
Fenylketonurie v dospělosti
Phenylketonuria in Adulthood
One hundred adult patients with phenylketonuria (PKU) and hyperphenylalaninemia (HPA) aged from 18 to 43 years were examined. 55% of patients with PKU were not on a low-protein diet. 45% of them kept the diet but 22.2 % had the phenylalanine blood level higher than 20 mg/dl.
The educational attainment of patients:
66.2% of patients were apprenticed, 21.7% of them finished their secondary school, 1.2% finished college and 1.2% finished university studies. 6% of patients attended special-needs school and 3.6 % of them were unable to educate.
The body mass index (BMI) of female patients was 23.6 ± 0.05 (16.8–43.6). BMI of male patients was 24.0 ± 0.8 (16.7–38.9). The 7.4 % of female patients and 13.0 % of male patients had the BMI >30.
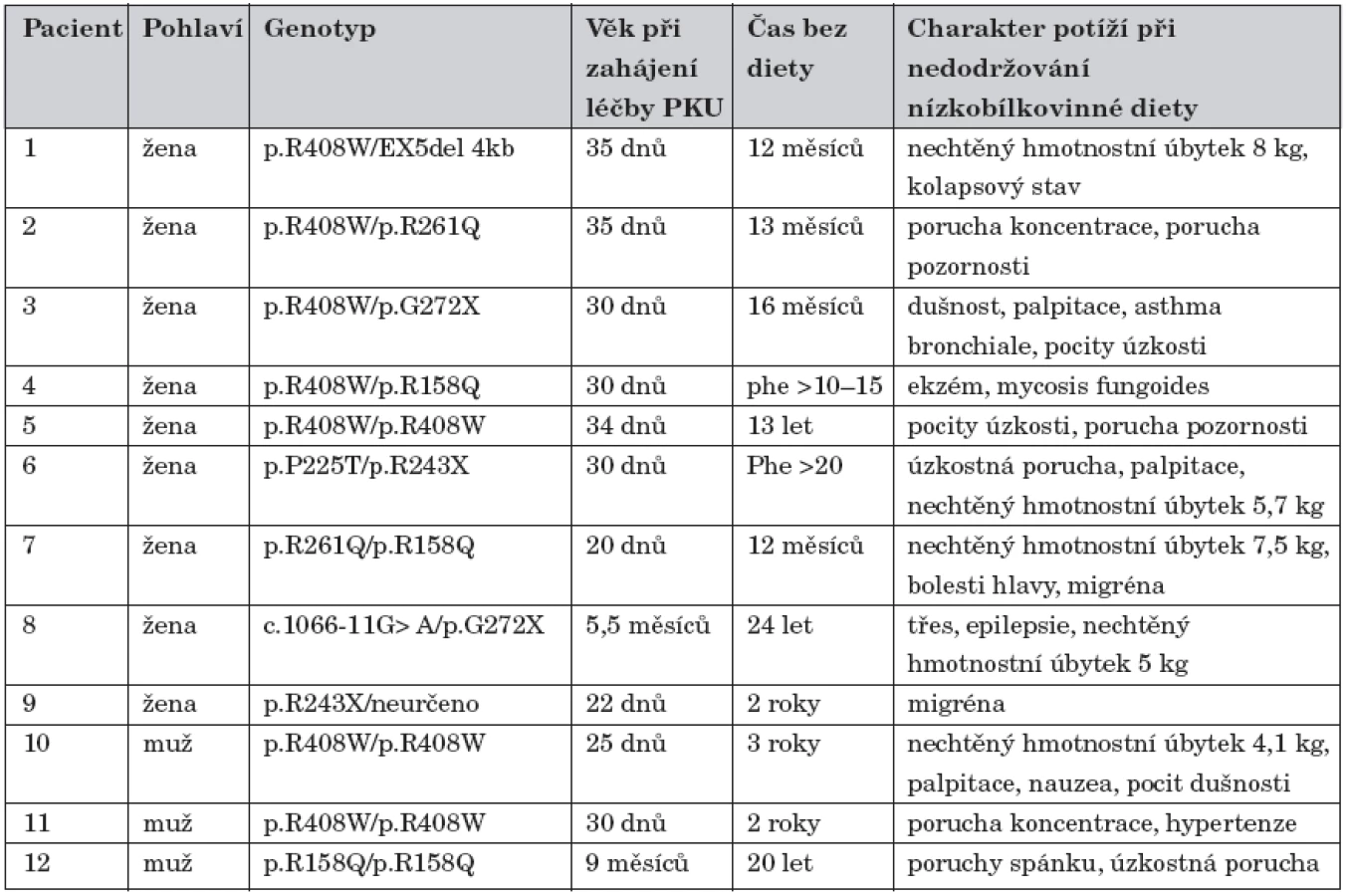
In the case of interruption or extenuation of the diet eleven patients (from 18 to 38 years old patients, 8 women and 3 men) restarted a low-protein diet because of following problems: body wasting, angst, breathlessness, anxious disorder and insomnia requiring psychiatric medication, activity disorder, migraine. One female patient was treated for mycosis fungoides. The most frequently observed mutations were p.R408W (41.7 % of patients), p.R158Q (16.7%) and p. R261Q (8.3%).
Key words:
PKU, adulthood, education, BMI, low-protein diet
:
D. Procházková 1; P. Konečná 1; L. Kolbová 1; E. Hrabincová 2; H. Vinohradská 3; H. Hrstková 1
:
1. dětská interní klinika LF MU a FN Brno
přednostka prof. MUDr. H. Hrstková, CSc.
1; Centrum molekulární biologie a genové terapie IHOK FN Brno
přednosta prof. MUDr. J. Vorlíček, CSc.
2; Oddělení klinické biochemie FN Brno
primář doc. MUDr. M. Dastych, CSc.
