
Kožní leiomyosarkom – popis případu
Cutaneous Leiomyosarcoma – Case Report
The authors describe a case of a very rare malignant mesenchymal tumor – cutaneous leiomyosarcoma. Clinically, it was a bland, benign looking lesion on the calf, manifested in men aged 62 years. After excision of the lesion the diagnosis was established by histological and immunohistochemical examination with desmin and alpha-SMA positivity. The authors provide an overview of current knowledge on the disease.
Key words:
leiomyosarcoma – histology - immunohistochemistr
Authors:
I. Strouhalová 1; L. Drlík 1; L. Pock 2
Authors‘ workplace:
Dermatovenerologické oddělení, Nemocnice Šumperk, a. s., prim. MUDr. Lubomír Drlík
1; Bioptická laboratoř, s. r. o., Plzeň, odborná vedoucí lékařka prof. MUDr. Alena Skálová, CSc.
2
Published in:
Čes-slov Derm, 93, 2018, No. 1, p. 17-19
Category:
Case interpretation
Overview
Autoři popisují klinický případ velmi vzácného maligního mezenchymálního tumoru – kožního leiomyosarkomu. Klinicky se jednalo o nevýraznou benigně vyhlížející lézi na lýtku, která se manifestovala u muže ve věku 62 let. Diagnóza byla stanovena po excizi léze na základě histologického a imunohistochemického vyšetření – desmin a alfa SMA pozitivní. Autoři poskytují přehled současných poznatků o tomto onemocnění.
