmoplantární keratodermie jsou heterogenní skupinou kožních poruch charakterizovaných ztluštěním kůže na dlaních a ploskách [12, 14]. Tato často nesprávně diagnostikovaná onemocnění představují pro pacienta významné omezení pracovních možností i kvality života [11, 15]. Správná diagnóza, respektive diferenciální diagnóza pomáhá stanovit vhodnou a účinnou terapii. V našem sdělení představujeme případ matky a dcery s keratosis punctata palmoplantaris typu 1 (KPPP-1).
POPIS PŘÍPADU
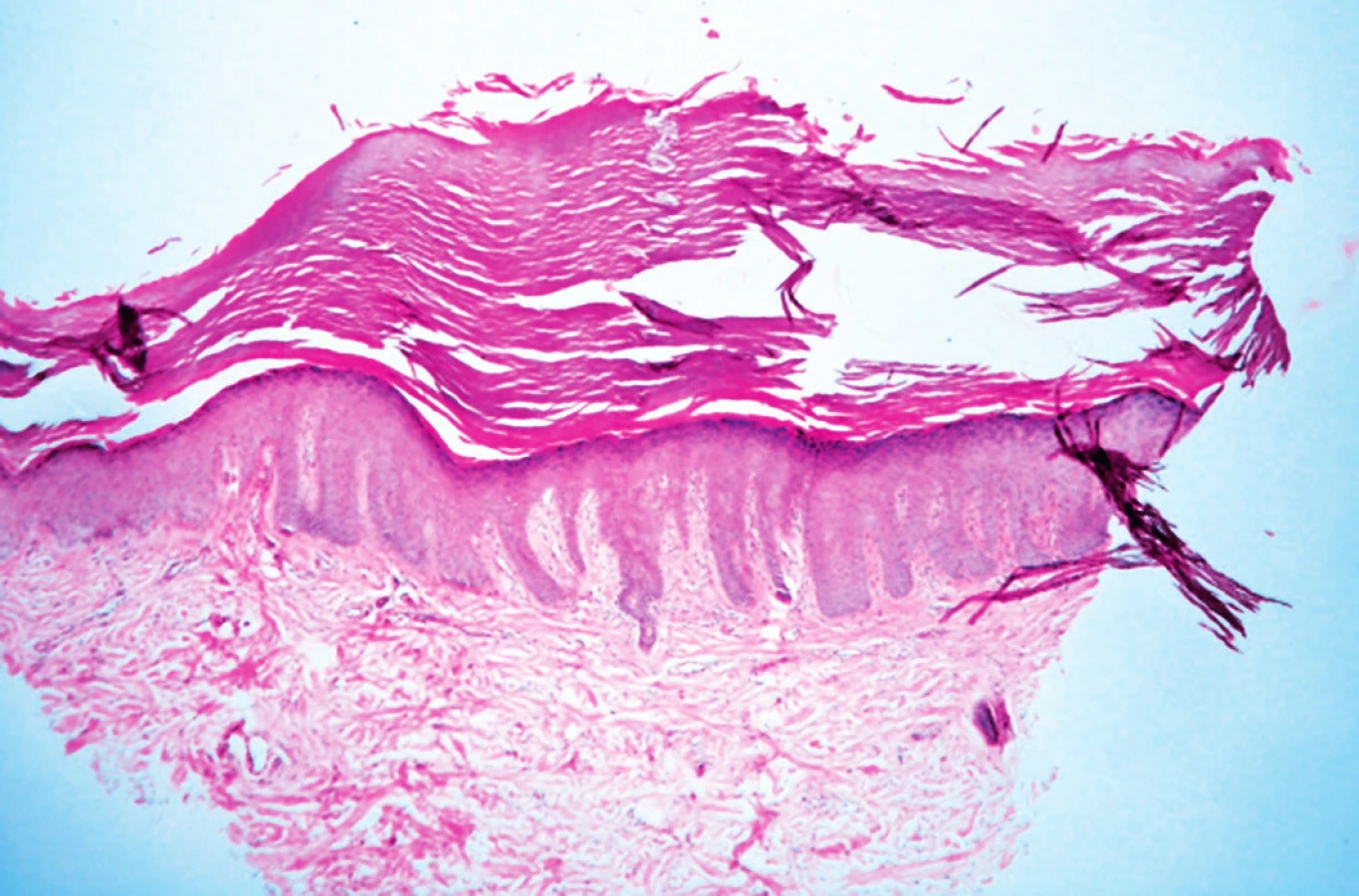
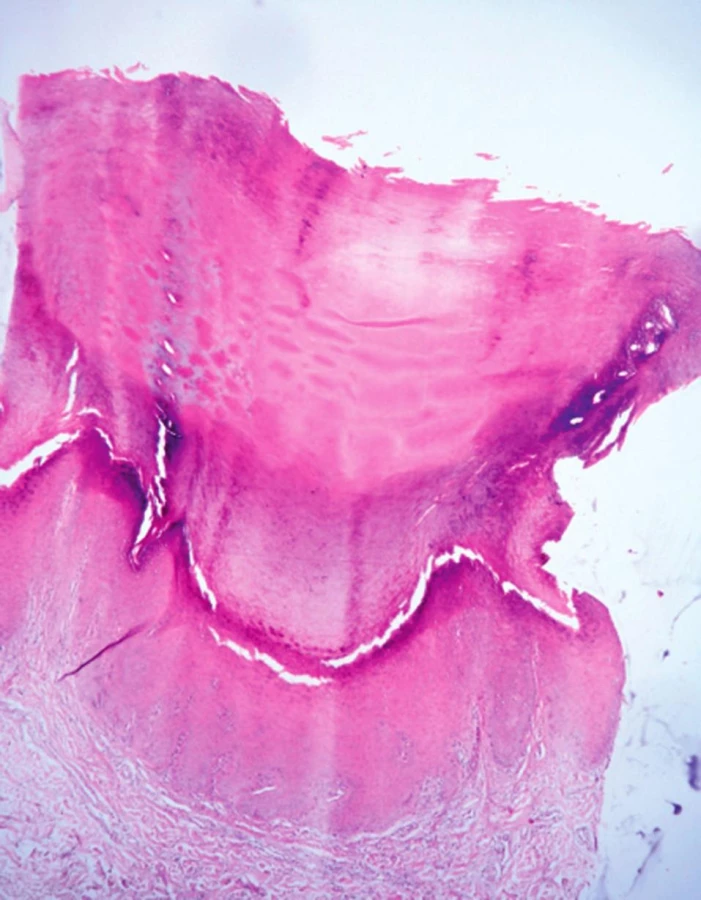
Žena, ve věku 48 let, navštívila v roce 2009 kožní ambulanci s nálezem hyperkeratotických papul na dlaních a ploskách (obr. 1a, 1b). Celkově byla pacientka v dobrém zdravotním stavu, léčila se s esenciální hypertenzí a endogenní depresí, užívala lercanidipin hydrochlorid, losartanum kalicum, hydrochlorothiazidum a sertralinhydrochlorid. Prodělala hysterektomii pro metroragie a operaci polypů paranazálních dutin. V rodinné anamnéze pacientka uvedla výskyt melanomu a karcinomu prsu u sestry, hematologické maligní onemocnění u matky a opakované cévní mozkové příhody u otce. Podobné kožní projevy u rodinných příslušníků neudávala. Standardní laboratorní vyšetření neprokázala, vyjma mírně zvýšené hladiny LDL cholesterolu, žádné patologie. Projevy se manifestovaly kolem puberty na dlaních, následně se objevily i na ploskách. Od 40. roku věku pozorovala značné zhoršení, papuly se zvětšily a přibyly na počtu. Histologické vyšetření materiálu z probatorní excize prokázalo mohutné stratum corneum, ve str. Malpighi bez průkazu zánětlivých změn. V lednu 2010 byl nasazen celkově acitretin v dávce 0,7 