
Rosai-Dorfmanova nemoc – kožní forma
Rosai-Dorfman Disease – Cutaneous Form
The authors describe case of a 67-year-old woman, who successively developed asymptomatic brownish cutaneous nodules during a two year period. Histological examination disclosed a Rosai-Dorfman disease, while an thorough examination ruled out other organ involvement. Total excision of lesions was an effective treatment. The article mentions a recent literature review on cutaneous variant of a Rosai-Dorfman disease.
Key words:
Rosai-Dorfman disease – cutaneous form
:
L. Drlík 1; L. Pock 2
:
Dermatovenerologické oddělení, Nemocnice Šumperk, prim. MUDr. Lubomír Drlík
1; Bioptická laboratoř Plzeň, s. r. o., odborná vedoucí lékařka prof. MUDr. Alena Skálová, CSc.
2
:
Čes-slov Derm, 92, 2017, No. 4, p. 180-183
:
Case Reports
Autoři popisují případ 67leté ženy, u které se v průběhu dvou let postupně vyvinuly hnědavé asymptomatické noduly. Histologické vyšetření prokázalo Rosai-Dorfmanovu nemoc, celkovým vyšetřením bylo vyloučeno postižení jiných orgánů. Léčebně byla dostatečná totální excize projevů. Je uváděn současný přehled literatury kožní varianty Rosai-Dorfmanovy nemoci.
Klíčová slova:
morbus Rosai-Dorfman – kožní forma
ÚVOD
Rosai-Dorfmanova nemoc je klasifikována jako histiocytóza z non-Langerhansových buněk, která byla popsána v roce 1965 Destombesem [3]. Rosai a Dorfman ji v roce 1969 určili jako nozologickou jednotku pod názvem „sinusová histiocytóza s masivní lymfadenopatií“ [7]. Onemocnění postihuje lymfatické uzliny a v některých případech současně i jiné orgány, jako např. respirační nebo urogenitální trakt, ústní dutinu, gastrointestinální oblast. Kožní postižení bývá asi v 10 % případů [1, 6]. Čistě kožní forma byla popsána v roce 1978, je extrémně vzácná a představuje méně než 3 % případů Rosai-Dorfmanovy nemoci [10]. Do roku 2015 bylo celosvětově publikováno přibližně 100 případů [4, 12], v naší dermatologické literatuře je uvedený případ první.
POPIS PŘÍPADU
K dermatologickému vyšetření se v roce 2015 dostavila 67letá žena s několik měsíců trvajícím asymptomatickým hnědočerveným částečně prominujícím kožní nodulem na levé hýždi velikosti 15 mm. Klinicky byl zvažován poinjekční granulom nebo panikulitida. Pacientka byla v péči naší ambulance od roku 2014 po operaci morbus Bowen genitální oblasti. V anamnéze pacientka udávala léčbu kompresí a venofarmaky primárního varikózního syndromu dolních končetin a operaci pravého prsu pro tumor v roce 2009 s následnou radioterapií a několik roků trvající léčbou letrozolem. Pro esenciální hypertenzi užívala losartanum kalicum.