3
:
Čes-slov Pediat 2008; 63 (11): 601-605.
:
Original Papers
Autoři vyšetřili 100 dospělých pacientů s PKU a HPA ve věku od 18 do 43 let. 55 % pacientů nízkobílkovinnou dietu při PKU nedodržuje. 45 % je na nízkobílkovinné dietě, ale 22,2 % z nich mělo hladinu fenylalaninu v krvi vyšší než 20 mg/dl.
Dosažené vzdělání:
66,2 % výuční list, 21,7 % úplné střední vzdělání s maturitou, nadstavbové 1,2 %, vysokoškolské 1,2 %. Pomocnou školu absolvovalo 6,0 % pacientů a 3,6 % je bez vzdělání.
#BMI ženy:
23,6 ± 0,05 (16,8–43,6), BMI muži: 24,0 ± 0,8 (16,7–38,9). 7,4 % žen a 13,0 % mužů mělo BMI >30.
Jedenáct pacientů (18–38 let, 8 žen, 3 muži) se vrátilo na nízkobílkovinnou dietu po jejím přerušení nebo výrazném uvolnění z následujících důvodů: hubnutí, pocit úzkosti, dušnost, úzkostná porucha a porucha spánku vyžadující psychiatrickou léčbu, porucha pozornosti, migréna. Jedna žena je léčena pro mycosis fungoides. Nejčastějšími mutacemi v této skupině byly p.R408W (41,7 %), p.R158Q (16,7 %) a p.R261Q (8,3 %).
Klíčová slova:
PKU, dospělost, vzdělání, BMI, nízkobílkovinná dieta
Úvod
Fenylketonurie (PKU; OMIM 261600) je autozomálně recesivně dědičná porucha metabolismu aminokyseliny fenylalaninu (Phe), která je způsobena deficitem fenylalaninhydroxylázy (PAH; EC 1.14.16.1) v játrech. V současnosti je popsáno více než 500 mutací v PAH genu (12q24.1). V české populaci je nejčastější mutace p.R408W, kterou má 47,2 % pacientů [1]. Incidence klasické PKU v naší populaci je 1 : 9461 živě narozených dětí [2]. PKU je diagnostikována v rámci novorozeneckého screeningu, ze zákona od roku 1975 a poté následuje dietní restrikce fenylalaninu ve stravě. Nízkobílkovinná dieta je doporučována jako tzv. „diet for life“ celoživotně.
