
Lokalizovaná sklerodermie – morfea:
současný stav a možnosti léčby
Localised Scleroderma and Morphea: Current State of Knowledge and Therapy
Localized scleroderma (LS) or morphea are terms encompassing a group of rare chronic inflammatory fibrosing disorders of the skin and underlying structures such as subcutaneous fat tissue, fascia, muscle and bone. As, in contrast to systemic sclerosis, internal organs like GIT, lungs, heart and kidney are not affected and the disease has normal life expectancy, some classifications use term morphea for all subtypes in order to avoid unnecessary confusion with systemic form, other use term LS. The subtypes of the disease include plaque, generalized, linear, deep and mixed variants, some classifications comprise also eosinophilic fasciitis and atrophoderma of Pasini-Pierini. The treatment should start early in the course of the disease before complications occur as these are difficult to treat. The main indication criteria for the treatment are based on disease activity, subtype of the disease and depth of involvement. Current treatment options for superficial forms include phototherapy and topical therapy with corticosteroids, tacrolimus, calcipotriol, alone or in combination with bethamethason dipropionate, some studies suggestt the use of imiquimod. The well - established systemic therapy for severe forms include systemic methotrexate alone or in combination with systemic corticosteroids, in treatment resistant cases the use of mycophenolate mofetil is promissing. It seems that prolonged systemic therapy with methotrexate for 1–2 years reduce relapses of the disease that are observed in one quarter of patients.
Key words:
localized scleroderma – morphea – classifications – therapy – course
:
J. Štork
:
Dermatovenerologická klinika 1. LF UK a VFN, Praha
přednosta prof. MUDr. Jiří Štork, CSc.
:
Čes-slov Derm, 91, 2016, No. 6, p. 258-271
:
Reviews (Continuing Medical Education)
Lokalizovaná sklerodermie (LS) a morfea jsou termíny zahrnující skupinu vzácných chronických zánětlivých fibrotizujících nemocí kůže, případně podkožní tukové tkáně, svalu až kosti. Na rozdíl od systémové sklerózy však nepostihují vnitřní orgány, jako je gastrointestinální trakt, srdce, plíce a ledviny a nejsou spojena se zkráceným přežitím nemocných, některé klasifikace proto užívají souhrnného termínu morfea pro všechny formy tohoto onemocnění, aby nedocházelo k zaměňování se systémovou formou, jiné užívají souhrnný termín LS. Typy onemocnění zahrnují ložiskovou, generalizovanou, lineární, hlubokou, a smíšenou formu, některé klasifikace zahrnují i eozinofilní fasciitidu a atrophodermia Pasini-Pierini. Léčba má být zahájena v časné fázi nemoci před vznikem komplikací, protože tyto se obtížně léčí. Hlavní ukazatele pro zahájení léčby představují známky aktivity nemoci, forma nemoci a hloubka postižení. Současné možnosti léčby povrchnějších forem zahrnují fototerapii, lokální aplikaci kortikoidů, takrolimu, kalcipotriolu v monoterapii či v kombinaci s betametazonem dipropionátem, některé práce uvádějí imiquimod. Pro těžší formy onemocnění je nejvíce zavedená systémová léčba metotrexátem v monoterapii či v kombinaci se systémovými kortikosteroidy, v rezistentních případech je příslibem mykofenolát mofetil. Zdá se, že protrahovaná systémová léčba metotrexátem po 1–2 roky snižuje riziko recidiv nemoci, které jsou pozorovány kolem čtvrtiny nemocných.
Klíčová slova:
lokalizovaná sklerodermie – morfea – klasifikace – terapie – průběh
ÚVOD
Sklerodermie jsou heterogenní skupinou onemocnění charakterizovaných vznikem tuhé kůže. Lokalizovaná sklerodermie (LS), též nazývaná morfea, je onemocnění postihující fibrotickým procesem kůži, případně podkožní tukovou tkáň, sval a kost. Mnozí odborníci doporučují užívání souhrnného termínu morfea pro všechny formy tohoto onemocnění, protože lokalizovaná sklerodermie, na rozdíl od systémové sklerózy (sklerodermie), zpravidla nepostihuje vnitřní orgány, jako je gastrointestinální trakt, srdce, plíce a ledviny, a není spojena se zkráceným přežitím nemocných [10, 11]. Důvodem je předejít zbytečnému zneklidnění a zaměňování těchto dvou chorob jak u nemocných, tak lékařů. I když se u obou onemocnění uplatňují podobné patogenetické pochody, jedná se o různá onemocnění, která se vzácně mohou vyskytovat současně, ale nepřechází jedno v druhé [25].
Jedná se o relativně vzácné onemocnění s incidencí výskytu 0,4–2,7/100 tisíc obyvatel [37, 28, 40] s častějším postižením žen v poměru k mužům 2,6–6 : 1 [44]. Průzkum juvenilní formy v Anglii a Irsku ukázal incidenci 0,34 případů na 100 tisíc dětí [15]. U dětí je nejčastější forma lineární vznikající u 90 % mezi 2.–14. rokem věku, u dospělých ložisková morphea „en plaque“ s nejčastějším výskytem mezi 40.–50. rokem věku.
Patogenetické mechanismy vedoucí ke zvýšené tvorbě kolagenu a extracelulární matrix nejsou jasné. Předpokládá se kombinace genetické predispozice a zevních vlivů, která vede k expresi adhezních molekul, jako např. adhezních molekul cévního endotelu (VCAM-1), intercelulárních adhezních molekul-1 (ICAM-1), aktivaci T lymfocytů a k tvorbě profibrotických cytokinů, jako je transformující růstový faktor beta (TGF-beta), destičkový růstový faktor, růstový faktor pojivové tkáně, interleukinů 4, 6 a 8 [16, 23, 24, 29]. Aktivace těchto prozánětlivých a profibrotických pochodů vede pak ke zvýšené tvorbě kolagenu a snížené tvorbě matrixových metaloproteináz odpovědných za degradaci kolagenu [24].
Na propuknutí nemoci se mohou podílet různé spouštěcí faktory. Z infekčních faktorů se zvažuje role borrelia sp., avšak zatímco některé evropské studie tyto mikroorganismy prokazují, americké studie tuto souvislost nepotvrzují, a proto zůstává patogenetická role borelií sporná [10, 24]. Role mechanického traumatu se pozoruje asi u 16 % nemocných [11, 14]. Postradiační morfea vzniká asi u 1 z 500 ozařovaných nemocných, přičemž u 20 až 25 % vzniká i mimo ozařovanou oblast [3, 45]. V případech, kde je nezbytná léčba chirurgická či radioterapie, nepředstavuje morfea jejich kontraindikaci. Spojitost vzniku morfey s aplikací léků byla pozorována u bleomycinu, D-penicillaminu, vitaminu K1, L-5 - hydroxytryptofanu v kombinaci s karbidopou, balicatibu aj. Pro autoimunitní povahu nemoci svědčí sdružení s autoimunitními chorobami – u dětí ve 2–5 % a u dospělých okolo 30 % se nachází choroby jako vitiligo, inzulin dependentní diabetes mellitus, Hashimotova tyreoiditida, Gravesova choroba, ulcerózní kolitida, psoriáza. Rodinná anamnéza bývá pozitivní ve 12–23,8 % u dětí a v 10,6 % u dospělých [30, 56]. Antinukleární protilátky (ANA) se nachází u 20 až 80 % nemocných, přičemž jejich titr nekoreluje s průběhem ani závažností nemoci. Jedná se především o protilátky antinukleární (ANA) s homogenním typem imunofluorescence, protilátky proti jednovláknové DNA (anti ss-DNA), protilátky antihistonové, revmatoidní faktor [10].
KLASIFIKACE
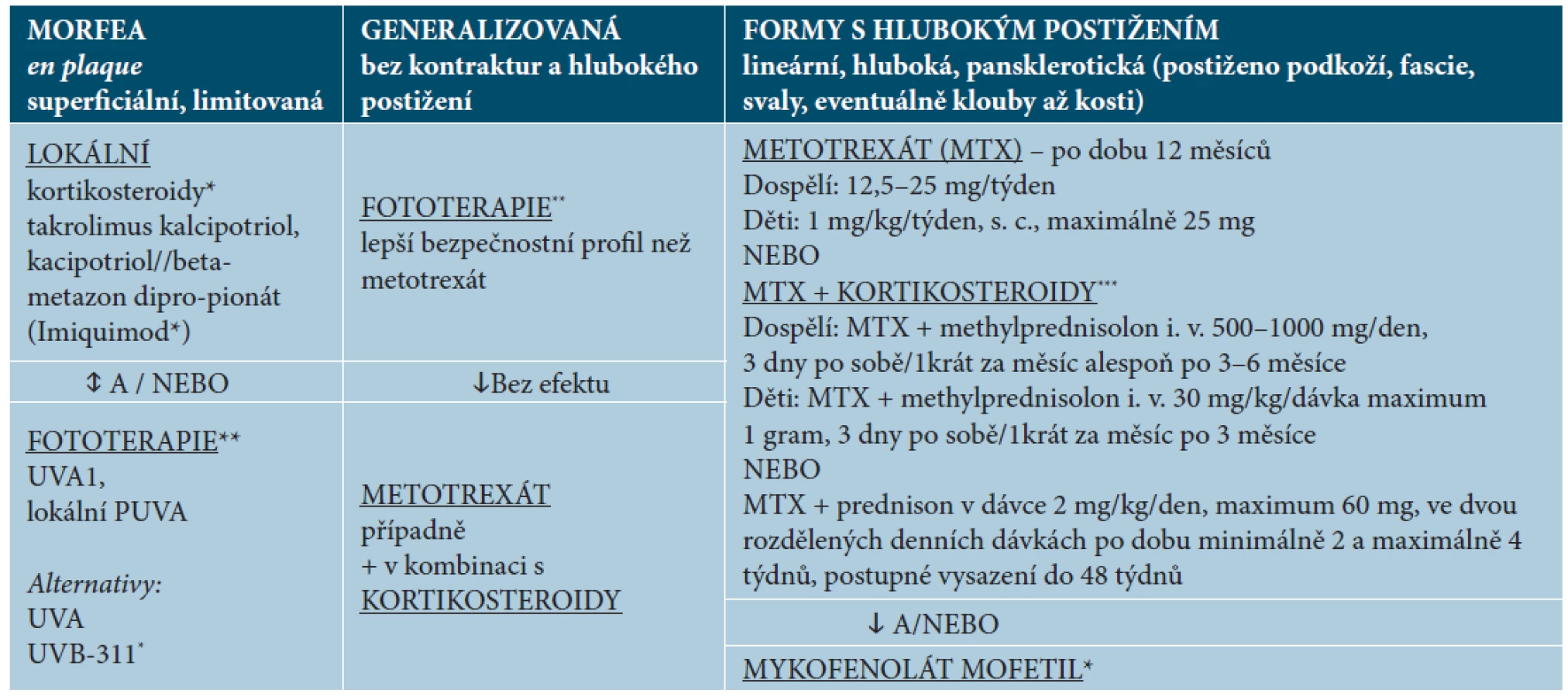
Termíny „lokalizovaná sklerodermie“ a „morfea“ bývají v praxi užívány jako synonyma a jsou stále předmětem diskuse. V současné době jsou nejvíce uplatňovány dvě klasifikace onemocnění. Jedna je zastáncem souhrnného termínu morfea pro všechny typy této choroby – tabulka 1 [28, 56]. Druhá klasifikace pocházející od německých autorů, používaná především v Evropě a v tomto sdělení, používá souhrnný termín „lokalizovaná sklerodermie“ (sclerodermia circumscripta) pro všechny formy tohoto onemocnění a termín morfea vyhrazuje jen pro povrchní ložiskové postižení typu „morphea en-plaque“. Toto členění je navrženo s ohledem na rozsah a hloubku postižení, léčbu a průběh rozlišovaných forem – tabulka 2 [25]. Limitované formy trvají do vyhasnutí aktivity choroby u 50 % nemocných kolem 2,7 roku, kdežto formy generalizované, lineární a hluboké kolem 5,5 roku [40].