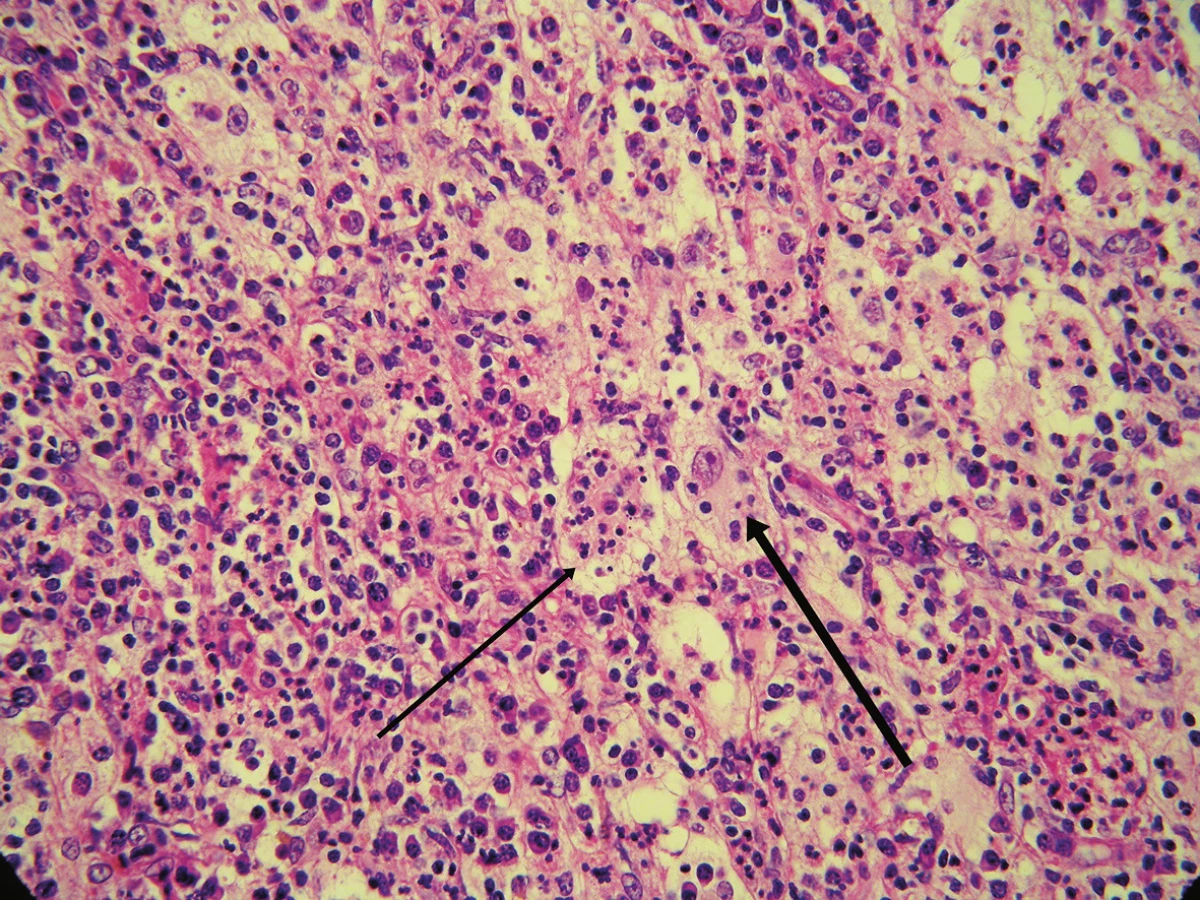
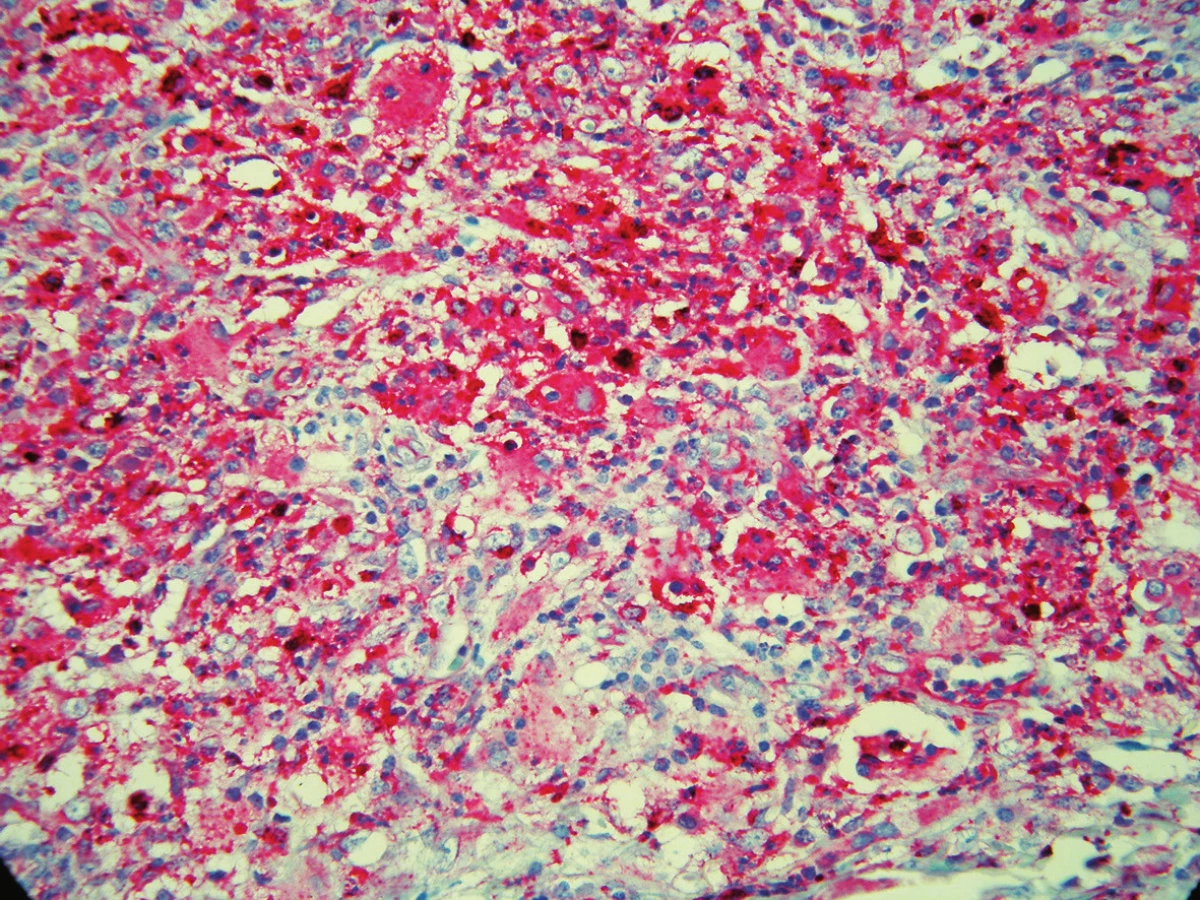
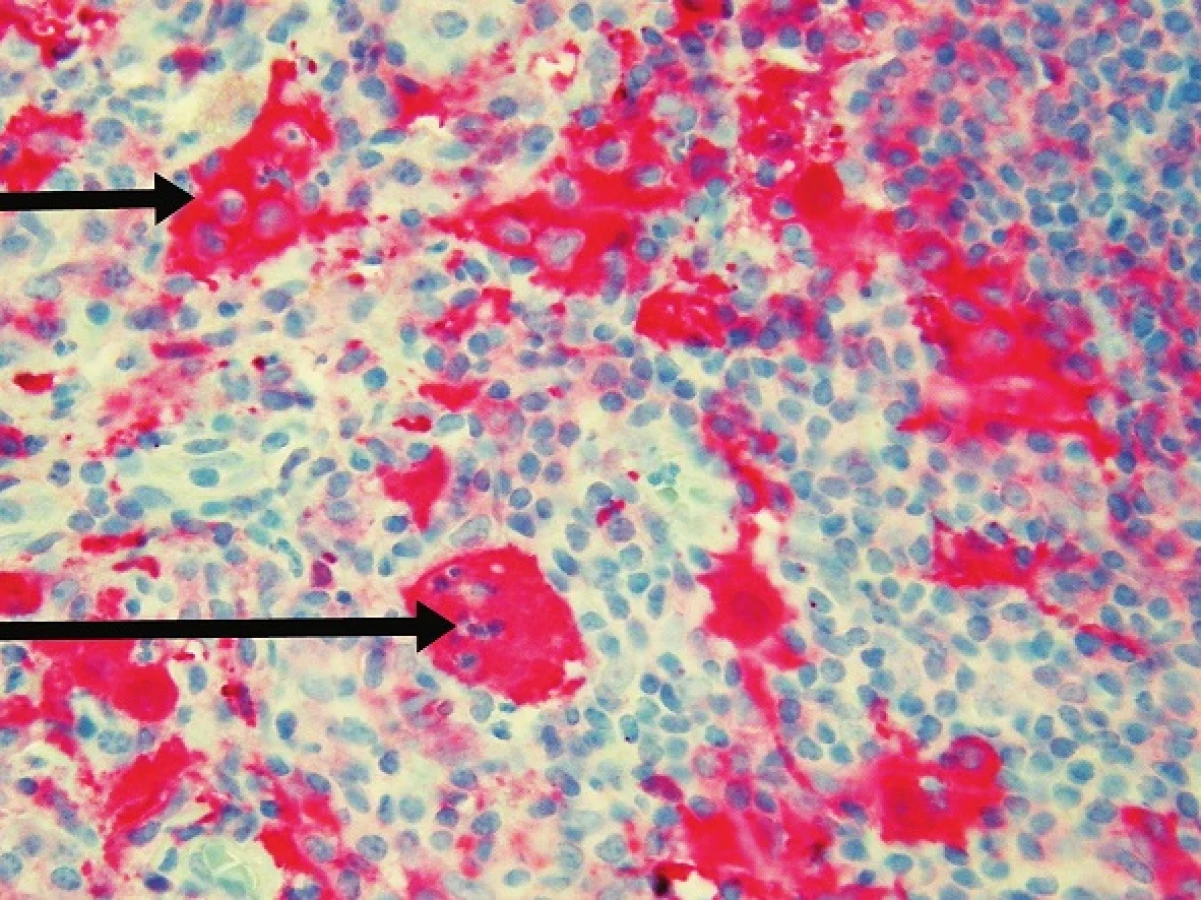
Byla provedena totální excize. Histologický nález vykázal v dolním koriu až subcutis poměrně ostře ohraničený hustý infiltrát s četnými zárodečnými lymfoidními folikuly. V tomto infiltrátu byly