mg/kg/den, který pacientka užívala po dobu necelého roku. Tato léčba měla zpočátku dobrý efekt, projevy byly plošší, ale později se objevily komplikace ve formě infekčních koutků a paronychií a léčba byla ukončena. V roce 2011 pacientka podstoupila druhou kúru léčby acitretinem v dávce 0,5–0,7 mg/ kg/den s podobně dobrým efektem, která ale v listopadu 2011 musela být ukončena pro komplikace – cheilitis sicca a zvýšení transamináz. Opakované histologické vyšetření v roce 2012 popsalo vkleslinu kožního povrchu o průměru 3 mm vyplněnou hyperkeratotickým čepem, na spodině mírně akantotickou epidermis (obr. 2). Tento histologický nález odpovídal klinické diagnóze punktátní keratodermie. Při hospitalizaci na kožním oddělení v roce 2014 byl nasazen lokální isotretinoin a vazelína s 10 % salicylové kyseliny, po kterých se kožní nález podstatně zlepšil. Tato léčba pokračuje spolu s pravidelnou pedikérskou péčí – obrušováním frézou, změkčováním koupelemi. Začátkem roku 2014 navštívila kožní ambulanci 26letá dcera naší pacientky, která od puberty pozorovala podobné, ale méně vyjádřené hyperkeratotické papuly na ploskách. Klinický obraz a histologické vyšetření (výrazná hyperkeratóza na povrchu epidermis, v centru miskovitá vkleslina kožního povrchu o průměru 1,5 mm, na jejíž spodině je rozšířené stratum granulosum i spinosum, nad ní pak mohutný hyperkeratotický čep) potvrdily diagnózu keratosis punctata palmoplantaris typu 1 (obr. 3). Byla nasazena lokální keratolytika – střídavě vazelína s 10 % salicylové kyseliny a ambiderman s 10 % urey v okluzích s podstatným zlepšením stavu – projevy byly méně nápadné, nevystupovaly nad povrch okolní kůže, nevadily při chůzi.