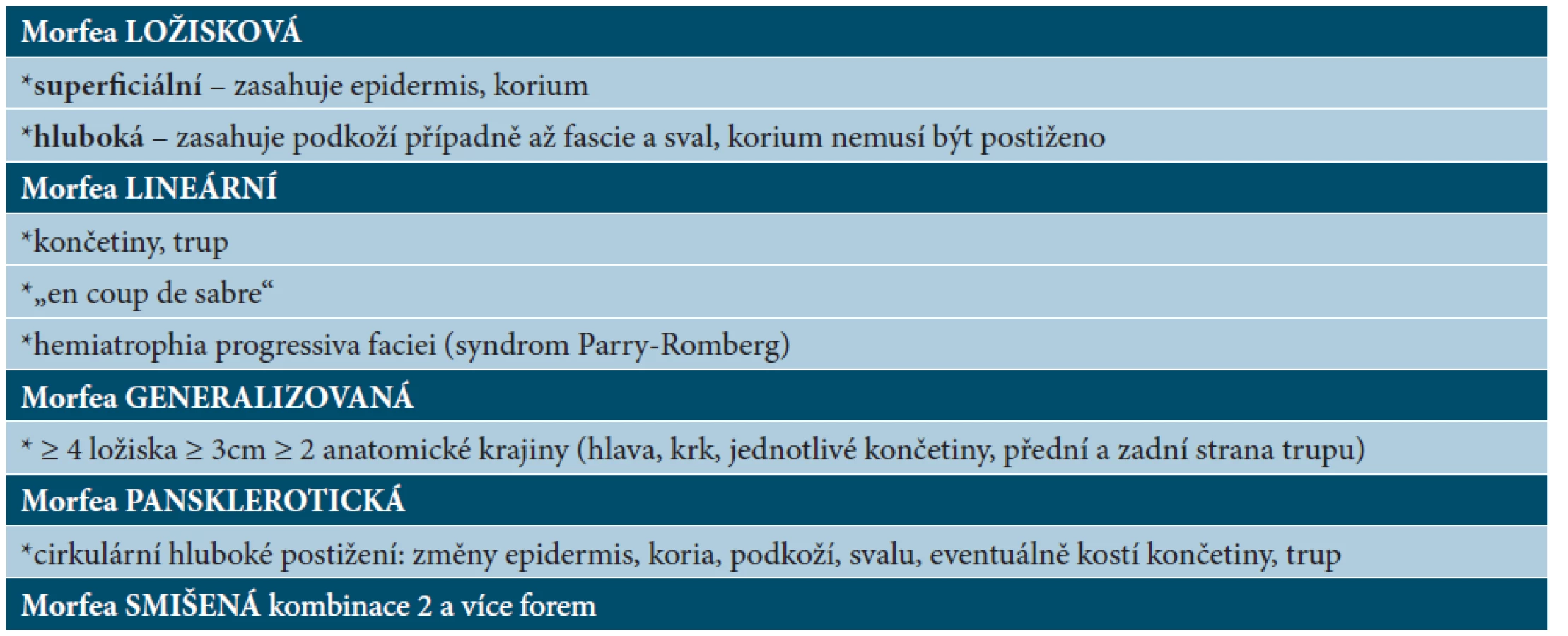

Nejčastější formou onemocnění u dospělých je ložisková morfea („en-plaque“), která je charakterizována superficiálním postižením epidermis a koria. Většinou začíná jako oválné červenofialové ložisko, často s povrchem vzhledu pomerančové kůry, v centru s postupným vznikem bělavé indurace, která je zpočátku lemována erytémem, než přejde v tužší bělavé ložisko v celém rozsahu. Během 3–5 let projev změkne a přejde v šedohnědavou, někdy lehce atrofickou, trvalou hyperpigmentaci (obr. 1) [10]. Nejčastěji postihuje trup jedné až dvou anatomických krajin. Gutátní morfea představuje superficiální indurované léze o průměru do 1 cm (obr. 2). Atrophodermia idiopathica Passini-Pierini, vzhledem k současnému výskytu s morfeou a nálezu mírného zhrubění vaziva koria, je v současné době mnohými odborníky považována za frustní formu morfey [25]. Projevuje se jednotlivými či mnohočetnými, symetrickými, ostře ohraničenými, šedohnědými, měkkými, někdy lehce vkleslými makulami, které jsou stejného vzhledu jako odeznělý projev morfey (obr. 3).

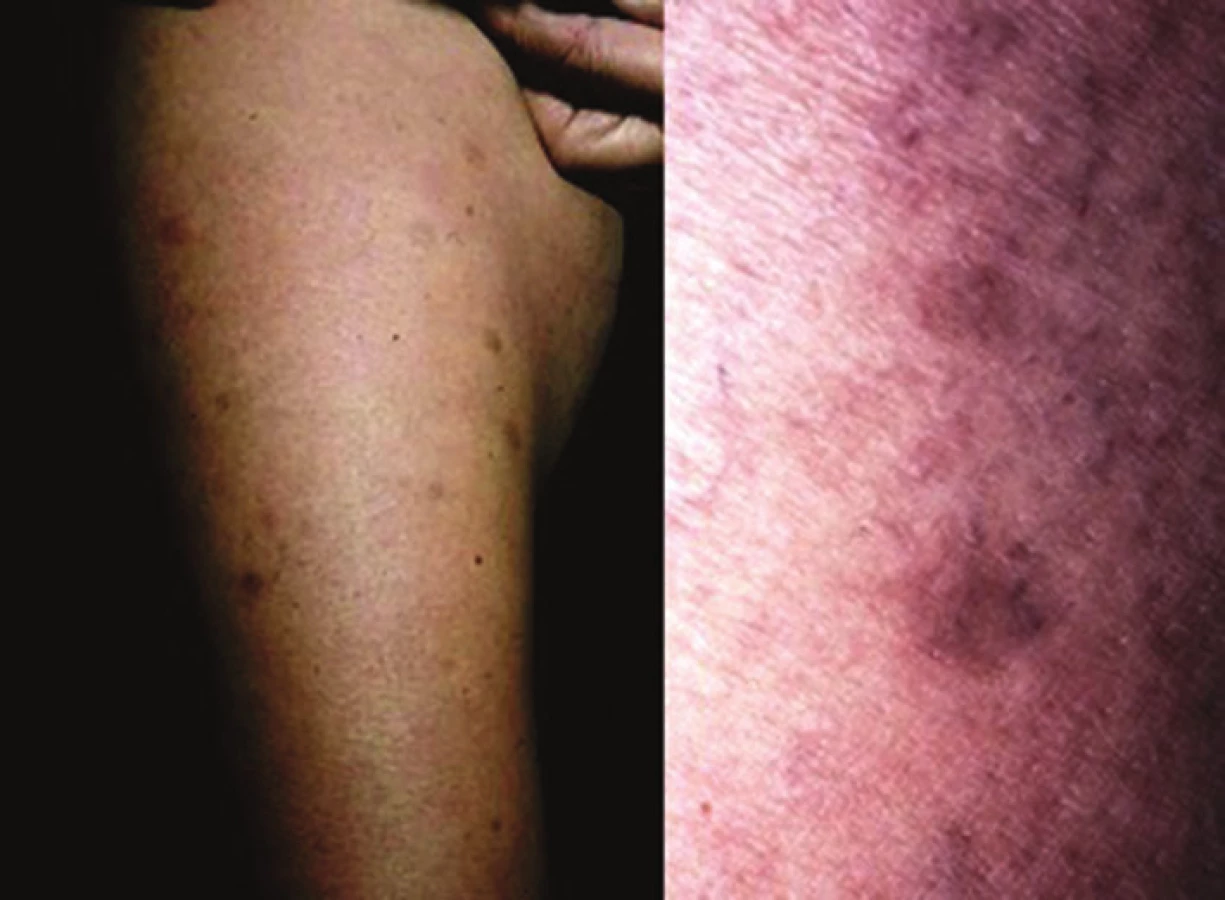

Generalizovaná morfea, podle Laxera [28], je charakterizována vznikem více než čtyř ložisek o průměru přes 3 cm ve dvou a více ze sedmi anatomických krajin (hlava, krk, jednotlivé končetiny, přední a zadní strana trupu). Podle německých autorů je generalizovaná LS charakterizována postižením tří a více anatomických oblastí [25]. Ložiska často symetricky postihují zejména trup a mohou splývat v rozsáhlé indurované plochy (obr. 4).
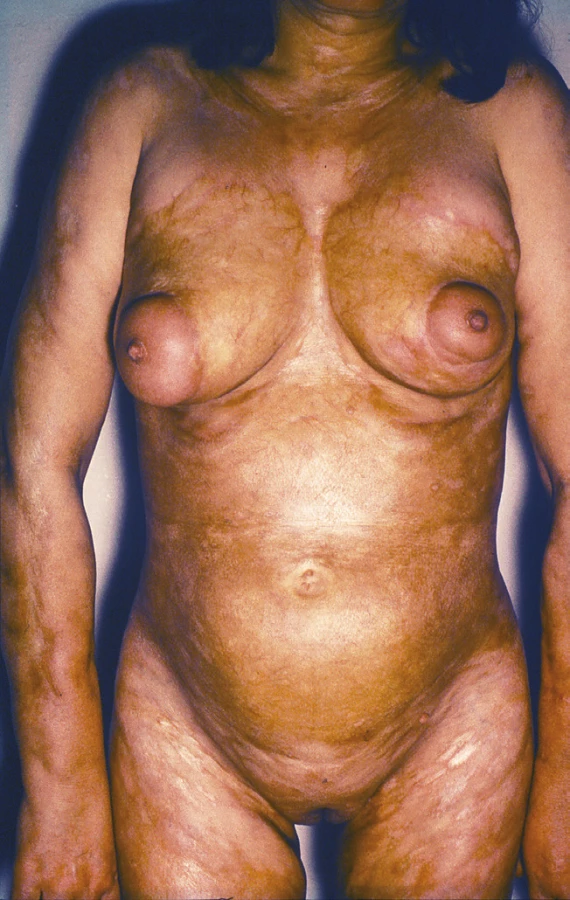
Pansklerotická invalidizující morfea je vzácná forma generalizované LS vznikající často jako smíšená lineární a diseminovaná forma s cirkulárním postižením končetin a/nebo trupu, bez postižení rukou a nohou, která je charakterizována hlubokým fibrotickým procesem koria, podkožní tukové tkáně, fascie, svalu až kosti se vznikem flekčních kontraktur, s možnou zástavou růstu končetiny, vznikem trofických vředů a s nevýrazným sklonem k regresi (obr. 5).