přítomny poměrně rozsáhlé oblasti s velkými buňkami s eozinofilní cytoplazmou, okrouhlými velkými jádry, které obsahovaly v cytoplazmě fragmenty jader pravděpodobně polynukleárů, místy i lymfocytů a celé plazmocyty. Šlo tedy o obraz charakteru emperipolesis. Tyto buňky byly CD64 a S100 pozitivní, CD1a negativní, kolem nich se nacházely hojné infiltráty tvořené plazmocyty (ty byly polyklonální – kappa, lambda v poměru 1 : 1), lymfocyty, ložiskovitě i polynukleáry, místy s extravazálními erytrocyty a xantomovými buňkami (obr. 1, 2, 3, 4).
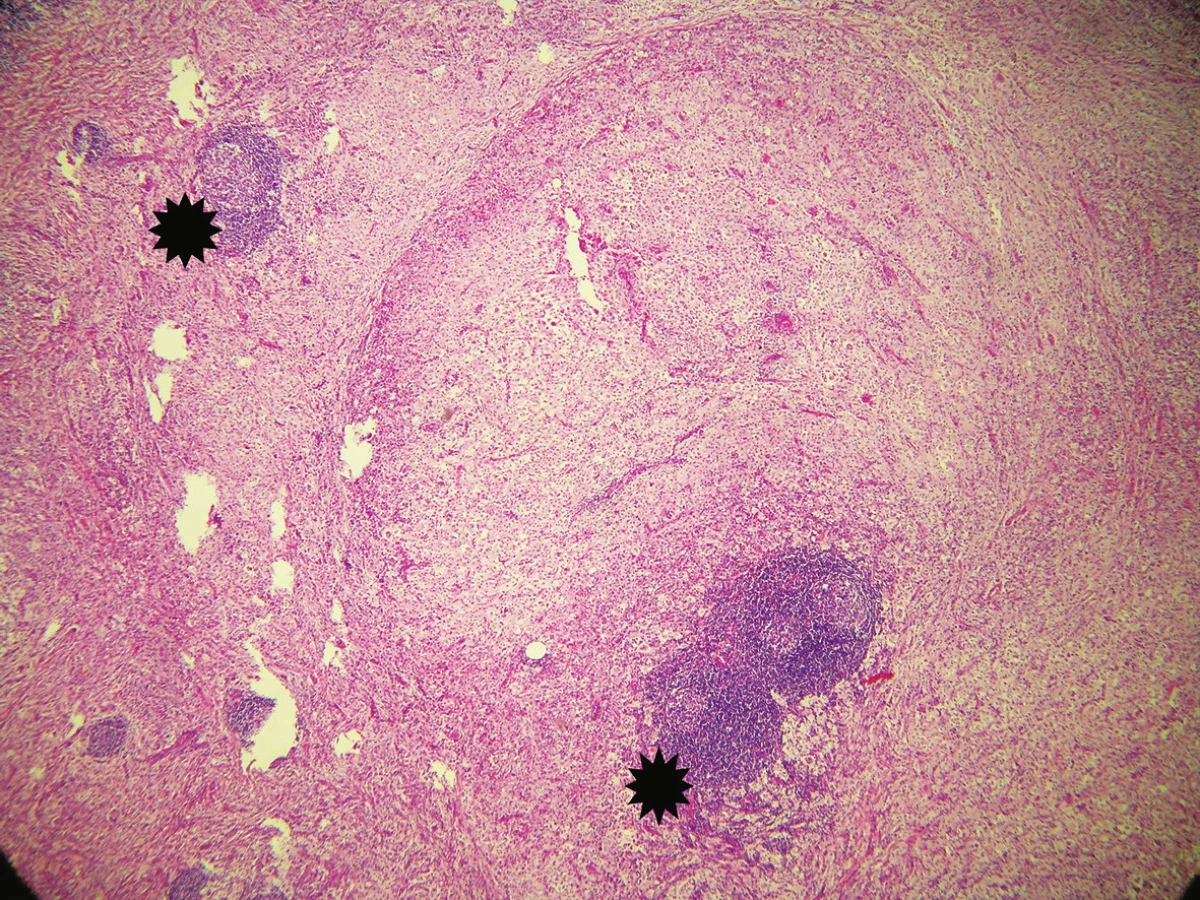
Následně proběhla excize dvou podobných uzlů z levého