Časné přerušení nízkobílkovinné diety nebo její nedodržování při PKU je asociováno s následujícími potížemi: pokles IQ, agresivita, poruchy koncentrace, náladovost, syndrom ADHD, neschopnost dokončit školu, udržet si zaměstnání, úzkostná porucha, migréna, poruchy spánku, pyrománie a impotence u mužů. Častěji se vyvíjí astma. Je pozorována obezita s poruchou metabolismu tuků, u jiných naopak nechtěné hubnutí. Při kostní denzitometrii mohou být nalezeny změny kostní denzity ve smyslu minus – osteopenie, osteoporóza [3, 4]. Na NMR mozku jsou popisovány změny v oblasti bílé hmoty – leukodystrofie. V graviditě je pak nedodržování nízkobílkovinné diety spojeno s hyperfenylalaninovou embryopatií – syndromem maternální PKU, který nejčastěji zahrnuje mentální postižení, mikrocefalii, vrozenou srdeční vadu, dysmorfii, nízkou porodní hmotnost a syndrom ADHD. Jsou popsány i rozštěpové vady [5, 6, 7].
V literatuře existuje přes dva tisíce sdělení o PKU, problematikou dospělého věku se zabývá dosud asi 300 publikací. V článku předkládáme výsledky práce s dospělými s PKU v regionu Moravy.
Metoda
Na 1. dětské interní klinice FN Brno, ambulanci dědičných poruch metabolismu, jsme vyšetřili 100 dospělých pacientů s klasickou PKU (88 osob) a hyperfenylalaninémií (HPA; 12 osob). Jednalo se o 54 žen a 46 mužů ve věku od 18 do 43 let.
Pacienti byli zváženi a změřeni, vypočten index tělesné hmotnosti (BMI), klienti či jejich rodiče odpověděli na dotaz dosaženého ukončeného vzdělání či zda ještě studují, zda dodržují nízkobílkovinnou dietu a bylo provedeno měření hladiny fenylalaninu, kyseliny močové, glykémie, celkového cholesterolu a triacylglycerolů v krvi na lačno. Byl stanoven genotyp pacienta s PKU a HPA.
Výsledky byly zpracovány pomocí počítačového programu Microsoft Office Excel 2003.
Výsledky
Zjistili jsme, že 55 % pacientů nízkobílkovinnou dietu nedodržuje (22 žen, 33 mužů), 45 % udávalo dodržování nízkobílkovinné diety (32 žen, 13 mužů), ale 22,2 % (10 osob) z nich mělo hladinu Phe v krvi vyšší než 20 mg/dl (norma 0,6–2,0 mg/dl nebo 40–120 μmol/l) a dietu tedy nedodržovali přísně.
Celkem 83 pacientů s PKU a HPA ukončilo vzdělávací proces. Dosažené vzdělání: 66,2 % střední vzdělání (výuční list), 21,7 % úplné střední vzdělání s maturitou, 1,2 % nadstavbové a 1,2 % vysokoškolské vzdělání. Pomocnou školu absolvovalo 6,0 % pacientů a 3,6 % bylo bez vzdělání. Jednalo se o pacienty s DMO anebo další dosud neléčitelnou metabolickou poruchou. Ve skupině pacientů, kteří již nízkobílkovinnou dietu nedodržují, neměl nikdo vysokoškolské vzdělání, jeden pacient vysokou školu studuje, ale má HPA. Devět pacientů mělo ukončené středoškolské vzdělání s maturitou, 42 výuční list a 3 pomocnou školu.