Eozinofilní fasciitida je onemocnění mnohými autory považované za formu generalizované LS [25], primárně postihující zánětlivým fibrotickým procesem fascii, případně podkožní tukovou tkáň, dermis a sval. Klinicky se projevuje náhlým vznikem bolestivých dolíčkujících symetrických otoků (zpravidla končetin, bez postižení rukou a nohou), které přechází v induraci. Retrakce fibrotizovaných vazivových sept podkožní tukové tkáně podmiňuje typický dolíčkovaný, „matracovitý“ vzhled (obr. 6) povrchu kůže v místech s větším tukovým polštářem (nejčastěji vnitřní strany paží, stehen). Laboratorně je provázena zvýšenou sedimentací krve, hypereozinofilií a hypergamaglobulinémií. Asi ve 30 % se nachází projevy ložiskové morfey, které často vznikají před či po akutní fázi fascitidy [33].
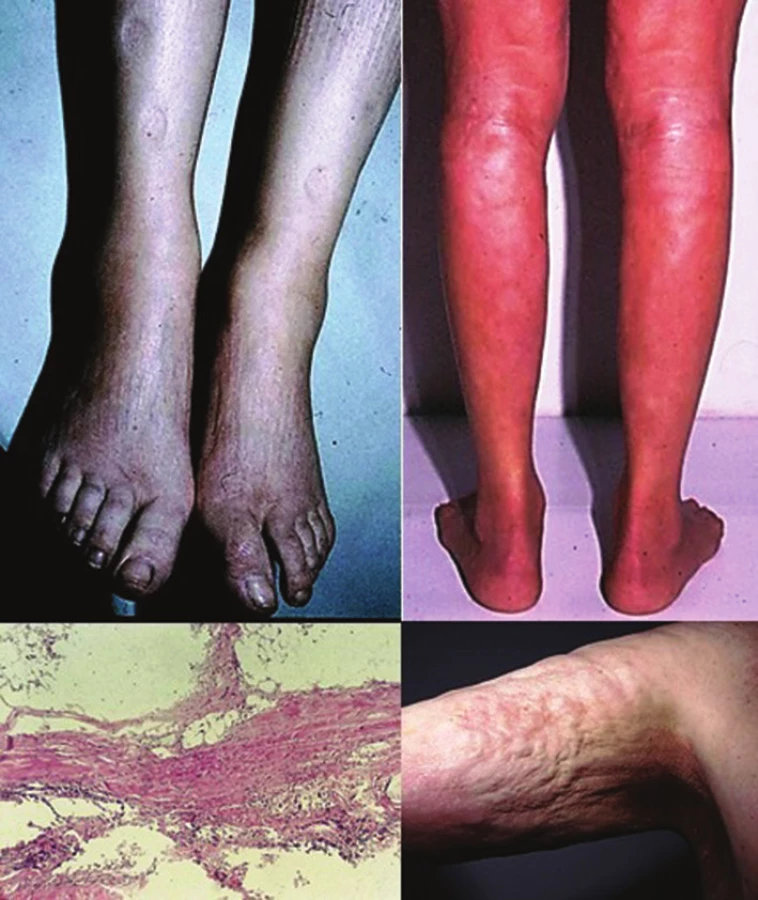
Generalizované formy a eozinofilní fascitida vstupují do diferenciální diagnózy se systémovou sklerózou, která většinou klinicky nečiní obtíže. LS, na rozdíl od systémové sklerózy, zpravidla nepostihuje symetricky ruce, nohy, vynechává podkolení a loketní jamky, oblast axil, hlavu, krk, areoly, nevykazuje kapilaroskopické změny kapilár proximálních nehtových valů, přítomnost teleangiektazií, typické orgánové postižení a nebývá provázena Raynaudovým fenoménem.
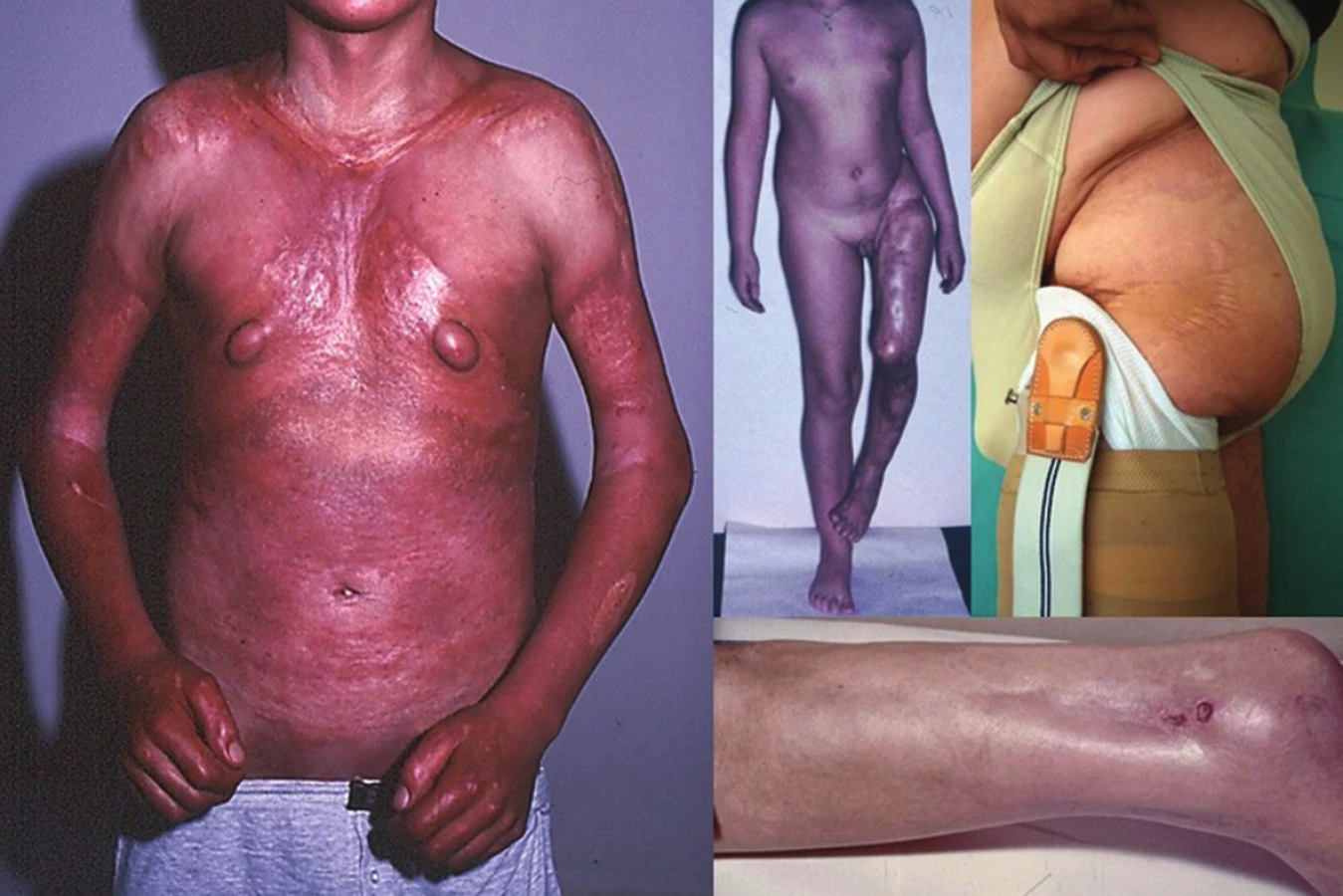
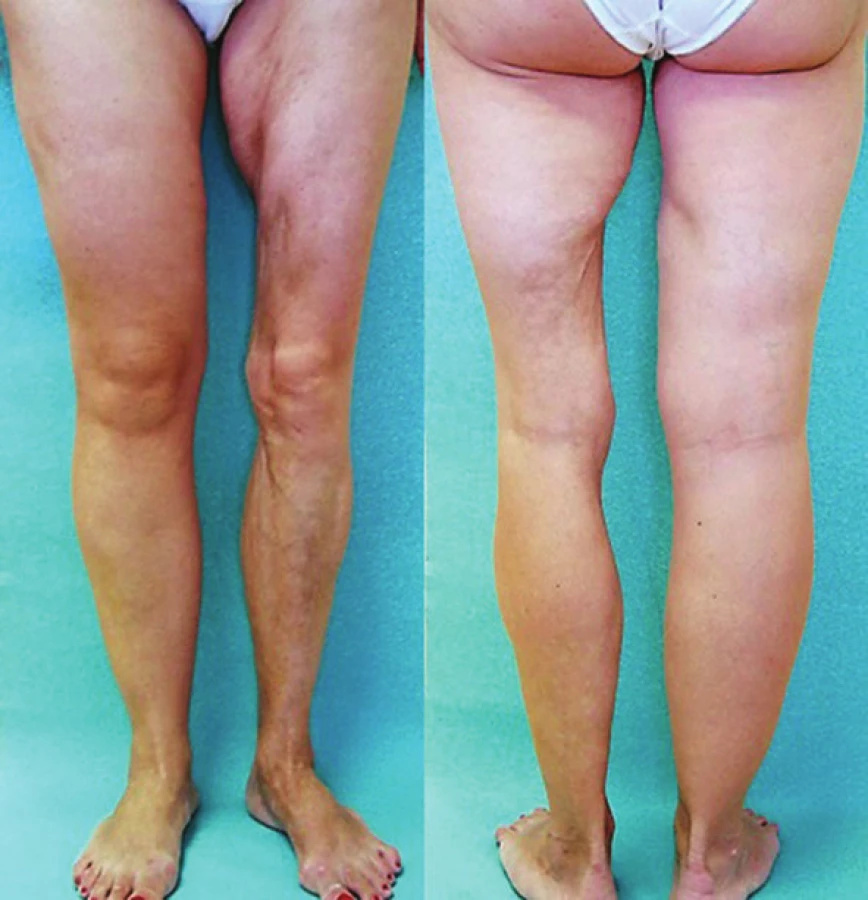
Lineární LS je nejčastější formou u dětí [56]. Projevuje se jako pruhy tuhé kůže, nejčastěji na končetinách, které mohou sledovat Blaschkovy linie. V případě hlubokého postižení může vést v místech přemosťujících klouby k flekčním kontrakturám (obr. 7), atrofii podkoží, případně svalů, a zpomalení růstu končetiny (obr. 8). U dětí má závažnější průběh než u dospělých a častěji vede k výrazným funkčním, kosmetickým a psychologickým následkům s výskytem artralgií a artritid postižené končetiny asi u 30–50 % nemocných. Nejznámější je frontoparietální forma („en coup de sabre“) projevující se sklerotickým pruhem probíhajícím paramediálně na čele s přesahem do kštice, se vznikem jizvící alopecie, vzácně zasahující pod úroveň obočí, která může být spojena s poruchami centrálního nervového systému (kalcifikacemi falx cerebri, cefalgiemi, epileptickými záchvaty) a očními komplikacemi (uveitidou, episcleritidou, změnami adnex víček a poruchou jejich hybnosti) u 3,2 % nemocných dětí (obr. 9). U osob se změnami centrálního nervového systému a známkami aktivity choroby jsou proto doporučovány pravidelné oční kontroly v intervalu čtyř měsíců po první tři roky [56, 55].