Klíčová slova:
leiomyosarkom – histologické a imunohistochemické vyšetření
ÚVOD
Primární kožní leiomyosarkom je maligní tumor z hladkých svalů. Literárně ho poprvé popsali Montgomery a Winkelmann v roce 1959 [14]. Jedná se o vzácné onemocnění, které představuje 0,04 % všech tumorů a 5–10 % sarkomů měkkých tkání [3, 8, 16]. V naší dermatologické literatuře byl leiomyosarkom naposledy zmíněn v roce 1982, proto uvádíme případ našeho nemocného [10].
POPIS PŘÍPADU
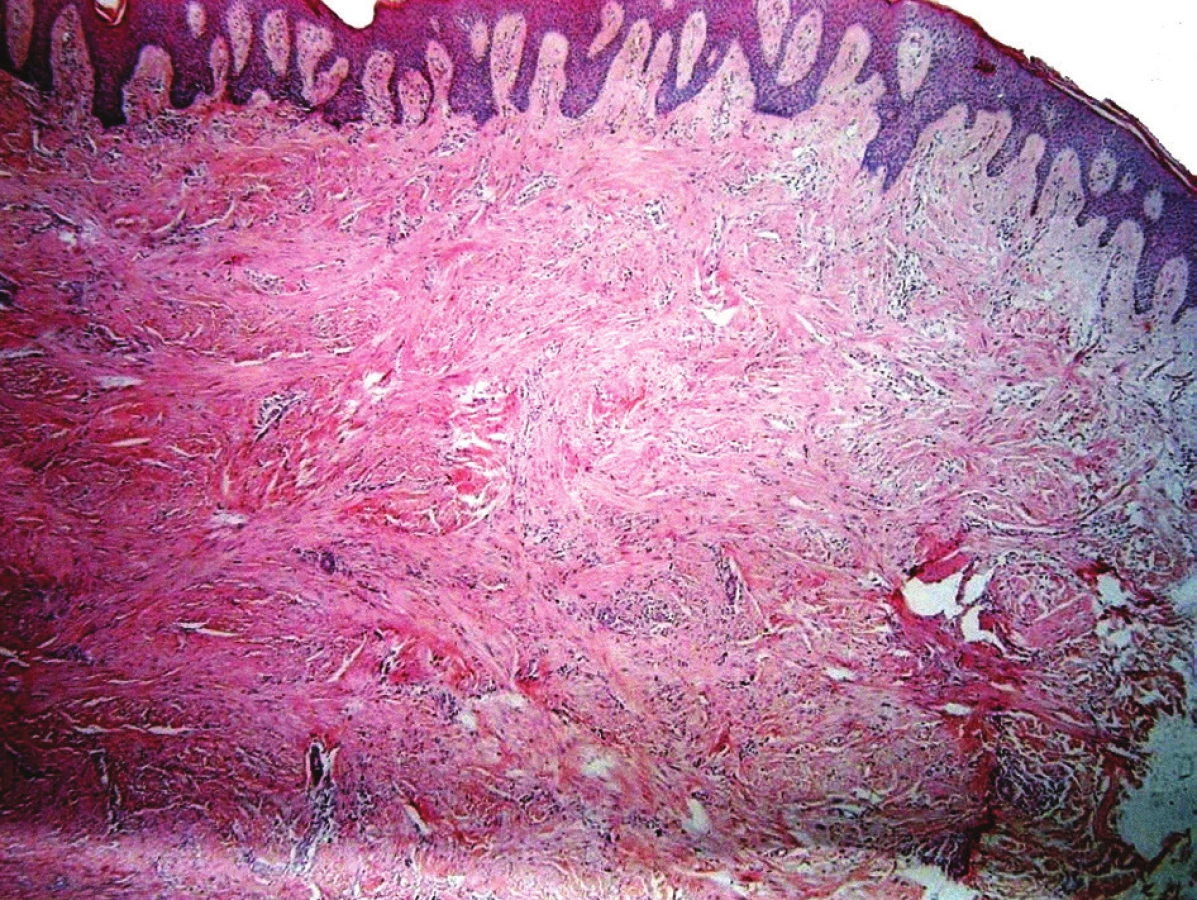
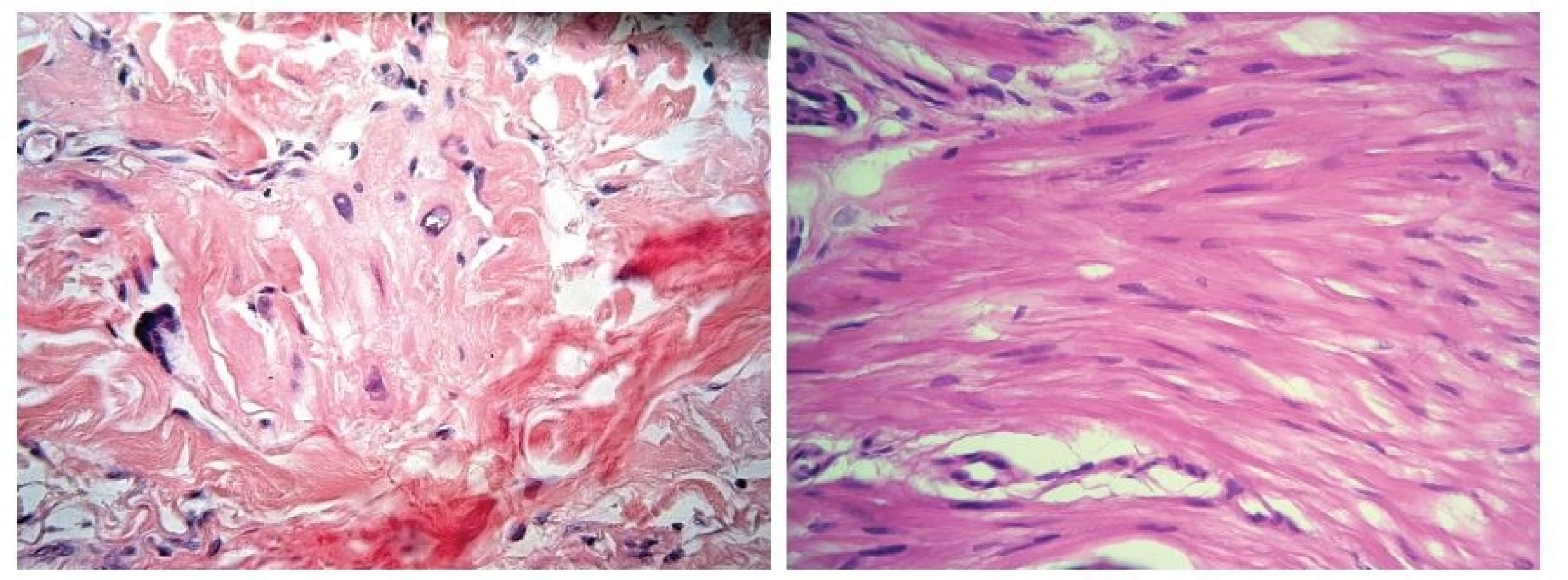
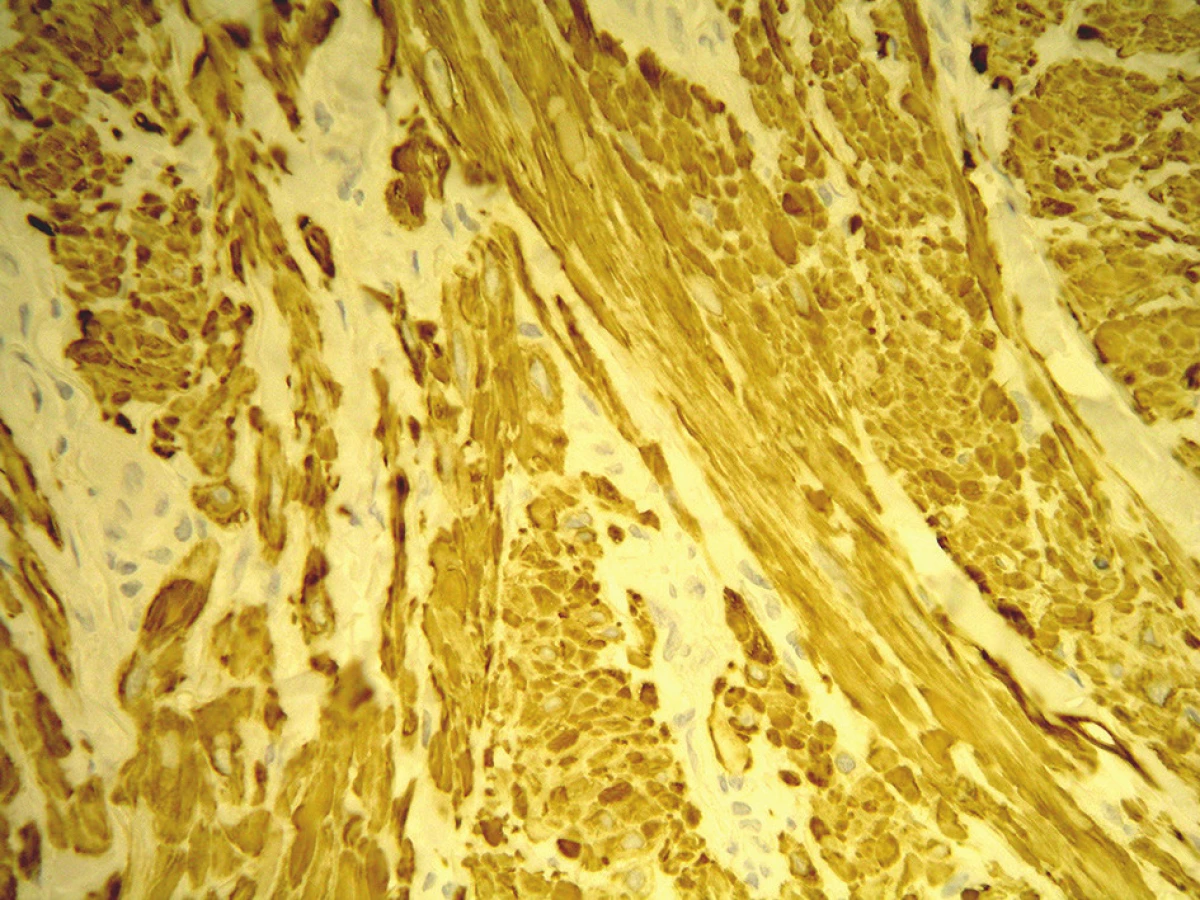
V březnu 2012 se dostavil k vyšetření muž (62 let) s projevem na pravém lýtku velikosti 1 cm, který měl minimálně 10 let, v posledních měsících se zvětšil a působil svědění. Tuber byl ohraničený, tuhý, hnědavého zbarvení, klinicky nejblíže dermatofibromu. Pacient se léčil s hypertenzní nemocí, operace neprodělal, rodinná anamnéza stran kožních chorob byla negativní, pracoval jako úředník. Tumor na lýtku byl exstirpován. Histologické vyšetření ukázalo pod mírně akantotickou epidermis v rozsahu celého koria fascikulárně uspořádaný tumor