Ve druhé skupině (s nízkobílkovinnou dietou) dokončila vysokou školu jedna pacientka, 6 dosud studuje. Jedna pacientka měla nadstavbové vzdělání, 9 dokončilo středoškolské vzdělání s maturitou a 10 dosud studuje. Výuční list získalo 13 probandů, 2 měli pomocnou školu a 3 byli bez vzdělání.
BMI ženy: 23,6 ± 0,05 (16,8–43,6), BMI >25–30 20,4 %, BMI >30 7,4 %, BMI <18 7,4 %. BMI muži: 24,0 ± 0,8 (16,7–38,9), BMI >25–30 30,4 %, BMI >30 13,0 %, BMI <18 6,5 %. Ve skupině pacientů bez diety byla průměrná hodnota BMI u žen 24,2 ± 4,4 a u mužů 25,4 ± 4,2 (od 19,2 do 35,7). 36,3 % pacientů mělo BMI >25–30 a u 9,1 % BMI dosahoval nad 30. Žádný pacient neměl BMI <18. V této skupině mělo 14 pacientů zvýšenou kyselinu močovou v krvi >339 µmol/l, 5 mělo glykémii na lačno >5,6 mmol/l, 7 pacientů vykazovalo hodnotu celkového cholesterolu v krvi >5,2 mmol/l a u 7 byla hodnota triacylglycerolů v krvi na lačno >2,0 mmol/l.
Ve skupině pacientů dodržujících nízkobílkovinnou dietu byla průměrná hodnota BMI u žen 23,0 ± 5,3, u mužů 23,4 ± 5,7 (od 16,7 do 43,6). BMI >25–30 mělo 11,1 % pacientů a 11,1 % mělo BMI >30. Oproti tomu BMI <18 mělo 15,5 % dospělých na nízkobílkovinné dietě. Vyšetřované biochemické parametry byly v normě.
Zvláštní pozornost jsme věnovali pacientům, kteří se na nízkobílkovinnou dietu vrátili po přerušení nebo výrazném uvolnění diety při PKU: 8 žen a 3 muži ve věku 18–38 let. K jejich rozhodnutí je vedly následující zdravotní potíže: nechtěný úbytek hmotnosti, pocit úzkosti, úzkostná porucha a porucha spánku vyžadující psychiatrickou léčbu, porucha pozornosti, porucha soustředění, palpitace, migréna a hypertenze. Jedna pacientka měla třes, epilepsii a hmotnostní úbytek, ale návrat na nízkobílkovinnou dietu se nezdařil. Další pacientka na nízkobílkovinné dietě (Phe v krvi <15 mg/dl) se léčila dva roky pro ekzém, rezistentní na terapii. Po dvou letech byl po nástupu otoku kůže a horečky diagnostikován kožní T-lymfom: mycosis fungoides. Tato forma lymfomu může mít na kůži zpočátku charakter psoriázy či erytrodermie.
Charakteristiku pacientů, kteří se vrátili na nízkobílkovinnou dietu při PKU, sumarizuje tabulka 1.
Diskuse
Studie ukazují, že celoživotní dodržování nízkobílkovinné diety může být pro některé pacienty s PKU problematické. Ve studii Waltera bylo 17 % vzorků krve desetiletých dětí nad doporučenou hladinou fenylalaninu v krvi, zatímco u dvacetiletých to bylo již 75 %. Pravidelnou kontrolu Phe v krvi podstupovalo 83 % pacientů v 10 letech, ve 20 letech již jen 51 % [8]. Ačkoli více než 50 % pacientů v dospělosti udávalo zlepšenou kvalitu života při dodržování nízkobílkovinné diety při PKU, 47 % nemělo dobrou dietní compliance [9]. Téměř polovina dospělých s PKU na kontroly nechodila pravidelně či vůbec [10].