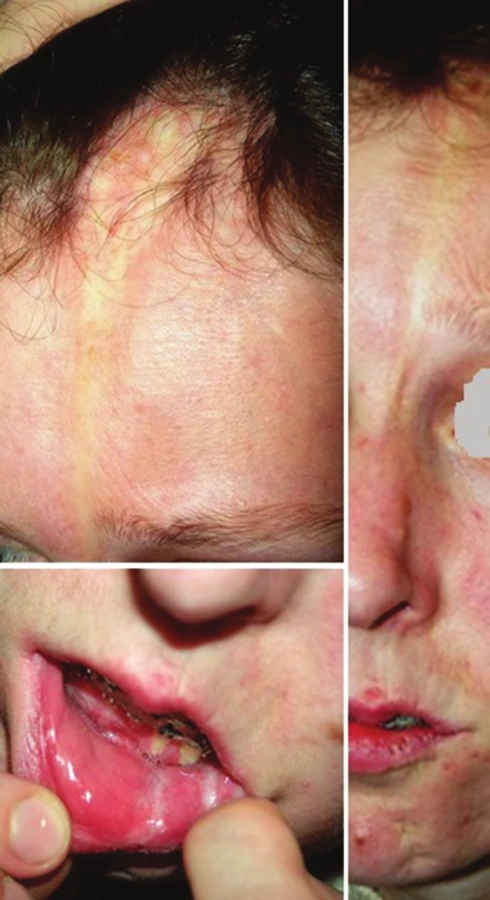
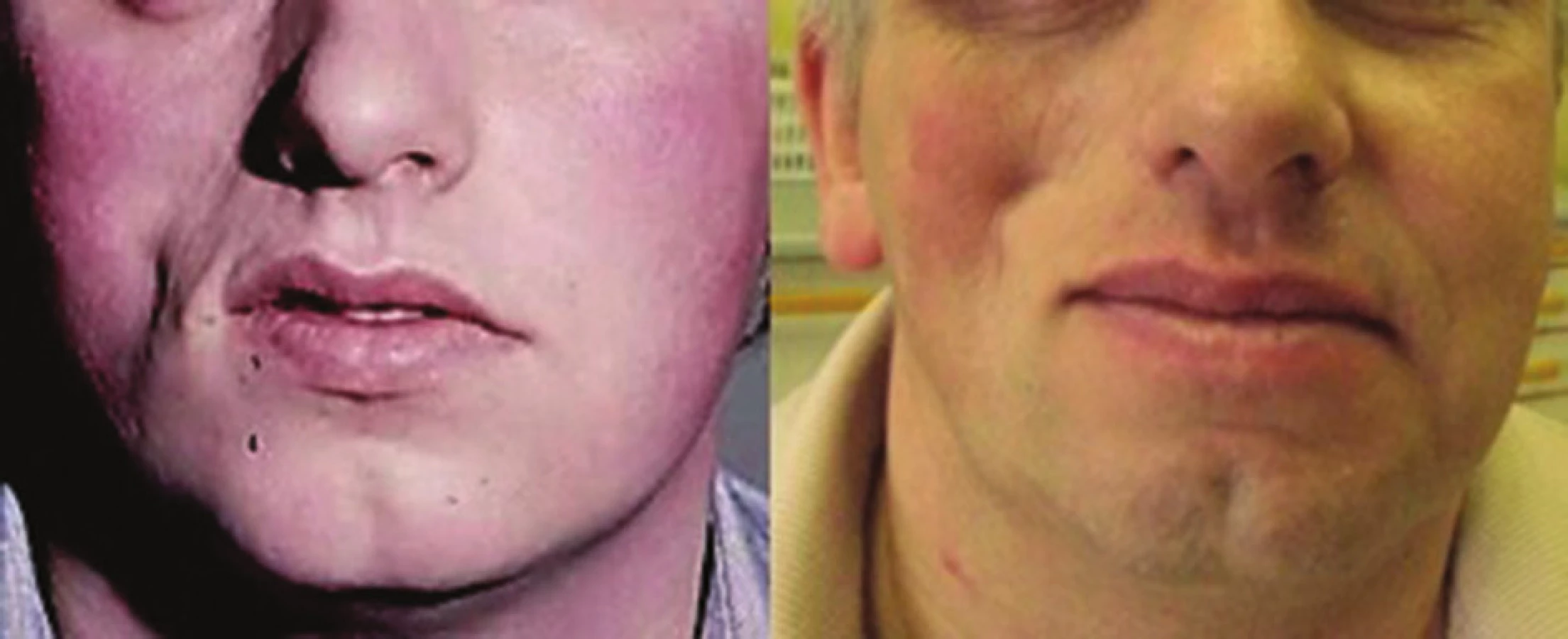
Progresivní faciální hemiatrofie (syndrom Parry-Romberg) (PFH) je onemocnění projevující se primární atrofií podkožní tukové tkáně, případně svalu, chrupavek až kostí vedoucí k těžké asymetrii obličeje, případně poruchám dentice a dentální okluze [47, 48] (obr. 10). Onemocnění vzniká zpravidla do 20 let věku, nejčastěji kolem desátého roku, je pozvolně progredující a vyhasíná do 2–10 let. Vzhledem k častému současnému výskytu lineární LS u PFH (až u 71 % nemocných s PFH) jsou většinou obě tato onemocnění považována za klinické formy stejné choroby [48].
Hluboká LS (morphea subcutanea, profunda) postihuje fibrotickým procesem primárně podkožní tukovou tkáň, případně hluboké korium, fascii až sval. Jedná se o vtažené, ke spodině fixované podkožní indurace, na povrchu je kůže často normálního vzhledu (obr. 11). Může se jednat o solitární projev postihující zejména trup či o léze symetricky lokalizované nejčastěji na končetinách [24, 33].
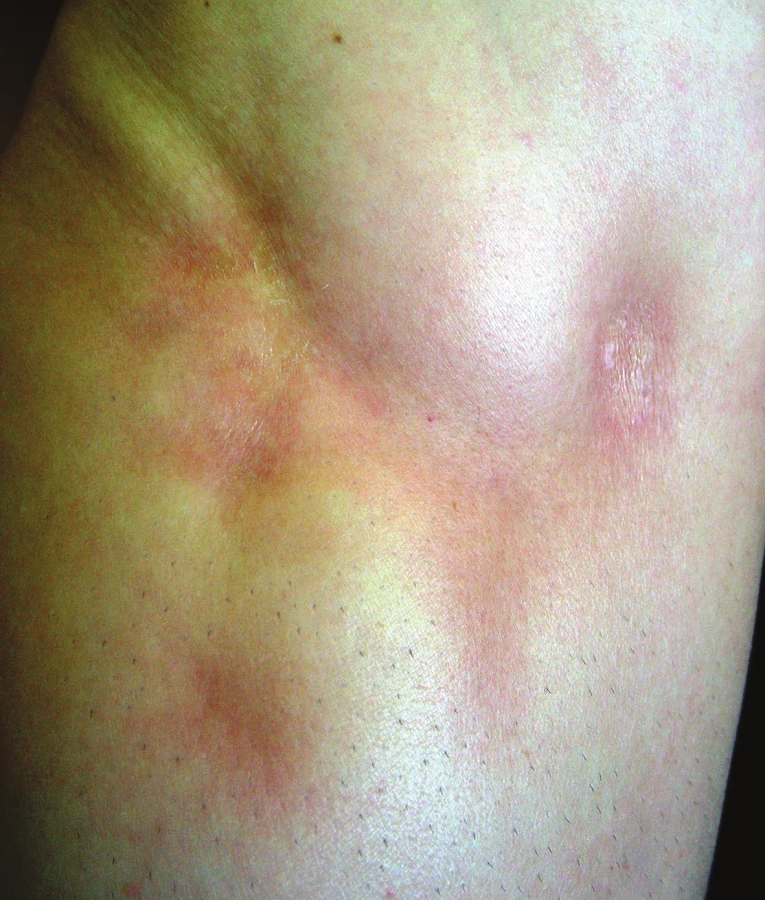
Smíšená forma představuje současný výskyt různých forem, nejčastěji lineární formy s jiným typem postižení, která se objevuje asi u 15 % nemocných [10, 56].
VYŠETŘENÍ
Zpravidla je dostatečné základní sérologické vyšetření zahrnující sedimentaci erytrocytů, C reaktivní protein, krevní obraz včetně diferenciálního počtu leukocytů, základní biochemii a antinukleární protilátky (ANA) (nepřímá imunoflurescence na Hep-2 buňkách) a u eozinofilní fasciitidy též elektroforézu bílkovin. Vyšetřování protilátek proti extrahovatelnému nukleárnímu antigenu (ENA) připadá v úvahu při podezření na systémové onemocnění. Specifický sérologický ukazatel aktivity nemoci není k dispozici [25]. Podle některých autorů byly zvýšené hladiny kreatinkinázy a aldolázy pozorovány v souvislosti s tvorbou nových projevů a mohly by být známkou aktivity nemoci u dětských nemocných [54]. V jedné studii u dětských nemocných vykázaly korelaci s aktivitou onemocnění sérové hladiny interferonem gamma indukovaného proteinu 10 (IP 10), tumor nekrotizujícího faktoru alfa (TNF-alfa) a kolonie stimulujícího faktoru granulocytů a makrofágů (GM-CSF) [50]. U forem s hlubším postižením mohou být zvýšeny transaminázy, kreatinkináza a aldoláza v důsledku svalového postižení, mohou být dále zvýšeny i zánětlivé ukazatele, jako je sedimentace erytrocytů a C reaktivní protein, hypereozinofilie, hypergamaglobulinémie, pozitivita ANA. Při podezření na postižení kloubů je vhodné vyšetření revmatoidního faktoru. V případě pozitivity a u závažnějších forem, zejména u dětských pacientů, je vhodné vyšetření ve spolupráci s revmatologem. Na rozdíl od systémové sklerózy není LS provázena nálezem specifických autoprotilátek, vzácně se nachází pozitivita protilátek anticentromerových a proti DNA-topoizomeráze I (anti-Scl70), jejich průkaz pak vyžaduje pečlivé sledování nemocných [56]. Vyšetřování protilátek na borélie je kontroverzní a většinou nebývá bez klinického podezření doporučováno [25].