tvořený eozinofilní tkání s buňkami s vřetenitým hyperchromním jádrem projevujícím ložiskovitě různě intenzivní míru pleomorfismu. Některé buňky byly až monstrózní a obsahovaly výrazné jadérko, mitózy byly nečetné. Nádorové buňky byly S100, CD34 negativní, desmin, alfa SMA pozitivní. V okolí se nacházely nepříliš velké infiltráty lymfocytů. Závěr – kožní leiomyosarkom zasahující do roviny excize (obr. 1, 2, 3). Na základě tohoto nálezu byla provedena doporučená reexcize, vzhledem k histologickému popisu reziduálního leiomyosarkomu bylo radikální operační řešení následně provedeno na klinice plastické chirurgie. U pacienta byla provedena vyšetření: RTG plic, sonografie břicha a uzlin, PET-CT, laboratorní vyšetření, onkologické vyšetření – vše s negativním nálezem. Pacient je nadále v dispenzární péči dermatologické a onkologické ambulance.
DISKUSE
Leiomyosarkomy se dělí do čtyř hlavních skupin. Nejčastější je leiomyosarkom postihující vnitřní orgány – uterus, retroperitoneum, břišní dutinu. Druhou skupinu tvoří leiomyosarkomy měkkých tkání, které mají lepší prognózu. Třetí skupina jsou kožní leiomyosarkomy, které pro povrchní uložení jsou příznivější nežli předchozí dvě. Čtvrtou skupinu tvoří cévní leiomyosarkomy [8].