stehna velikosti 10 a 8 mm, histologické vyšetření prokázalo také Rosai-Dorfmanovu nemoc (obr. 5).
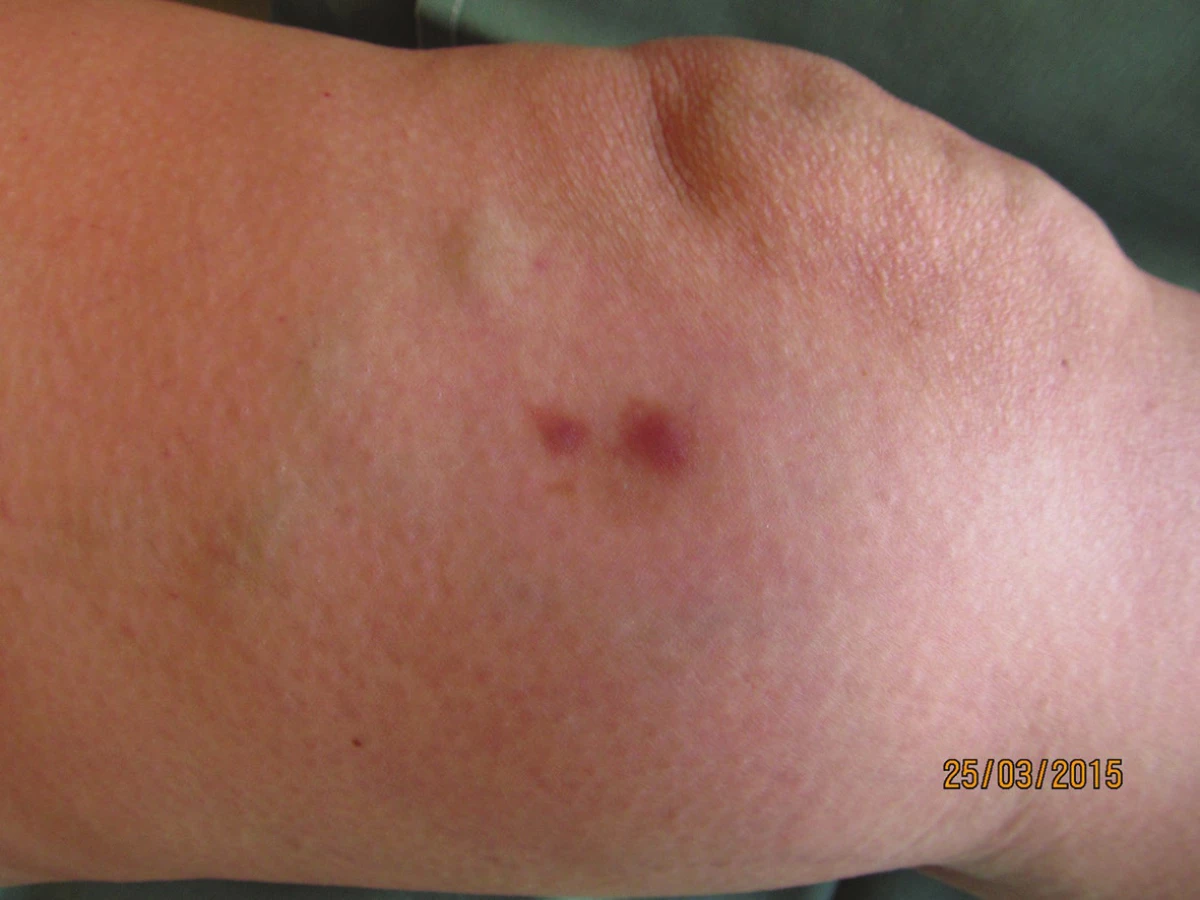
Klinický nález – lymfatické uzliny třísel, axil, krku a šíje nebyly zvětšeny, játra byla v oblouku, další kožní nález negativní, fyzicky byla pacientka ve velmi dobrém stavu.