Histologické vyšetření je vhodné při nejasném nálezu, nerozliší však morfeu od systémové sklerózy. Biopsie by vždy měla zavzít i podkožní tukovou tkáň. Při hlubším postižení je nutné provést hlubokou biopsii skalpelem, případně až s odběrem fascie. Histologický nález je závislý na charakteru místa odběru biopsie (údaj o odběru z centra indurace či zánětlivého lemu by měl být uveden na histologické průvodce), vývojové fázi a hloubce projevu. Zánětlivé léze vykazují perivaskulární a intersticiální infiltrát z lymfocytů, plazmocytů, případně eozinofilů, mastocytů a makrofágů, se zhruběním kolagenních snopců. Postupně zánětu ubývá a dochází ke zmnožení zhrubění až homogenizaci kolagenních snopců vaziva koria, případně vazivových sept podkožní tukové tkáně. Zhrubělé kolagenní snopce v koriu těsně obepínají adnexa. Přítomnost zánětu svědčí pro aktivitu onemocnění. Nález zánětu a/nebo sklerózy vaziva pouze ve vyšších partiích koria ukazuje na možnost použití povrchněji působících léčebných přístupů (fototerapie, lokální terapie), postižení hlubokého koria až podkožní tukové tkáně pak na uplatnění systémové či intralezionální léčby [38].
Zobrazovací metody, zejména magnetická rezonance (MRI), mají význam při diagnostice hlubokého postižení jako např. u eozinofilní fasciitidy, při plánování operace nebo při podezření na postižení centrálního nervového systému. Při postižení hlavy může být přítomna atrofie mozkové kůry či lbi, kalcifikace, léze bílé hmoty, dilatace komor, rozšíření leptomening, anomálie intrakranialních cév. Alternativou pro vyšetření měkkých tkání je sonografie. U lineárních forem postihujících hlavu a u progresivní faciální hemiatrofie s neurologickými příznaky je možné zvážit vyšetření neurologické. U lineárních forem s postižením hlavy jsou nutné oční kontroly (viz výše), případně vyšetření ortodontické. Nemocní s LS jen vzácně trpí Raynaudovým fenoménem či vykazují abnormální nálezy na plicích a jícnu. Jedná se většinou o postižení asymptomatická, prokazatelná pouze při cíleném vyšetřování, a proto není jejich podrobné rutinní vyšetřování bez přítomnosti klinických příznaků doporučováno [25, 56].
LÉČBA
V současné době není k dispozici kauzální, plně uspokojivá léčba LS. Liší se přístup k léčbě mezi dermatology, kteří léčí všechny formy, a to nejčastěji lokálními kortikoidy a fototerapií, a dětskými revmatology, kteří léčí především lineární a generalizovanou formu obvykle systémovou léčbou metotrexátem a kortikoidy [17]. I když používání níže uvedených léčiv v léčbě morfey je podpořeno různými studiemi, nejsou pro léčbu těchto onemocnění schváleny [25].
Léčbu onemocnění je nutné zahájit v aktivní fázi, aby se zabránilo vzniku komplikací, které jsou léčebně špatně ovlivnitelné. Volba a zahájení terapie se proto řídí aktivitou nemoci, její formou, hloubkou postižení a rizikem funkčního či kosmetického postižení.
Pro stanovení aktivity a monitorování průběhu onemocnění nejsou k dispozici sérologické ukazatele (viz vyšetření). Klinickými známkami aktivity nemoci je přítomnost erytému, přibývání nových či rozšiřování již přítomných projevů. Pro sledování průběhu onemocnění je k dispozici validovaná metoda klinického hodnocení, tzv. LoSCAT (Localized Scleroderma Cutaneous Assessment Tool). Skóre LoSCAT zahrnuje tři parametry [1, 2]:
- hodnocení aktivity nemoci (mLoSSI – modified localized scleroderma severity index), vycházející z přítomnosti erytému, indurace a tvorby nových projevů v bodové škále 0–3 v 18 anatomických krajinách;
- hodnocení vzniklého poškození (LoSDA – localized scleroderma skin damage assessment): přítomnost atrofie dermis, atrofie podkožní tukové tkáně a poruchy pigmentace;
- celkové hodnocení lékařem (PGA – physician global assessment).
LoSCAT představuje slibný, snadno použitelný nástroj hodnocení průběhu a léčby nemoci bez nutnosti použití zvláštního přístrojového vybavení [19, 25]. Ke sledování aktivity a průběhu choroby se používá i řada metod (kutometr, durometr, elastometr, systém hodnotící poměr kožního skóre, laserové dopplerovské zobrazovací techniky, sonografie 20-MHz, termografie aj.), které však vyžadují příslušné přístrojové vybavení a zkušené hodnotitele.
Při léčbě sklerotických projevů lze účinek hodnotit po 2–3 měsících léčby [25]. Navrhované schéma léčby pro naše podmínky je uvedeno v tabulce 3 – je členěné podle formy onemocnění a hloubky postižení a vychází z modifikovaných publikovaných doporučení [11, 25, 31].
Lokální léčba
Lokální léčba vzhledem k omezenému průniku do hloubky přichází v úvahu u méně rozsáhlých a povrchnějších projevů LS. V lokální léčbě se stále uplatňují lokální kortikosteroidy. Přestože nejsou v literatuře dostatečné důkazy o jejich účinnosti, jsou nadále doporučovány u povrchních forem onemocnění, zejména v akutní zánětlivé fázi onemocnění. Zpravidla se doporučuje použití velmi silných kortikosteroidů 1krát denně po dobu jednoho měsíce, případně silných kortikoidů 1krát denně po dobu tří měsíců, aplikovaných případně v okluzi [25]. Při dlouhodobějším použití se doporučuje intervalová terapie. Výjimečně se kortikoidy aplikují intralezionálně.
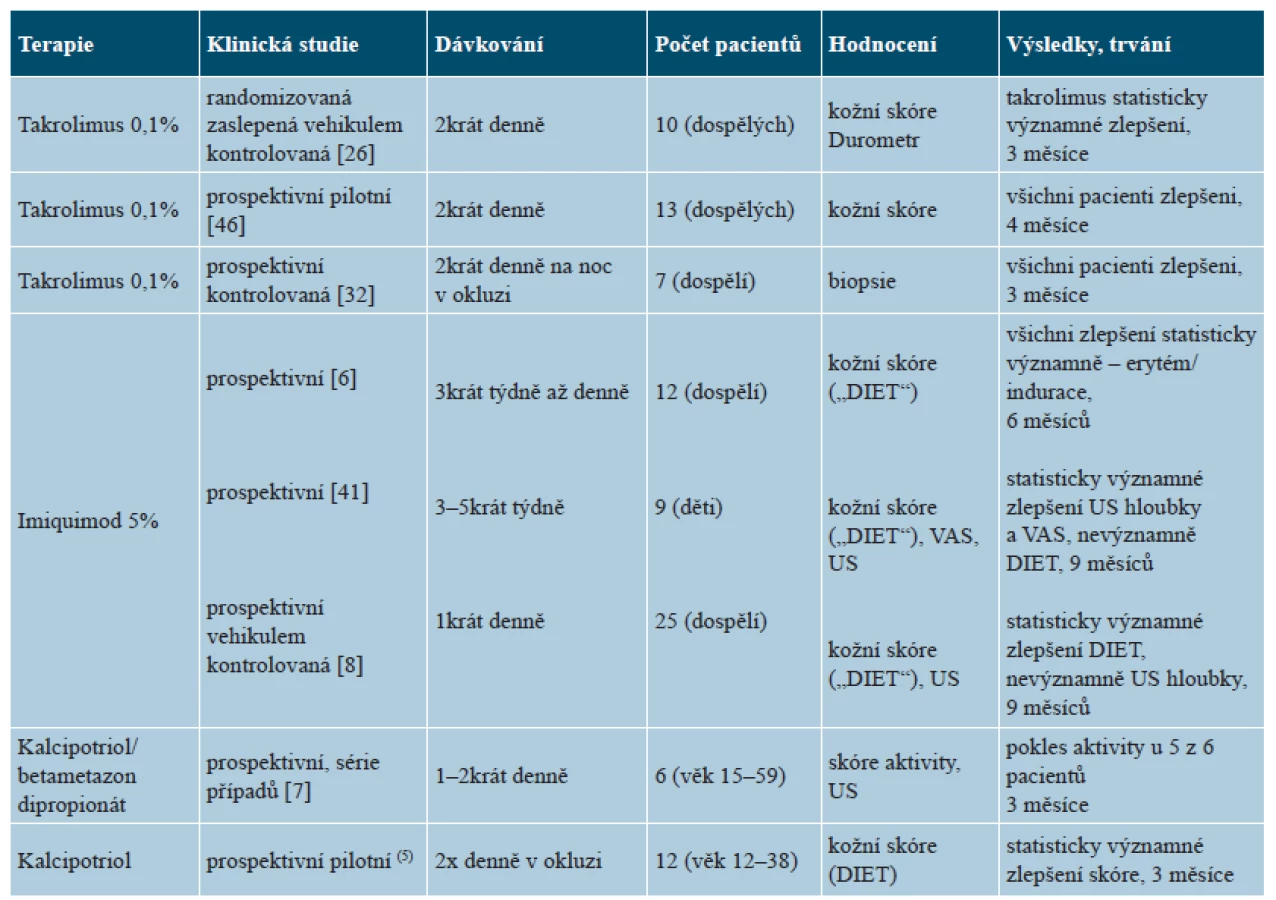
V literatuře jsou k dispozici tři studie na malém počtu pacientů dokladujících příznivý účinek kalcipotriolu aplikovaného 1–2krát denně v monoterapii [5], v kombinaci s betametazonem či UVA1 fototerapií (tab. 4) [7, 20]. Aplikace se doporučuje provádět 2krát denně, pokud možno v okluzi, alespoň po dobu tří měsíců [25]. Účinnost lokální aplikace takrolimu 0,1% se ukázala ve třech prospektivních studiích, zejména na akutní zánětlivou a indurativní fázi nemoci [27, 32, 46]. Takrolimus se doporučuje jako alternativa lokálních kortikoidů nebo i v první linii léčby, 2krát denně, případně na noc v okluzi [11, 25]. Lokální imunomodulátor imiquimod vede indukcí tvorby interferonu gamma (IFN-γ) k inhibici transformujícího růstové faktoru beta (TGF-β), a tak k antifibrotickému účinku. Tři studie na dětských i dospělých nemocných vykázaly příznivý účinek 5% imiquimodu aplikovaného po dobu 6–9 měsíců 1krát denně po 3–7 dní v týdnu [6, 8, 41]. Někteří autoři však jeho použití nedoporučují [25].