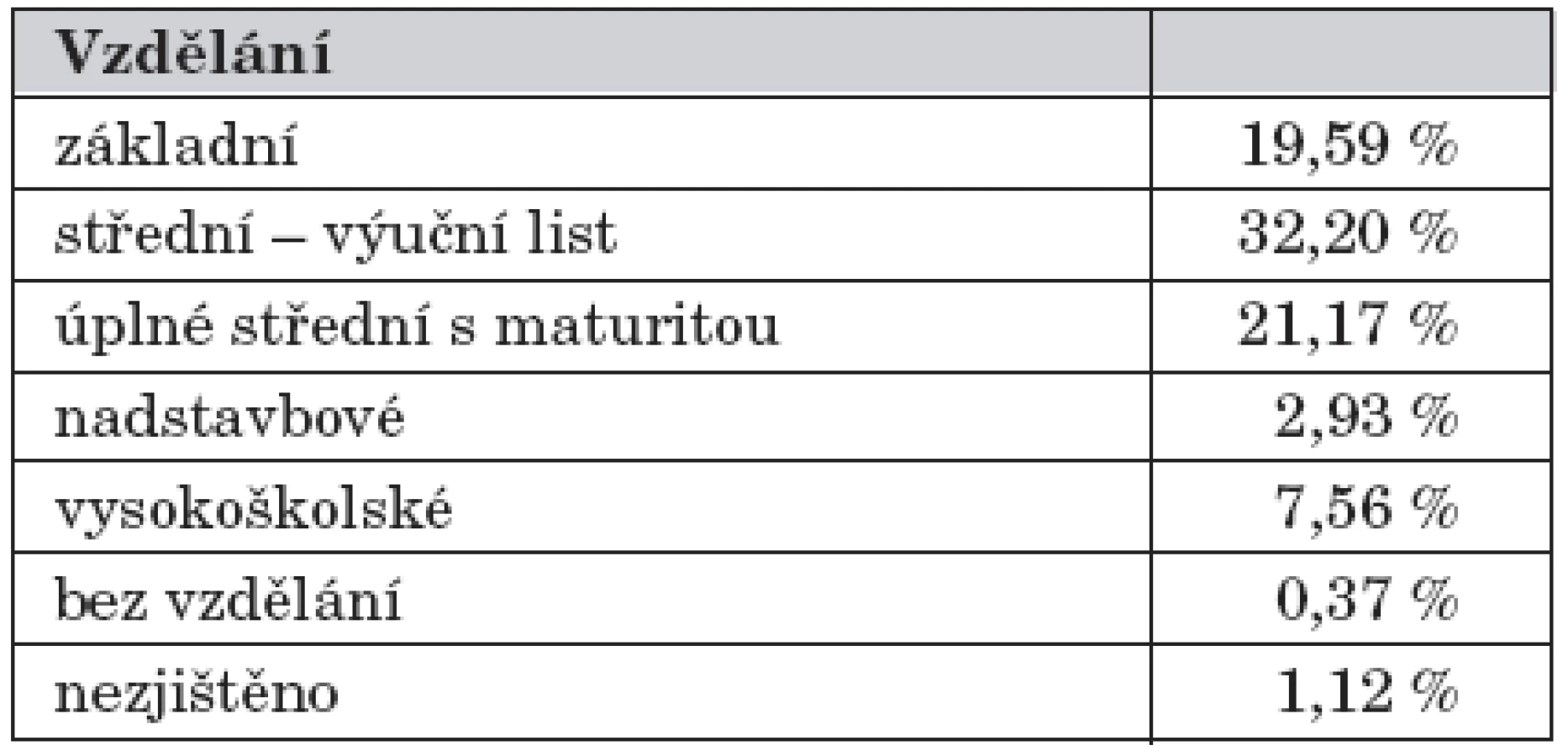
Při srovnání dosaženého vzdělání pacientů s PKU a HPA s údaji Českého statistického úřadu o dosaženém vzdělání české populace (tab. 2) jsme zjistili, že u našich pacientů převažovalo střední vzdělání s výučním listem (66,2 %) a procento pacientů s nadstavbovým (1,2 %) a vysokoškolským vzděláním (1,2 %) bylo nižší. Oproti tomu procentuálně vyšší byl výskyt pacientů s pomocnou školou (6 %) a bez vzdělání (3,6 %). Na druhé straně můžeme říci, že se v současné době zvýšil podíl pacientů studujících vysokou školu ve skupině dodržujících nízkobílkovinnou dietu do 25 let věku.