DISKUSE
Palmoplantární keratodermie (PPK) představují velkou skupinu klinicky podobných kožních poruch s rozdílnou etiologií. Klasifikace se vyvíjela během minulého století, ale matoucí termíny a nomenklatura omezovaly její konsenzuální použití [2]. Základní rozdělení, které rozlišuje palmoplantární keratodermie do tří hlavních skupin – difuzní, fokální a punktátní, je založeno na klinickém obraze, ale nemoci v rámci jedné skupiny mohou mít různý etiopatogenetický základ. U každé skupiny se rozlišují získané a dědičné varianty PPK (tab. 1).
![Klinické varianty punktátních palmoplantárních keratodermií [21]](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f4267afb2957ccef56954279ec1d28a1.jpg)
Keratosis punctata palmaris et plantaris typ 1 (KPPP-1) (syn. Buschke-Fischer-Brauerova nemoc, keratoderma dissipatum, keratoderma punctata – papulosa, diseminovaný clavus, papulotranslucentní acrokeratodermie) byla popsána německými dermatology Buschkem a Fischerem v roce 1910, o tři roky později Brauer potvrdil způsob její dědičnosti [3]. KPPP-1 je autozomálně dominantní onemocnění s variabilní penetrancí, incidencí přibližně 1/100 000, se stejnou distribucí mezi muži a ženami, existují však i sporadické případy na podkladě spontánních mutací [2]. Jde tedy o velmi vzácné onemocnění na rozdíl od jiných forem palmoplantárních keratodermií [8]. Klinické příznaky se obvykle manifestují v druhém až třetím deceniu malými hyperkeratotickými papulami na dlaních a ploskách [11, 16]. Projevy začínají jako špendlíčkovité tvrdé průsvitné papuly velikosti od 1 do 10 mm, které mohou zůstat průsvitné nebo se časem stanou neprůsvitnými a verukózními. Někdy se v centru nachází keratotické jádro, které po odstranění zanechá centrální depresi. Vlivem mechanického tření se projevy zvětšují až do mozolovitých projevů [19]. Postižení plosek (a také dlaní u manuálních pracovníků) bývá výraznější v důsledku většího tlaku v těchto lokalitách [6, 12]. Pacienti mohou v postižených místech pociťovat svědění, méně často i bolestivost, onemocnění se může v rámci jedné rodiny projevovat širokým spektrem fenotypických variant. V literatuře byly popsány asociace KPPP-1 s dalšími poruchami – lipomatózou, spastickou paralýzou, ankylozující spondylitidou nebo sebaceózní hyperplazií [5, 21]. Schöpf-Schulz-Passarge syndrom je raritní autozomálně recesivní ektodermální dysplazie, kde kromě punktátní PPK jsou přítomny palpebrální apokrinní hidrocystomy, hypotrichóza, hypodontie a dystrofické změny nehtů. V nejasných případech pomáhá stanovit správnou diagnózu histologické vyšetření. V histologickém obraze je hlavním projevem výrazná kompaktní sloupcovitá ortohyperkeratóza omezená na ostře ohraničený okrsek epidermis a ztluštění stratum granulosum [1, 2, 4, 12]. V diferenciální diagnostice je nutno vyloučit hlavně ostatní dědičné a získané keratodermie, virové bradavice, callus, pitted keratolysis a névoidní bazaliom [21]. Mezi získané formy patří arzénové keratózy, PPK asociovaná s vyšším věkem a malignitami vnitřních orgánů, idiopatická filiformní porokeratotická PPK (spojena s karcinomy prsou, ledvin, plic a kolorekta) a punktátní keratodermie palmárních záhybů [7], která se vyskytuje téměř výlučně u černochů a projevuje se tuhými bradavičnatými lézemi na záhybech prstů nebo v dlaňových rýhách s hyperkeratózou, je lokalizovaná v místech