Fototerapie
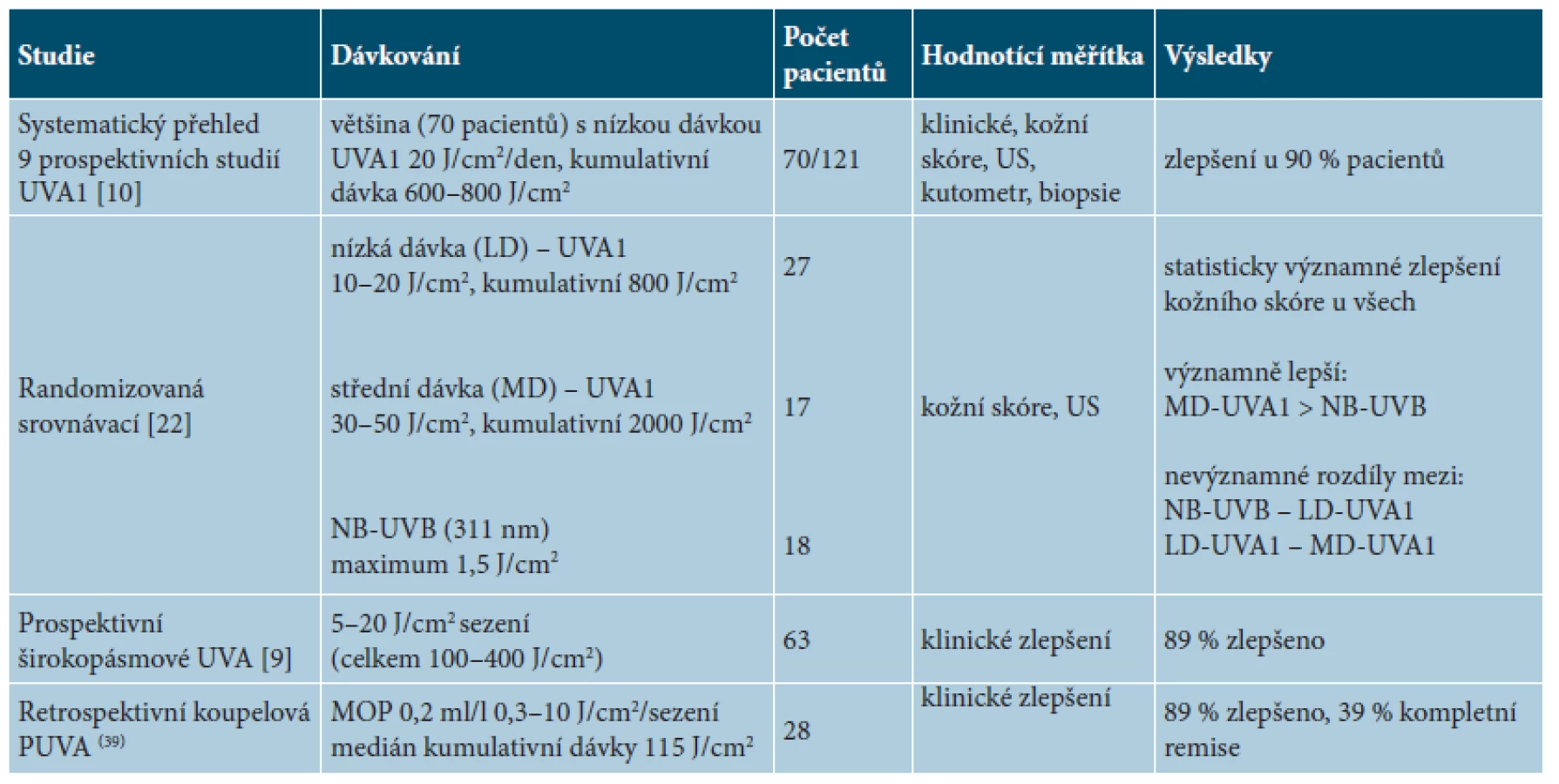
Fototerapie se v léčbě LS uplatňuje přes 20 let a je předmětem velkého počtu klinických studií (tab. 5). Ultrafialové záření má protizánětlivý a antifibrotický účinek prostřednictvím indukce různých matrixových metaloproteináz vedoucích k inhibici syntézy kolagenu. Vzhledem k omezenému průniku tohoto záření do hloubky (úzkopásmové NB-UVB 311nm dosahuje k dermoepidermálnímu rozhraní, UVA/UVA1/PUVA pak do středního až dolního koria) své uplatnění nalézá zejména u povrchnějších forem LS. Největší randomizovaná studie porovnávala účinnost nízké a střední dávky UVA1 a NB-UVB u 64 nemocných. Nízká dávka UVA1 byla aplikována v režimu 20 J/cm2 na sezení s celkovou kumulativní dávkou 800 J/cm2, střední jednotlivá dávka UVA1 byla 50 J/cm2 na sezení s celkovou kumulativní dávkou 2 000 J/cm2, léčba NB-UVB probíhala v iniciální dávce 0,1 J/cm2 pro fototyp II a 0,2 J/cm2 pro fototyp III, zvyšované podle snášenlivosti o 0,1–0,2 J/cm2 do maximální dávky 1,3 J/cm2 pro fototyp II a dávky 1,5 J/cm2 pro fototyp III. Ozařování probíhalo 5krát týdně po dobu osmi týdnů, celkem ve 40 sezeních [22]. Statisticky významné zlepšení podle modifikovaného kožního skóre, bylo pozorováno u všech skupin nemocných. Střednědávková UVA1 byla významně účinnější než NB-UVB. Nevýznamný byl rozdíl mezi střední a nízkou dávkou UVA1 a mezi nízkou dávkou UVA1 a NB-UVB. Menší dostupnost zářičů UVA1 vedla ke studiím terapií širokospektrým UVA zářením v monoterapii či v kombinaci s psoraleny. Studie používající širokospektré UVA (320–400 nm) na 63 pacientech ukázala klinické zlepšení kůže u 89 % nemocných léčených ve 20 sezeních jednotlivými dávkami 5, 10 a 20 J/cm2 s kumulativní dávkami 100, 200 a 400 J/cm2. Nižší jednotlivé dávky (5, 10 J/cm2) byly stejně účinné jako dávky vyšší (20 J/cm2) [9].
Fotochemoterapie PUVA se vzhledem ke gastrointestinálním potížím po systémovém podání psoralenu doporučuje jako lokální aplikace psoralenu ve formě krému či koupelí [11]. Nedávno publikovaná retrospektivní studie koupelové formy PUVA terapie u 28 nemocných vykázala klinické zlepšení u 89 % nemocných a kompletní remisi u 39 %. Fotochemoterapie probíhala 3krát týdně, medián kumulativní dávky činil 115 J/cm2 a medián počtu sezení 71 [39].
Fototerapie UVA1, PUVA, širokospektré UVA a NB-UVB jsou prospěšné u nemocných při léčbě LS bez hlubokého postižení kůže. Němečtí autoři doporučují jako první volbu střednědávkovou UVA1, dále pak lokální PUVA terapii. Fototerapii širokospektrým UVA považují za méně účinnou než lokální PUVA terapii a UVA1 [25]. Použití NB-UVB je možné zvážit u povrchních forem morfey [11, 13].
Systémová léčba
Systémová léčba je nutná u aktivních forem nemoci postihujících hlubší struktury, hrozících funkčním postižením, zejména flekčními kontrakturami.
Systémové kortikosteroidy jsou užívány při léčbě závažnějších a hlubších forem nemoci. Jejich užití v monoterapii bylo publikováno v jediné nekontrolované studii zahrnující převážně dospělé nemocné. Prednison se podával p. o. v dávce 0,5–1,0 mg/kg denně po průměrnou dobu 18 měsíců u 17 nemocných, což vedlo ke zlepšení u 13 nemocných, ale po vysazení došlo u 6 z nich k exacerbaci nemoci (tab. 6) [18]. Kortikosteroidy jsou lékem první volby u eozinofilní fasciitidy, která zpravidla na tuto léčbu dobře reaguje a k ústupu nemoci je monoterapie kortikosteroidy často dostatečná [25]. U LS se kortikosteroidy používají většinou v kombinaci s metotrexátem. V monoterapii jsou vzhledem k vedlejším účinkům doporučovány pouze pro akutní fázi těžkých forem nemoci.
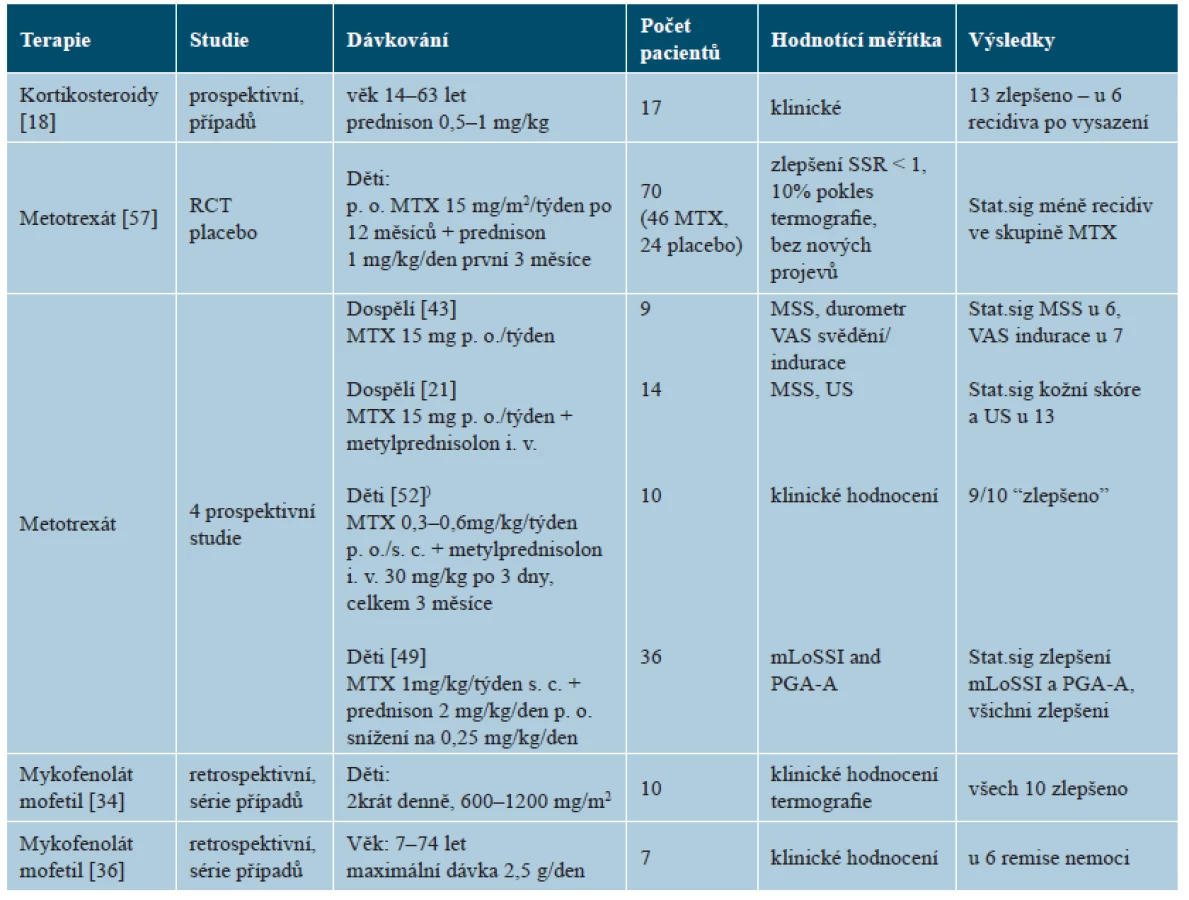
Metotrexát (MTX) představuje pilíř systémové léčby LS. K dispozici je jediná randomizovaná placebem kontrolovaná studie léčby juvenilní formy po dobu 12 měsíců u 70 nemocných. Perorálním MTX bylo léčeno 46 z nich (v dávce 15 mg/týden, maximálně 20 mg) a placebem 24 nemocných. Všichni nemocní byli léčeni první tři měsíce prednisonem (v dávce 1 mg/kg/den, maximálně 50 mg) [57]. Ve skupině léčené MTX došlo ke zlepšení u 67,4 % nemocných, oproti 29,2 % ve skupině placeba. K exacerbaci došlo u 32,6 % ze skupiny s MTX oproti 70,8 % u placeba, vznik nových lézí byl pozorován u 6,5 % s MTX a 16,7 % s placebem. Mírné vedlejší příznaky léčby byly pozorovány u 56,5 % nemocných s MTX a 45,8 % s placebem, u nikoho však nevedly k ukončení léčby. Prospektivní studie léčby MTX v monoterapii či v kombinaci s kortikosteroidy ukazují zlepšení klinických projevů u valné většiny nemocných [21, 43, 49, 51] – viz tabulka 6. Nedávno publikovaný přehled výsledků čtyř retrospektivních studií 119 nemocných (monoterapie MTX u 52 osob – u dospělých v dávce 15–25 mg/týden a dětí 0,3–0,4 mg/kg/týden, a u 67 osob v kombinaci MTX s kortikoidy podávanými v počátku intravenózně a posléze při pokračování léčby perorálně) ukázal zlepšení u 97 % nemocných [4, 12, 27, 25, 53].