Je všeobecně akceptováno, že pacienti s PKU mají nižší IQ oproti populaci a pro jejich konečné IQ je rozhodující věk 2 roky [11]. Ukazuje se, že i časně léčení pacienti s PKU mají specifickou kognitivní poruchu, která zahrnuje exekutivní funkce, pozornost, verbální paměť, vyjadřovací schopnost a verbální fluenci. Výsledky psychologických testů kognitivních funkcí ukazují souvislost s dietní compliance v dětství [4, 10].
Ve skupině pacientů s pomocnou školou či bez vzdělání byli pacienti s různým stupněm mentálního postižení. Získat adekvátní odpověď od rodičů těchto pacientů byla citlivá záležitost. Setkali jsme se s tím, že pacient školu dokončil, ale ani jednoduchou práci nezvládal nebo studoval již několikáté zvláštní učiliště za sebou, protože neměl kde trávit čas. Je důležité, zda jsou do vyšetřované skupiny zahrnuti i pacienti s mentálním postižením. Studie s vybranou skupinou pacientů s PKU mohou vést k výsledkům, kdy např. procentuální zastoupení vysokoškolsky vzdělaných pacientů dosahuje procentuálního zastoupení v populaci [12].
Pro srovnání indexu tělesné hmotnosti našich pacientů jsme vycházeli ze dvou zdrojů. Ze široké studie české populace, zahrnující 3053 vyšetřovaných osob Wagenknechta [13], která udávala: BMI muži 26,04 ± 3,84 (průměr ± SD), BMI ženy 24,83 ± 4,8 (průměr ± SD). Dalším zdrojem byla data Českého statistického úřadu: Česká republika v mezinárodním srovnání stran obezity z roku 2002. Ve studii bylo zjištěno, že 14,8 % žen a 13,4 % mužů starších 15 let mělo BMI >30.
Lze říci, že český pacient s PKU bez ohledu na pohlaví má průměrný BMI se sklonem k nadváze a obezitě. Tento stav může souviset se zvýšeným podílem sacharidů v nízkobílkovinné dietě při PKU či s dalším dosud neobjasněným metabolickým defektem v oblasti tukového metabolismu. V našem souboru mělo 7 % pacientů zvýšenou hladinu celkového cholesterolu a triacylglycerolů v krvi na lačno. V jiných studiích byla prokázáno zvýšení celkového cholesterolu, LDL cholesterolu a triacylglycerolů v krvi na lačno až u 15 % dospělých s PKU. Z těchto důvodů doporučujeme monitorovat lipidový profil u dospělých s PKU vzhledem k možnému zvýšenému riziku pro arteriosklerózu [14].
Ve skupině pacientů s PKU, kteří se vrátili po přerušení nebo uvolnění zpět na nízkobílkovinnou dietu, převažovaly ženy. Nejčastější mutací v souboru byla p.R408W (42,7 %), ale ta je i nejčastější v populaci (47,2 %). Dalšími nejčastějšími byly p.R158Q (16,7 %) a p.R261Q (8,3 %). Jejich výskyt v české populaci činí 4,9 % a 1,6 % [1]. U těchto dvou mutací byla popsána potencionálně možná senzitivita na léčbu tetrahydrobiopterinem, který je kofaktorem fenylalaninu [15].