Primární kožní leiomyosarkom se vyskytuje ve dvou základních formách, dermální a subkutánní, které se významně liší prognózou. Dermální forma vychází z mm. arrectores pillorum nebo genitálních dartoických svalů, subkutánní forma z hladkých svalů stěn krevních cév (maligní varianta angioleiomyomu) [2, 4, 8, 11].
Dermální kožní leiomyosarkom je obvykle lokalizován na končetinách s predilekcí v ochlupených místech (50 až 75 % dolní končetiny, 20–30 % na pažích) a jen 10–15 % lézí se objevuje na trupu [1, 8]. Tumory tváře (1–5 %), scrota a rtů jsou vzácné. Poměr postižení muži : ženy je 2–3 : 1, vyskytuje se nejčastěji v 5.–7. dekádě, byl ale zaznamenán i u 5měsíčního dítěte [1, 2, 15]. Patogeneze není známá, někdy předchází výskyt leiomyomu, trauma, radiace, popálení, očkování [6, 8, 12]. U imunosuprimovaných mohou vznikat v souvislosti s infekcí virem Epstein-Barrové [7 ]. V 80–95 % případů je pacienty udávaná spontánní nebo indukovaná bolest, dalšími subjektivními příznaky mohou být parestezie, pruritus [1, 6]. Klinicky se jedná většinou o solitární nodulus červené nebo hnědé barvy do velikosti 2 cm s pravidelnými nebo i nepravidelnými okraji, mohou se vyskytovat umbilikace či ulcerace.