Skupina dětských revmatologů a dermatologů sdružených pod označením CARRA („Childhood Arthritis and Rheumatology Research Alliance“) doporučuje pro léčbu aktivní, velmi závažné juvenilní formy LS (generalizovanou, pansklerotickou, kraniofaciální lineární „en coup de sabre“) nebo závažné formy s výraznou morbiditou (s příznaky neurologickými, s rizikem zkrácení končetin a vzniku flekčních kontraktur) tři léčebná schémata [31]. První schéma představuje monoterapii MTX v dávce 1 mg/kg/týden s. c., maximum 25 mg/týden, po dobu 12 měsíců. Druhé schéma je kombinace MTX (v uvedeném dávkování) po dobu 12 měsíců s intravenózně podávanými kortikosteroidy (IV-CS). Metylprednisolon se aplikuje v dávce 30 mg/kg (maximum 1 gram) v intravenózních pulzech 3 dny po sobě /1krát za měsíc po 3 měsíce (celkem 9krát) nebo intervalově 1krát týdně po 12 týdnů, celkem 12krát. Třetí schéma kombinuje MTX (v uvedeném dávkování) s perorálním prednisonem. Prednison v dávce 2 mg/kg/den (maximum 60 mg) se podává ve dvou rozdělených denních dávkách po dobu minimálně 2 a maximálně 4 týdnů, s postupným snižováním dávky po dosažení klinické odpovědi – tzn. ztrátě aktivity nemoci (netvoří se nové projevy), snižení intenzity erytému a indurace. V 8. týdnu se dosahuje snížení na 50 % dávky prednisonu (ve dvou denních dávkách), v 16. týdnu na 25 % dávky (1krát denně), ve 24. týdnu na 12,5 % až do vysazení ve 48. týdnu. U nemocných rezistentních na léčbu je možno zvážit kombinaci mykofenolát mofetilu p. o. v kombinaci s kortikosteroidy, případně i současně s MTX.
Mykofenolát mofetil (MMF) vykázal příznivý efekt ve dvou klinických studiích série případů. V retrospektivní studii 10 dětí neodpovídajících na léčby MTX a kortikosteroidy bylo pozorováno klinické zlepšení u všech nemocných po léčbě mykofenolát mofetilem (MMF) v dávce 600–1200 mg/den, který byl podáván současně s MTX u 6 nemocných, u pěti nemocných léčba vedla k významnému snížení dávky kortikosteroidů [34]. Ve studii 7 nemocných věku od 7 do 74 let, rezistentních či netolerujících léčbu MTX, nasazení MMF vedlo ke klinické remisi u 4 nemocných, u jednoho nemocného musel být MMF přes příznivý účinek vysazen pro elevaci jaterních testů, u jednoho nemocného nedošlo k exacerbaci, u jednoho došlo k progresi nemoci [36]. Přestože údaje o této léčbě jsou sporé, MMF je považován za příslib léčby druhé linie tohoto onemocnění [11, 31, 25].
Jiná systémová léčba LS zahrnuje celou řadu léčiv, která se používala či používá. V kontrolovaných randomizovaných studiích se nepotvrdila účinnost kalcitriolu a IFN-γ. D-penicilamin nebývá doporučován pro vedlejší účinky a sporný efekt. Stejně nebývá doporučováno i užití penicilinu pro nepotvrzenou účinnost [25]. Celá řada kazuistických zpráv popisuje příznivý účinek různých léčebných postupů, jejichž užití připadá v úvahu v jednotlivých individuálních případech. Jedná se o léky jako cyklosporin A, azathioprin, fenytoin, chlorochin a hydroxychlorochin, kolchicin, retinoidy, extrakorporální fotoferézu, plazmaferézu, intravenózní imunoglobuliny, abatacept, infliximab, rituximab, imatinib aj. [25]. Příslibem je biologická léčba fresolimumabem, což je humánní IgG4-kappa monoklonální protilátka proti TGFß, který vedl v klinické studii u systémové sklerodermie ke zlepšení fibrózy, modifikovaného Rodnanova skóre a k rychlé inhibici exprese genů regulujících TGFß [42].
V současné době, u případů vyžadujících systémovou léčbu, je lékem volby MTX podávaný po dobu aspoň 12 měsíců (někteří autoři doporučují 24 měsíců) v akutní fázi případně kombinovaný s celkovými kortikosteroidy, jako alternativa bývá doporučován MMF [25].
Fyzikální a rehabilitační léčba
Fyzikální a rehabilitační léčba jsou považovány za velmi důležitou součást terapie ve sklerotické fázi nemoci, i když dosud nebyly předmětem řádných klinických studií. Zahrnují rehabilitační cviky a posilující svalová cvičení, které jsou významné zejména u flekčních kontratur, dále masáže a manuální lymfodrenáže. Lázeňská léčba zaměřená na pohybový aparát má též příznivý vliv.
Chirurgická léčba
Chirurgická léčba připadá v úvahu u onemocnění bez známek aktivity, nejlépe několik let po vyhasnutí choroby. V úvahu připadají ortopedické korekce kontraktur, korekce rozdílné délky končetin, dentálních abnormalit, aplikace výplní a plastických výkonů [25].
PRŮBĚH, RECIDIVY, PROGNÓZA
LS je onemocnění se sklonem k recidivám po ukončení léčby. Retrospektivní studie se vznikem nemoci v dětském i dospělém věku prokázala recidivu téměř u čtvrtiny nemocných. Hlavním rizikových faktorem byla lineární sklerodermie končetin, bez ohledu na věk v době vzniku [35]. Autoři předpokládají, že prodloužení celkové léčby by mohlo vést ke snížení recidiv, což podpořily i práce jiných autorů a i výsledky otevřeného prodloužení předchozí randomizované placebem kontrolované studie léčby metotrexátem [57]. V této studii z 65 pacientů jich na léčbu MTX odpovědělo 73,8 %. Během 24 měsíců od zahájení léčby recidivovalo 15,4 % (většinou v prvních 12 měsících léčby), ke kontrolám se nedostavilo 10,8 % nemocných. Ze skupiny nemocných reagujících na léčbu MTX (průměrně 27,5 měsíce, medián 24, rozmezí 18 až 30), jich 73 % bylo v klinické remisi od ukončení léčby po dobu 25,6 měsíce a více (medián 24, rozmezí 6–48), 27 % sledovaných pak v průměru po 20,5 měsíců (medián 15,5, rozmezí 6–45) [58]. Pouze u jednoho pacienta z 39 nemocných v kompletní remisi na léčbě MTX došlo ve 24. měsíci sledování k recidivě. Recidiva nebyla pozorována u žádného nemocného léčeného déle než 24 měsíců. Autoři proto k dosažení dlouhodobé remise onemocnění doporučují délku léčby MTX alespoň po dobu 24 měsíců. V práci nebyl nalezen žádný ukazatel předurčující recidivující průběh, větší pravděpodobnost recidiv měli nemocní s exacerbacemi v prvních dvou letech léčby.
ZÁVĚR
LS je chronické onemocnění, recidivující až u čtvrtiny nemocných, a proto je vhodné jejich dlouhodobé sledování. I když zpravidla nepostihuje vnitřní orgány a nevede ke zkrácení délky života nemocných, může vést k výrazným funkčním kosmetickým a psychologickým následkům. Včasná diagnóza a dostatečná léčba nemoci v aktivní fázi či při exacerbaci mohou zabránit vzniku komplikací, protože již vzniklá poškození jsou obtížně léčitelná, zejména v dětském věku a u lineárních forem. Volba a zahájení léčby se řídí aktivitou nemoci, rychlostí progrese, její formou, hloubkou postižení a přítomností mimokožních příznaků. Systémová léčba metotrexátem a kortikosteroidy připadá v úvahu u závažných forem s rizikem vzniku invalidizace. U hlubokých forem a u závažného postižení musí být léčba metotrexátem dostatečně dlouhá (1–2 roky) pro snížení rizika nového vzplanutí nemoci. Fyzioterapie, zejména u pacientů s rizikem vzniku flekčních kontraktur, je důležitou součástí léčby.
Do redakce došlo dne 19. 7. 2016.
Adresa pro korespondenci:
prof. MUDr. J. Štork, CSc.
Dermatovenerologická klinika 1. LF UK a VFN
U Nemonice 499/2,
128 00 Praha 2
e-mail: jiri.stork@lf1.cuni.cz
Sources
1. ARKACHAISRI, T., VILAIYUK, S., LI, S., O’NEIL, K. M. et al. The localized scleroderma skin severity index and physician global assessment of disease activity: a work in progress toward development of localized scleroderma outcome measures. J. Rheumatol., 2009, 36, p. 2819–2829.
2. ARKACHAISRI, T., VILAIYUK, S., TOROK, K. S., MEDSGER, T. A. jr. Development and initial validation of the localized scleroderma skin damage index and physician global assessment of disease damage: a proof-of-concept study. Rheumatology (Oxford), 2010, 49, p. 373–381.
3. BLEASEL, N. R., STAPLETON, K. M., COMMENS, C., AHERN, V. A. Radiation-induced localized scleroderma in breast cancer patients. Australas J. Dermatol., 1999, 40, p. 99–102.