Terapie našich pacientů byla zahájena později oproti dnešku, kdy děti odcházejí v novorozeneckém věku domů s kompenzovanou PKU. Skupina zahrnovala dva pacienty s pozdě diagnostikovanou PKU před zahájením novorozeneckého celoplošného screeningu. Pokles hladiny fenylalaninu v krvi na terapeutickou hladinu po stanovení diagnózy PKU byl ve vyšetřované skupině dosažen během 19–44 dnů. Dietní compliance v dětském věku lze u tří žen a jednoho muže označit za velmi dobrou. U ostatních vyšetřovaných osob hladiny Phe v krvi překračovaly doporučené hodnoty.
Doba od opuštění či uvolnění nízkobílkovinné diety k počátkům klinických potíží nebyla konstantní. U některých pacientů ani jednoznačně nedošlo k přerušení diety, ale na tento stav lze usuzovat z poklesu preskribce směsí aminokyselin bez fenylalaninu, snížení frekvence kontrol v ambulanci, ze snížení frekvence zasílání kontrolních suchých skvrn na hladinu fenylalaninu v krvi z místa bydliště či zvýšení hladin fenylalaninu v krvi při kontrolách v naší ambulanci. Pokud pacient dietu při PKU opustil, vrátil se na dietu sám jen na základě výrazných zdravotních potíží. Oproti dětství pak nízkobílkovinnou dietu dodržoval striktně.
Je otázkou, proč u některých pacientů s PKU k zdravotním potížím bez nízkobílkovinné diety dochází a u jiných nikoli. V současné době se předpokládá, že tento stav ovlivňuje interindividuální prostupnost molekul fenylalaninu přes hematoencefalickou bariéru a tedy rozdílná koncentrace hladin fenylalaninu v krvi a v mozku [16]. Uvažuje se také o interindividuální toleranci fenylalaninu ve stravě, kdy pacienti se stejným genotypem při stejné dodávce přirozených bílkovin mají rozdílnou hladinu fenylalaninu v krvi. K řešení těchto otázek přispěly metody molekulární genetiky a studie sourozenců s PKU a HPA se stejným genotypem [17]. Možným vysvětlením je i tzv. negativní intraalelový komplementační efekt, kdy je pacient nosičem alely PAH genu, u jejíhož produktu předpokládáme vyšší zbytkovou enzymatickou aktivitu v in vitro studiích, ale ve skutečnosti je hladina fenylalaninu v krvi vyšší oproti očekávání. Tato pozorování ukazují, že mutace, které kompletně nedestruují gen pro PAH, mohou vést k variabilnímu fenotypu, který nelze dopředu odhadnout [18]. Na závěr lze uzavřít, že každý pacient s PKU má fenotyp, který nelze jednoduše a jediným způsobem vysvětlit a podle toho by se mělo přistupovat k jeho léčbě [19].
Závěr
Pacientům doporučujeme celoživotní nízkobílkovinnou dietu při PKU. Hladina fenylalaninu v krvi by měla být v rozmezí 2–4 mg/dl ve věku od 0 do 6 let, v rámci těhotenské přípravy a po celou dobu gravidity. Ve školním věku posunujeme horní hranici na 6–8 mg/dl. Doporučené hladiny fenylalaninu v krvi pro jednotlivá věková období a maternální PKU jsou předmětem diskusí na celém světě a liší se v jednotlivých státech Evropské unie. Podle našich zkušeností by hladina fenylalaninu v krvi neměla v dospělosti převyšovat 15 mg/dl.
Došlo: 7. 11. 2007
Přijato: 1. 3. 2008
MUDr. Dagmar Procházková, Ph.D.
1. dětská interní klinika FN Brno
Černopolní 9
625 00 Brno
e-mail: prochazkovad@fnbrno.cz
Sources
1. Kozák L, Blažková M, Kuhrová V, et al. Mutation and haplotype analysis of phenylalanine hydroxylase alleles in classical PKU patients from the Czech Republic: identification of four novel mutations. J. Med. Genet. 1997;34 : 893–898.
2. Čechák P, Hejcmanová L, Procházková D, et al. Výsledky screeningu hyperfenylalaninémií v českých zemích v letech 1970–2000. Čes.-slov. Pediat. 2001;56 : 667–670.