Sonografické vyšetření prokázalo steatózu jater, oboustranně uzly štítné žlázy, nezvětšené lymfatické uzliny krku, axil a třísel benigního vzhledu. Nález rentgenového vyšetření nitrohrudních orgánů byl v rámci normy. Vyšetření na virus Ebstein-Barrové a parvovirus B19 prokázalo přítomnost jen anamnestických protilátek, protilátky proti cytomegaloviru byly negativní, HHV6 IgG slabě pozitivní. Beta 2 mikroglobulin 2,23 mg/l (ref. do 2,0 mg/l), anti-streptolyzin O, C-reaktivní protein, krevní obraz s diferenciálním rozpočtem bílých krvinek a biochemické vyšetření séra byly v normě.
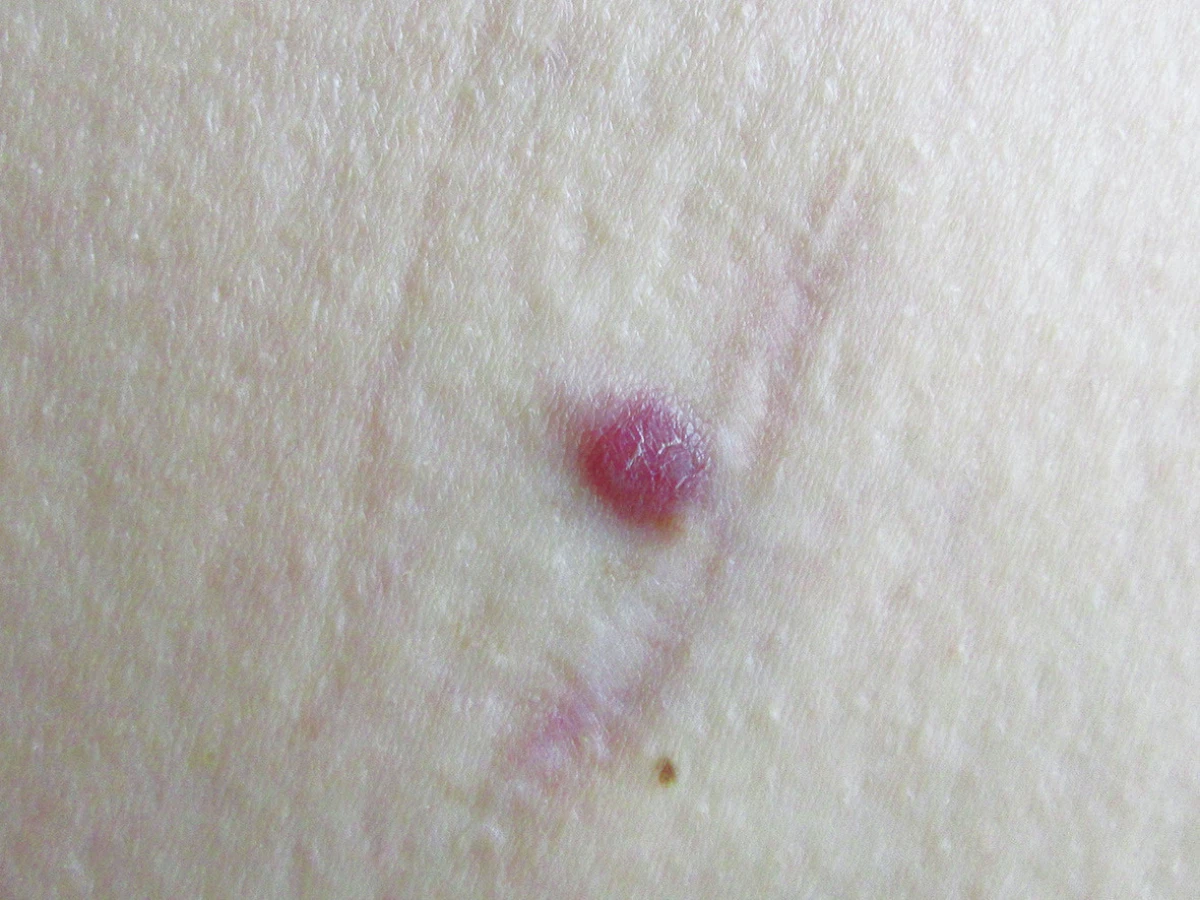
Při kontrole za čtyři měsíce byla pacientka bez projevů nemoci. Za sedmnáct měsíců se dostavila na pozvání k další kontrole, pozorovala asi dva měsíce v okraji jizvy na levé hýždi drobný částečně zanořený asymptomatický nodulus hnědočervené barvy (obr. 6). Lymfatické uzliny nebyly zvětšeny, pacientka byla v celkově dobrém stavu. Nodulus byl excidován, histologické vyšetření prokázalo lokální recidivu Rosai-Dorfmanovy nemoci. Sonografické vyšetření spádových i mimospádových lymfatických uzlin, nitrobřišních orgánů a retroperitonea bylo negativní.
DISKUSE
Kožní forma Rosai-Dorfmanovy nemoci postihuje častěji ženy v poměru 2 : 1 v průměrném věku 43,5 roků, má tedy pozdější výskyt než forma lymfadenopatická, která se objevuje kolem 20. roku života. Vyšší prevalence je u Asiatů a kavkazského etnika, naopak lymfadenopatická forma se vyskytuje o něco častěji u mužů a Afričanů [2, 4, 5, 6].