Podkožní primární kožní leiomyosarkomy jsou rezistence velikosti 2–4 cm, méně často vícečetné, vyklenutá kůže nad nimi má nezměněný charakter [3, 6]. Klinický vzhled je nespecifický, klinická diagnóza není možná. V kůži se také mohou vyskytovat metastázy leiomyosarkomů vnitřních orgánů [16]. Sonograficky se nachází hypoechogenní tumorózní projev, větší tumory jsou heterogenní, mohou se vyskytnout nekrózy, hemoragie nebo cystické změny [16]. Další zobrazovací možností je magnetická rezonance.
Histologicky se v koriu nacházejí propletené snopce vřetenitých buněk s eozinofilní cytoplazmou s hyperchromními, často vakuolizovanými, vřetenitými, tupě zakončenými jádry. Mitózy, zvýšená buněčnost, pleomorfní a bizarní buňky svědčí pro maligní proces [4, 11]. Někteří autoři rozlišují dva histologické typy:
- nodulární s vysokou buněčností, atypiemi jader a mitózami;
- plošný (difuzní, infiltrativní) méně buněčný, dobře diferencovaný s nečetnými mitózami [9].
K potvrzení diagnózy je nezbytné imunohistochemické vyšetření, které vykazuje pozitivitu markerů hladké svaloviny. Velmi senzitivní (100%) je marker SMA (smooth muscle actin, alfa hladkosvalový aktin), pozitivní bývá také desmin a h-caldesmon, CD34 bývá pozitivní v cévách, negativní v nádorových buňkách [3, 4, 5, 17, 18]. Klinická a histologická diferenciální diagnóza je velmi široká. Zahrnuje leiomyom, fibrosarkom, maligní fibrózní histiocytom, rhabdomyosarkom, maligní schwannom, dermatofibrom, dermatofibroma protuberans, proliferující a nodulární fasciitis, neurofibrom, vřetenobuněčný melanom, vřetenobuněčný spinaliom, plexiformní neurofibrom, angiosarkom, synoviální sarkom a další nozologické jednotky [2, 4, 11].
Terapeuticky je nutná časná excize s okraji 2–5 cm a hloubkou zahrnující podkoží a fascii, při neadekvátním postupu hrozí recidivy a metastazování (recidivující nádory mají horší biologické chování nežli původní tumor). Radioterapie a chemoterapie mají velmi neuspokojivé výsledky [4, 6, 8, 11, 12].
Prognóza dermální formy je příznivá. Vznik metastáz u této formy není jednoznačně doložený, recidivuje v 30 až 50 % a vykazuje pětileté přežití v 95 % případů. U podkožní formy jsou recidivy více než 50%, metastázy se objevují u 30–60 % postižených a pětileté přežití je 60–70% [3, 5, 6].
Nádor metastazuje nejčastěji hematogenní cestou do plic, lymfogenní šíření je méně obvyklé [6]. Nepříznivé prognostické faktory jsou velikost tumoru více než 5 cm, podkožní a akrální lokalizace, vysoká mitotická aktivita, výskyt nekrózy, pronikání do fascie a intratumorózní vaskulární invaze [2]. Při velikosti tumoru menší než 2 cm je pětileté přežití 95 %, nad 5 cm 30 % [6].
ZÁVĚR
Kožní leiomyosarkom je velmi vzácně se vyskytující nádor. Klinicky není charakteristický, diagnóza se stanoví prostřednictvím histologického a zejména imunohistochemického vyšetření. Terapeutický postup je jednoznačný – provedení radikální excize. Následně je nutné dlouholeté sledování pacienta [13].
Do redakce došlo dne 16. 2. 2017.
Adresa pro korespondenci:
MUDr. Ivana Strouhalová
Dermatovenerologické oddělení Nemocnice Šumperk, a. s.
Nerudova 41
787 01 Šumperk
e-mail: ivana@strouhal.cz
Sources
1. ABBASI, F., MAHMUDLU, R., NIKNIAZ, Y. et al. Primary Cutaneous Leiomyosarcoma in a Young Patient Previously Misdiagnosed as Pleomorphic Fibroma. Iran J. Pathol., 2015, 10(1), p. 69–73.