4. COX, D., O’ REGAN, G., COLLINS, S. et al. Juvenile localised scleroderma: a retrospective review of response to systemic treatment. Ir. J. Med. Sci., 2008, 177, p. 343–346.
5. CUNNINGHAM, B. B., LANDELLS, I. D., LANGMAN, C. et al. Topical calcipotriene for morphea/linear scleroderma. J. Am. Acad. Dermatol., 1998, 39, p. 211–215.
6. DYTOC, M., TING, P. T., MAN, J. et al. First case series on the use of imiquimod for morphoea. Br. J. Dermatol., 2005, 153, p. 815–820.
7. DYTOC, M., KOSINTSEVA, I, TING, P. T. First case series on the use of calcipotriol-betamethasone dipropionate for morphoea. Br. J. Dermatol., 2007, 157, p. 615–618.
8. DYTOC, M., WAT, H., CHEUNG-LEE, M. et al. Evaluation of topical imiquimod 5% for plaque-type morphea: a multicenter, prospective, vehicle-controlled trial. J. Cutan. Med. Sur., 2015, 19, p. 132–139.
9. EL-MOFTY, M., MOSTAFA, W., EL-DAROUTY, M. et al. Diffferent low doses of broad-band UVA in the treatment of morphea and systemic sclerosis. Photodermatol. Photoimmunol. Photomed., 2004, 20, p. 148–156.
10. FETT, N., WERTH, V. P. Update on morphea: part I. Epidemiology, clinical presentation, and pathogenesis. J. Am. Acad. Dermatol., 2011, 64, p. 217–228.
11. FETT, N., WERTH, V. P. Update on morphea: part II. Outcome measures and treatment. J. Am. Acad. Dermatol., 2011, 64, p. 231–242.
12. FITCH, P. G., RETTIG, P., BURNHAM, J. M. et al. Treatment of pediatric localized scleroderma with methotrexate. J. Rheumatol., 2006, 33, p. 609–614.
13. GORDON, S., PRATT, E. A., GORCEY, L. V., SOTER, N. A. et al. Phototherapy, photodynamic therapy and photopheresis in the treatment of connective tissues diseases: a review. Brit. J. Dermatol., 2015, 173, p. 19–30.
14. GRABELL, D., HSIEH, C., ANDREW, R. et al. The role of skin trauma in the distribution of morphea lesions: A cross-sectional survey of the morphea in adults and children cohort IV. J. Am. Acad. Dermatol., 2014 71, p. 493–498.
15. HERRICK, A. L., ENNIS, H., BHUSHAN, M. et al. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res. (Hoboken), 2010, 62, p. 213–218.
16. IHN, H., SATO, S., FUJIMOTO, M., KIKUCHI, K., TAKEHARA, K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch. Dermatol. Res., 1995, 287, p. 193–197.
17. JOHNSON, W., JACOBE, H. Morphea in adults and children cohort II: Patients with morphea experience delay in diagnosis and large variation in treatment. J. Am. Acad. Dermatol., 2012, 67, 5, p. 881–889.
18. JOLY, P., BAMBERGER, N., CRICKX, B. et al. Treatment of severe forms of localized scleroderma with oral corticosteroids: follow-up study on 17 patients. Arch. Dermatol., 1994, 130, 5, p. 663–664.
19. KELSEY, C. E., TOROK, K. S. The Localized Scleroderma Cutaneous Assessment Tool: responsiveness to change in a pediatric clinical population. J. Am. Acad. Dermatol., 2013, 69, p. 214–220.
20. KREUTER, A., GAMBICHLER, T., AVERMAETE, A. et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet A1 phototherapy in childhood morphea. Pediatr. Dermatol., 2001, 18, p. 241–245.
21. KREUTER, A., GAMBICHLER, T., BREUCKMANN, F. et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch. Dermatol., 2005, 141, p. 847–852.
22. KREUTER, A., HYUN, J., STÜCKER, M. et al. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J. Am. Acad. Dermatol., 2006, 54, 3, p. 440–447.
23. KREUTER, A., HYUN, J., SKRYGAN, M. et al. Ultraviolet A1-induced downregulation of human beta-defensins and interleukin-6 and interleukin-8 correlates with clinical improvement in localized scleroderma. Br. J. Dermatol., 2006, 155, p. 600–607.
24. KREUTER, A. Localized scleroderma. Dermatol. Therapy, 2012, 25, p. 135–147.
25. KREUTER, A., KRIEG, T., WORM, M. et al. German guidelines for the diagnosis and therapy of circumscribed scleroderma. J. Dtsch. Dermatol. Ges., 2016, 14, 2, p. 199–216.
26. KROFT, E. B., GROENEVELD, T. J., SEYGER, M. M. B. et al. Efficacy of topical tacrolimus 0.1% in active plaque morphea. Randomized, double-blind, emollient-controlled pilot study. Am. J. Clin. Dermatol., 2009, 10, p. 181.
27. KROFT, E. B., CREEMERS, M. C., VANDEN HOOGEN, F. H. et al. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study. Br. J. Dermatol., 2009, 160, p. 1075–1082.
28. LAXER, R. M., ZULIAN, F. Localized scleroderma. Curr. Opin. Rheumatol., 2006, 18, p. 606–613.
29. LEASK, A., ABRAHAM, D. J. TGF-beta signaling and the fibrotic response. FASEB J, 2004, 18, p. 816–827.
30. LEITENBERGER, J. J., CAYCE, R. L., HALEY, R. W., ADAMS-HUET, B., BERGSTRESSER, P. R., JACOBE, H. T. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch. Dermatol., 2009, 145, 5, p. 545.
31. LI, S. C., TOROK, K, S., POPE, E. et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken) 2012, 64, p. 1175–1185.
32. MANCUSO, G., BERDONDINI, R, M. Localized scleroderma: response to occlusive treatment with tacrolimus ointment. Br. J. Dermatol., 2005, 152, p. 180–182.
33. MARSOL, I. B. Update on the classification and treatment of localized scleroderma. Acta Dermatosilifiliogr., 2013, 104, 8, p. 654–666.
34. MARTINI, G., RAMANAN, A. V., FALCINI, F. et al. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology (Oxford), 2009, 48, p. 1410–1413.
35. MERTENS, J. S., SEYGER, M. M. B., KIEVIT, W. et al. Disease recurrence in localized scleroderma: a retrospective analysis of 344 patients with paediatric - or adult-onset disease. Br. J. Dermatol., 2015, 172, p. 722–778.
36. MERTENS, J. S., MARSMAN, D. S., VAN DE KERKHOF, P. C. M. et al. Use of mycophenolate mofetil in patients with severe localized scleroderma resistant or intolerant to methotrexate. Acta Dermato-Venereologica, 2016, 96, p. 510–513.
37. MURRAY, K. J., LAXER, R. M. Scleroderma in children and adolescents. Rheum. Dis. Clin. N. Am., 2002, 28, 3, p. 603–624.
38. NOURI, S., JACOBE, H. Recent developments in diagnosis and assessment of morphea. Curr. Rheumatol. Rep., 2013, 15, p. 308–315.
39. PAVLOTSKY, F., SAKKA, N., LOZINSKI, A. et al. Bath psoralen-UVA photochemotherapy for localized scleroderma: experience from a single center. Photodermatol. Photoimmunol. Photomed., 2013, 29, p. 247–252.
40. PETERSON, L. S., NELSON, A. M., SU, W. P. et al. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960–1993. J. Rheumatol., 1997, 24, 1, p. 73–80.
41. Pope, E., Doria, A.S., Theriault, M., et al. Topical imiquimod 5% cream for mediatric plaque morphea. Dermatology, 2011, 223, p. 363-369.
42. RICE, M., PADILLA, C. M., MCLAUGHLIN, S. R. et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Invest., 2015, 125, 7, p. 2795–2807.
43. SEYGER, M. M., VANDEN HOOGEN, F. H., DEBOO, T., DEJONG, E. M. Low-dose methotrexate in the treatment of widespread morphea. J. Am. Acad. Dermatol., 1998, 39, p. 220–225.
44. SILMAN, A., JANNINI, S., SYMMONS, D., BACON, P. An epidemiological study of scleroderma in the West Midlands. Br. J. Rheumatol., 1988, 27, p. 286–290.
45. SPALEK, M., JONSKA-GMYREK, J., GAŁECKI, J. Radiation-induced morphea – a literature review. J. Eur. Acad. Dermatol. Venereol., 2015, 29, p. 197–202.
46. STEFANAKI, CH., STEFANAKI, K., KONTOCHRISTOPOULOS, G. et al. Topical tacrolimus 0.1% ointment in the treatment of localized scleroderma. J. Derm., 2008, 35, p. 712–718.
47. TOLKACHJOV, S. N., PATEL, N. G., TOLLEFSON, M. M. Progressive hemifacial atrophy: a review. Orphanet Journal of Rare Diseases, 2015, DOI 10.1186/s13023-015-0250-9.
48. TOLLEFSON, M. M., WITMAN, P. M. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J. Am. Acad. Dermatol., 2007, 56, p. 257–263.
49. TOROK, K. S., ARAKACHAISRI, T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single center study. J. Rheumatol., 2012, 39, 2, p. 286–294.
50. TOROK, K. S., KURZINSKI, K., KELSEY, H. C. et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Sem. Arthritis Rheum., 2015, 45, p. 284–293.
51. UZIEL, Y., KRAFCHIK, B. R., SILVERMAN, E. D. et al. Localized scleroderma in childhood: a report of 30 cases. Semin. Arthritis Rheum., 1994, 23, p. 328–340.
52. UZIEL, Y., FELDMAN, B., KRAFCHIK, B. et al. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J. Pediatr., 2000, 136, p. 91–95.
53. WEIBEL L., SAMPAIO, M. C., VISENTIN, M. T. et al. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br. J. Dermatol., 2006, 155, 1013–1120.
54. WU, E. Y., LI, S. C., TOROK, K. S. et al. A28: Description of the Juvenile Localized Scleroderma Subgroup of the CARRA Registry. Arthritis Rheumatol., 2014, 66, S3, p. S43–S44.
55. ZANNIN, M. E., MARTINI, G., ATHREYA, B. H. et al. Ocular involvement in children with localised scleroderma: a multi-centre study. Br. J. Ophthalmol., 2007, 91, p. 1311–1314.
56. ZULIAN, F., ATHREYA, B. H., LAXER, R. et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford), 2006, 45, p. 614–620.
57. ZULIAN, F., MARTINI, G., VALLONGO, C. et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum., 2011, 63, p. 1998–2006.
58. ZULIAN, F., VALLONGO, C., PATRIZI, A. et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J. Am. Acad. Dermatol., 2012, 67, p. 1151–1156.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2016 Issue 6
Most read in this issue
-
Lokalizovaná sklerodermie – morfea:
současný stav a možnosti léčby - Bilateral Giant Cell Temporal Arteritis – Case Report
- Relation of the Severity of Atopic Dermatitis and Occurrence of Respiratory Allergy to Dust, Animal Dander, Mites, Feather and IgE Food Allergy to Cow´s Milk, Egg, Soy, Peanuts and Wheat