Jedná se o reaktivní proces, jehož etiologie není známa, zvažují se virové a imunitní příčiny [2, 4, 12, 13]. Laboratorní nálezy – normocytární či mikrocytární anémie, polyklonální gamaglobulinémie bývají spíše u systémového lymfadenopatického postižení, dále se literárně udává pozitivita EBV nebo HHV-6 viru [5, 6, 11, 13].
Klinické příznaky čistě kožního postižení jsou variabilní, není přítomna lymfadenopatie. Jedná se o solitární či vícečetné papuly, noduly, plaky nebo jejich kombinace, byly popsány i akneiformní a granulomatózní léze, pustuly, projevy napodobující panikulitidu či vaskulitidu [1, 4, 11, 13].
V histologickém nálezu nebývá epidermis postižena, v dermis se nacházejí difuzní infiltráty histiocytů, lymfocytů a plazmocytů. Typický je fenomén emperipolesis – výskyt zejména intaktních lymfocytů, ale i plazmocytů, neutrofilů a erytrocytů v histiocytech. Mitózy a buněčné atypie jsou řídké, nebývají ani nekrózy. Histiocyty jsou S100 a CD68 pozitivní, CD1a negativní. Histiocyty mohou být uspořádány ve shlucích, které připomínají sinusy lymfatické uzliny. V elektronové mikroskopii se neprokazují Bierbeckova granula, tím je vyloučena histiocytóza z Langerhansových buněk [1, 2, 4, 5, 6, 12, 13].
Diferenciální diagnóza klinická zahrnuje Hodgkinův lymfom, histiocytózu z Langerhansových buněk, histiocytární sarkom, tezaurizmózy (Gaucherova nemoc), infekce – histoplazmózu, mykobakteriální infekty, dále metastazující melanom či karcinom a sarkoidózu.