2. AGALE, S. V., GROVER, S., ZODE, R. et al. Primary cutaneous leiomysarcoma. Indian J. Dermatol., 2011, 56(6), p. 728–730.
3. ANEIROS-FERNANDEZ, J., RETAMERO, J. A., HUSEIN-ELAHMED, H. et al. Primary cutaneous and subcutaneous leiomyosarcomas: evolution and prognostic factors. Eur. J. Dermatol., 2016, 26, 1, p. 9–12.
4. BALI, A., KANGLE, R., ROY, M. et al. Primary cutaneous leiomyosarcoma: A rare malignant neoplasm. Indian Dermatol. Online J., 2013, 4, 3, p. 188–190.
5. CARPINTERO ANDRÉ, M., VEIGA ANTUNES, J., DUARTE REIS, M. et al. Cutaneous leiomyosarcoma on the trunk, An. Bras. Dermatol., 2011, 86, 5, p. 1–5.
6. CIUREA, M. E., GEORGESCU, C. V., RADU, C. C. et al. Cutaneous leiomyosarcoma – Case report. J. Med. Life, 2014, 7, 2, p. 270–273.
7. DEKATE , J., CHETTY, R. Epstein-Barr Virus Associated Smooth Muscle Tumor. Arch. Pathol. Lab. Med., 2016, 140, 7, p. 718–722.
8. EKEN, H., KARAGUL, S.,TOPGÜL, K. Giant Cutaneous Leiomyosarcoma Originating From the Abdominal Wall: A Case Report. Am. J. Case Rep., 2016, 17, p. 35–38.
9. KADDU, S., BEHAM, A., CERRONI, L. et al. Cutaneous leiomyosarcoma. Am. J. Surg. Pathol., 1997, 21, 9, p. 979–987.
10. KASTL, J., HORÁČEK, J. K. problematice primárních myogenních nádorů (mnohočetný piloleiomyom, podkožní leiomyosarkom). Čes.-slov. Derm., 1982, 57, 4, s. 205–207.
11. LEE, K. C., KIM, M. S., CHOI, H. et al. Rapid growing superficial cutaneous leiomyosarcoma of the face. Ann. Dermatol., 2013, 25, 2, p. 237–241.
12. LIMAIEM, F., CHELLY, I., BELLIL, S. et al. Primary Cutaneous leiomyosarcoma: A histological and immunochistochemical study of 4 cases. Egypt. Dermatol. Online J., 2007, 3, 1, p. 1–7.
13. MASSI, D., FRANCHI, A., ALOS, L. et al. Primary cutaneous leiomyosarcoma: clinicopathological analysis of 36 cases. Histopathology, 2010, 56, 2, p. 251–262.
14. MONTGOMERY, H., WINKELMANN, R. K. Smooth--muscle tumors of the skin. Arch. Dermatol., 1959, 79, p. 32–41.
15. POL, R. A., DANNENBERG, H., ROBERTUS, J. L. et al. Cutaneous leiomyosarcoma arising in a smallpox scar. World J. Surg. Oncol., 2012, 10, p. 148.
16. SALEMIS, N. S. Recurrent subcutaneous trunk leiomyosarcoma: Management and review of the literature. J. Nat. Sci. Biol. Med., 2013, 4, 1, p. 238–242.
17. WATANABE, K., KUSAKABE, T., HOSHI, N. et al. h-Caldesmon in leiomyosarcoma and tumors with smooth muscle cell-like differentiation: its specific, expression in the smooth muscle cell tumor. Hum.Pathol., 1999, 30, 4, p. 392–396.
18. WOLF, K., GOLDSMITH, L. A. Fitzpatrick’s Dermatology in General Medicine, The McGraw-Hill Companies, 2008, p. 1173-1174. ISBN 978-0-07-146690-5.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2018 Issue 1
Most read in this issue
- Diagnostika melanomu a současná doporučení pro léčbu a sledování
- Kožní leiomyosarkom – popis případu
- Rothmundův-Thomsonův syndrom sdružený s anaplastickým velkobuněčným T lymfomem – popis případu