3. Zeman J, Bayer M, Stepan J. Bone mineral density in patients with phenylketonuria. Acta Pediatr. 1999;88 : 1348–1351.
4. Koch R, Buton B, Hoganson G, et al. Phenylketonuria in adulthood: a collaborative study. J. Inherited Metab. Dis. 2002;25 : 333–346.
5. Antshel KM, Waisbren SE. Developmental timing of exposure to elevated levels of phenylalanine is associated with ADHD symptom expression. J. Abnorm. Child Psychol. 2003;31 : 565–574.
6. Hyanek J, Kozak L, Hrabincova E, et al. Maternal hyperphenylalaninemias in healthy Czech population of pregnant women: 30 years experience with screening, prevention and treatment. Bratisl. lek. Listy 2004;105 : 291–298.
7. Procházková D, Konečná P, Kozák L, et al. Maternální fenylketonurie (PKU) v regionu Moravy. Čes.-slov. Pediat. 2005;60 : 251–256.
8. Walter JH, White FJ. Blood phenylalanine control in adolescents with phenylketonuria. Int. J. Adolesc. Med. Health 2004;16 : 41–45.
9. Gassio R, Campistol J, Vilaseca MA, et al. Do adult patients with phenylketonuria improve their quality of life after introduction/resumption of a phenylalanine-restricted diet? Acta Pediatr. 2003;92 : 1474–1478.
10. Ürge O, Strnová J, Mosendzová B. Fenylketonúria adolescentov a dospelých na Slovensku. Čes.-slov. Pediat. 2003;58 : 423–425.
11. Griffiths PV, Demelweek C, Fay N, et al. Wechsler subscale IQ and subtest profile in early treated phenylketonuria. Arch. Dis. Child. 2000;82 : 209–215.
12. Bosch AM, Tybout W, van Spronsen FJ, et al. The course of life and quality of life of early and continuosly treated Dutch patients with phenylketonuria. J. Inherit. Metab. Dis. 2007;30 : 29–34.
13. Wagenknecht M, Gauner V, Kunešová M, et al. Vztahy mezi faktory ,,Dotazníku jídelních zvyklostí“, socioekonomickým stavem a zdravotními riziky u české populace. Čas. Lék. čes. 2007;146 : 284–291.
14. Moseley R, Koch R, Moser AB. Lipid status and long-chain polysaturated fatty acid concentrations in adults and adolescents with phenylketonuria on phenylalanine restricted diet. J. Inherit. Metab. Dis. 2002;25 : 56–64.
15. Muntau AC, Röschinger W, Habich M, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N. Engl. J. Med. 2002;26 : 2122–2132.
16. Miller HE, Weglage J, Bick U, et al. Brain imaging and proton magnetic resonance spectroscopy in patients with phenylketonuria. Pediatrics 2003;112 : 1580–1583.
17. Weglage J, Wiedermann D, Denecke J, et al. Individual blood-brain barier phenylalanine transport in siblings with clasiccal phenylketonuria. J. Inherit. Metab. Dis. 2002;25 : 431–436.
18. Burgard P, Rupp A, Konecki DS, et al. Phenylalaninhydroxylase genotypes, predicted residua enzyme activity and phenotypic parameters of diagnosis and treatment of phenylketonuria. Eur. J. Pediatr. 1996;155(Suppl 1): 11–15.
19. Scriver ChR. The PAH gene, phenylketonuria, and a paradigma shift. Human Mutation 2007;28 : 831 – 845.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2008 Issue 11
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Phenylketonuria in Adulthood
- Choledochal Cyst – Clinical Manifestations, Surgical Technique and Results
- Pitfalls of Prenatal Ultrasound Screening in Diagnostics of Serious Inborn Developmental Defects of the Kidney and Urinary Pathways
- Case Report of Injury in Rectum-Sigmoid Region in a Fifteen-Year Boy