Histologická diferenciální diagnóza zahrnuje xantom, xantogranulom, fibrohistiocytom, histiocytózu z Langerhansových buněk, maligní histiocytózu, dermatofibrosarkoma protuberans [11].
Léčba u poloviny pacientů není nutná, protože dochází ke spontánním remisím.
U lymfadenopatické formy jsou spontánní remise udávány pouze v 5–11 % případů. Prognóza kožní formy je dobrá, byly však popsány asociace s lymfomem, revmatoidní artritidou, HIV infekcí, hypothyreoidismem, systémovým lupus erythematodes či uveitidou [4, 6]. Léčba je chirurgická – excize, může být využita radioterapie a kryoterapie. Dalšími možnostmi léčby jsou sulfony, intralezionálně nebo systémově podávané kortikosteroidy, retinoidy, eventuálně metotrexát [4, 5, 6, 9, 11, 13].
ZÁVĚR
Popsaný případ měl necharakteristické klinické příznaky, diagnóza byla stanovena na základě histologického vyšetření. Postupně byly excidovány uzly na hýždi, stehně a recidiva na hýždi. Ve všech histologických nálezech byl popsán stejný charakteristický obraz. Pacientka během dvouletého sledování neměla známky systémového postižení.
Do redakce došlo dne 28. 11. 2016.
Adresa pro korespondenci:
MUDr. Lubomír Drlík
Dermatovenerologické oddělení
Nemocnice Šumperk
Nerudova 41
787 52 Šumperk
Sources
1. BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. Dermatology, 2nd Edition, 2008, p. 1407–1408, ISBN 978-1-4160-2999-1.
2. BRENN, T., CALONJE, E., GRANTER, S. R. et al. Cutaneous rosai-dorfman disease is a distinct clinical entity. Am. J. Dermatopathol., 2002, 24, 5, p. 385–391.
3. DESTOMBES, P. Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases). Bull. Soc. Pathol. Exot. Filiales, 1965, 58, 6, p. 1169–1175.
4. FANG, S., CHEN, A. Facial cutaneous Rosai-Dorfman disease: A case report and literature review. Exp. Ther. Med., 2015, 9, 4, p. 1389–1392.
5. KOMARAGIRI, M., SPARBER, L. S., SANTOS-ZABALA, M. L. et al. Extranodal Rosai-Dorfman disease: a rare soft tissue neoplasm masquerading as a sarcoma. World J. Surg. Oncol., 2013, 11, p. 63.
6. MOLINA-GARRIDO, M. J., GUILL0N-PONCE, C. Extranodal Rosai - Dorfman Disease with Cutaneous and Periodontal Involvement: A Rare Presentation. Case Rep. Oncol., 2011, 4, 1, p. 96–100.
7. ROSAI, J., DORFMAN, R. F. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch. Pathol., 1969, 87, p. 63–70.
8. SHI, X. Y., MA, D. L., FANG, K. Cutaneous Rosai - -Dorfman disease presenting as a granulomatous rosacea-like rashs. Chin. Med. J., 2011, 124, 5, p. 793–794.
9. SUN, N. Z., GALVIN, J., COOPER, K. D. Cutaneous Rosai-Dorfman disease successfully treated with low-dose methotrexate. JAMA Dermatol., 2014, 150, 7, p. 787–788.
10. THAWERANI, H., SANCHEZ, R. L, ROSAI, J., DORFMAN, R. F. The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Arch. Dermatol., 1978, 114, 2, p. 191–197.
11. XIA, J. X., JIN, X. H., MOU, Y. et al. Combined treatment for cutaneous Rosai-Dorfman disease: a report of 2 cases. Int. J. Clin. Exp. Med., 2013, 25, 6, 9, p. 822–827.
12. YANG, M., CHANG, J. Cutaneous Rosai-Dorfman disease in a middle-aged man: A case report. Exp. Ther. Med., 2015, 10, 3, p. 1199–1201.
13. WOLF, K., GOLDSMITH, L. A. et al. Fitzpatrick‘s Dermatology in general medicine, 7th edition, McGrawHill, 2008, p. 1433–1434, ISBN 978-0-07-146690-5.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2017 Issue 4
Most read in this issue