akrosyringií, proto je často považována za nemoc potních žláz [17]. Mezi další dědičné formy keratodermie patří KPPP typ 2 (syn. porokeratosis Mantoux, keratoderma spinosum, porokeratosis punctata palmaris et plantaris), která se projevuje nejdříve ve druhém deceniu. Klinický obraz je tvořený drobnými keratotickými papulami, které pokrývají povrch celé plosky a dlaně [10]. V histologickém obraze je typická přítomnost sloupcovité parakeratózy, čímž se odlišuje od KPPP-1. Punktátní palmoplantární keratodermie typ 3, fokální akrální hyperkeratóza, se vyznačuje tvorbou oválných nebo polygonálních kráterovitých keratotických papul na okrajích plosek, dlaní a zápěstí [22]. Vývoj moderních molekulárních genetických metod umožnil postupně objasnit etiopatogenezi palmoplantárních keratodermií [13, 14]. Na základě klinického obrazu se předpokládalo, že poruchy budou lokalizovány v genech kódujících keratiny. Později byly skutečně prokázány mutace v genech keratinů 1 a 9 u některých typů keratolytických difuzních keratodermií, jako např. nemoci Unna-Thostové, ale bylo zjištěno, že paleta genetických poruch je stejně pestrá jako rozmanitost klinických variant [13]. U KPPP-1 byla molekulární příčina objasněna až v roce 2012 pomoci vazebné analýzy a sekvenování celého exonu. Tyto metody prokázaly heterogenní nulové mutace v genu AAGAB, který je zodpovědný za vznik KPPP-1 [17]. Gen AAGAB kóduje alfa a gama adaptin vázající protein p34, který hraje důležitou roli v buněčném vezikulárním transportu. Dodnes bylo zjištěno 20 nulových variant ve skotských, irských, anglických, německých, tuniských, čínských, mexických a japonských populacích [9]. Hlubší analýza genového pozadí umožní vypracovat genetický klasifikační systém, který by v budoucnosti nahradil stávající klasifikaci [18, 20].
Terapie je pouze symptomatická a po vysazení léčby dochází k relapsu projevů. Ke změkčení lézí se používají keratolytika. S úspěchem jsou podávány také lokální i systémové retinoidy, nicméně nežádoucí účinky těchto léků značně omezují jejich rutinní používaní [16]. Etretinát a acitretin jsou stejně účinné. Pro vyvarování se dlouhodobých nežádoucích účinků je vhodná přerušovaná terapie. Důležité je mechanické snížení hyperkeratotické epidermis dermabrazí nebo pomocí CO2 laseru. V průběhu dlouhodobého ošetřování nesmí být také opomenuta případná možnost mykotické nebo bakteriální superinfekce. Své zastoupení v rámci ošetřovatelské péče má i pravidelná pedikúra.
Do redakce došlo dne 3. 11. 2015.
Adresa pro korespondenci:
MUDr. Lilla Smižanský-Bari
Dermatologická ambulance Mohelnice
Nádražní 35
789 85 Mohelnice
e-mail:barililla@hotmail.com
Sources
1. ASADI, M. D. Type I hereditary punctate keratoderma. Dermatol. Online J., 2003, 9, 4, p. 38.
2. BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. Dermatology. 2nd ed., vol. 1, 2008, p. 787–788, ISBN 9781416029991.
3. BUSCHKE, A., FISCHER, W. Keratodermia maculosa disseminata symmetrica palmaris et plantaris. Ikonographia Dermatologica, 1910, 5, p. 183–192.
4. CALONJE, J. E., BRENN, T., LAZAR, A. J., MCKEE, P. H. McKee’s Pathology of the Skin. 4th ed., 2014, p. 75, ISBN 13-9781416056492.
5. CASTORI, M., RUGGIERI, S., GIANNETTI, L. et al. Schöpf-Schulz-Passarge syndrome: further delineation of the phenotype and genetic considerations. Acta Derm. Venereol., 2008, 88, 6, p. 607–612.
6. EMMERT, S., KÜSTER, W., ZUTT, M. et al. A new family with the rare genodermatosis keratosis punctata palmoplantaris Buschke-Fischer-Brauer. JAAD, 2003, 49, 6, p. 1166–1169.
7. ENA, P., COTTONI, F., CERIMELE, D. et al. Association of keratoderma punctata palmaris et plantaris with other morbid conditions (early grayness, carcinoma of the colon). Study of 3 families. G. Ital. Dermatol. Venereol., 1986, 121, p. 45–54.
8. GAMBORG NIELSEN, P., HOFER, P. A., LAGERHOLM, B. The dominant form of hereditary palmoplantar keratoderma in the northernmost county of Sweden (Norrbotten). Dermatology, 1994, 188, 3, p. 188–193.
9. GIEHL, K. A., ECKSTEIN, G. N., PASTERNACK, S. M. et al. Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type Buschke-Fischer-Brauer. Am. J. Hum. Genet., 2012, 91, 4, p. 754–759.
10. GRILLO, E., PÉREZ-GARCÍA, B., GONZÁLEZGARCÍA, C. et al. Spiny keratotic projections on the palms and fingers. Spiny keratoderma. Dermatol. Online J., 2012, 18, 6, p. 8.
11. ITIN P. H., FISTAROL S. K. Palmoplantar keratodermas. Clin. Dermatol., 2005, 23, 1, p. 15–22.
12. JUDGE, M. R., McLEAN, W. H. I., MUNRO, C. S. Disorders of Keratinization. In Burns, T., Breathnach, S., Cox, N. Griffiths, C., editors. Rook’s Textbook of Dermatology. 8th ed., 2012, vol. 1, p. 9.93–19.119, ISBN 978-1-4051-6169-5.
13. KELSELL, D. P., STEVENS, H. P., RATNAVEL, R. et al. Genetic linkage studies in non-epidermolytic palmoplantar keratoderma: evidence for heterogeneity. Hum. Mol. Genet., 1995, 4, 6, p. 1021–1025.
14. KIMYAI-ASADI, A., KOTCHER, L. B., JIH, M. H. The molecular basis of hereditary palmoplantar keratodermas. JAAD, 2002, 47, 3, p. 327–343.
15. LUCKER, G. P., VAN DE KERKHOF, P. C., STEIJLEN, P. M. The hereditary palmoplantar keratoses: an updated review and classification. Br. J. Dermatol., 1994, 131, 1, p. 1–14.
16. OZTAS, P., ALLI, N., POLAT, M. et al. Punctate palmoplantar keratoderma (Brauer-Buschke-Fischer syndrome). Am. J. Clin. Dermatol., 2007, 8, 2, p. 113 – 116.
17. PIÉRARD-FRANCHIMONT, C., PIÉRARD, G. E., MELOTTE, P. et al. Keratosis punctata of the palmar creases. Ann. Soc. Belg. Med. Trop., 1989, 69, 3, p. 257–261.
18. POHLER, E., HUBER, M., BOONEN S. E. et al. New and recurrent AAGAB mutations in punctate palmoplantar keratoderma. Br. J. Dermatol., 2014, 171, 2, p. 433–436.
19. RATNAVEL, R. C., GRIFFITHS, W. A. The inheritedpalmoplantar keratodermas. Br. J. Dermatol., 1997, 137, 4, p. 485–490.
20. STEVENS, H. P., KELSELL, D. P., BRYANT, S. P. et al. Linkage of an american pedigree with palmoplantar keratoderma and malignancy (palmoplantar ectodermal dysplasia type III) to 17q24. Arch. Dermatol., 1996, 132, 6, p. 640–651.
21. TORRES, G., BEHSHAD, R., HAN, A. et al. I forgot to shave my hands: A case of spiny keratoderma. JAAD, 2008, 58, 2, p. 344–348.
22. VAN STEENSEL, M. A., FRANK, J. Focal acral hyperkeratosis and acrokeratoelastoidosis: birds of a feather? JEADV, 2009, 23, 9, p. 1113–1114.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2015 Issue 6
Most read in this issue
- Up-to-Date Review on Pustular Psoriasis and its Management
-
Keratodermia punctata palmaris et plantaris typ 1:
popis případu matky a dcery - Psoriasis Treatment with Adalimumab in Clinical Practice: Long-Term Experience of a Center for Biological Therapy of Psoriasis