
„Café-au-lait“ makuly v diagnostike detských nádorových ochorení
„Café-au-lait“ Maculae in the Diagnostics of Child Tumorous Diseases
Café-au-lait maculae are the most frequently associated with neurofibromatosis type 1 disorder. Patients with NF1 develop light - to dark-brown CAL spots with smooth or irregular margins. The diagnosis is confirmed by presence of multiple spots, and at the prepubertal age the finding of six or more CAL maculae larger than 0.5 cm in diameter and at the postpubertal age those larger than 1.5 cm in diameter are required.
However, the finding of hyperpigmentations may represent lots of other genetic or genetically determined disorders. Many of them may predispose to a multiorgan injury or to development of malignancy.
This work describes a group of disorders characterized by CAL maculae that are also associated with:
1. multisystemic affection, 2. inherited diseases and malignances, 3. chromosomal instability syndromes and malignances, 4. neurofibromatosis and malignances, 5. a new syndrome of constitution deficit “ DNA repair system” (CMMR-D). The data about the character of CAL maculae, typical symptoms and type of malignances in individual syndromes serve to improve the differential diagnosis of the tumor disorders in childhood. Molecular diagnostics of germinal mutations may detect very effectively families at high risk for malignancy and help provide primary prevention.
Key words:
café-au-lait, neurofibromatosis type 1, Watson syndrome, NF like, CMMR-D, malignance in childhood
Authors:
D. Ilenčíková 1,2; D. Ďurovčíková 2; L. Copáková 1; D. Štrbová 1; J. Šuvada 1; A. Rybárová 4; A. Hlavatá 4; K. Wimmer 3
Authors‘ workplace:
Oddelenie onkologickej genetiky, Národný onkologický ústav, Bratislava
primárka MUDr. D. Ilenčíková, PhD.
1; Katedra klinickej genetiky SZU, Bratislava
vedúca doc. MUDr. D. Ďurovčíková, CSc.
2; Oddelenie biológie a genetiky, Viedenská univerzita, Viedeň
vedúca prof. RNDr. Ch. Fonatsch
3; II. Detská klinika LFUK a DFNsP, Bratislava
prednosta prof. MUDr. L. Kovács, DrSc.
4
Published in:
Čes-slov Pediat 2009; 64 (9): 406-414.
Category:
Review
Overview
„Cafe-au-lait“ (CAL) sú najčastejšie asociované s ochorením Neurofibromatóza 1 (NF1). CAL majú u NF1 slabo až tmavohnedé zafarbenie a hladké alebo nepravidelné okraje. Diagnózu potvrdzuje prítomnosť mnohopočetných fľakov, v predpubertálnom veku je podmienkou nález 6 a viac CAL väčších než 0,5 cm a v postpubertálnom období väčších ako 1,5 cm.
Nález hyperpigmentácií však môže skrývať mnoho ďalších genetických alebo geneticky podmienených ochorení. Mnohé z nich predisponujú ku vzniku multiorgánového postihnutia alebo vzniku malignity.
Predložená práca opisuje skupinu ochorení charakteristickú pre CAL:
1. multisystémovým postihnutím, 2. dedičnými ochoreniami a malignitami, 3. syndrómami chromozómovej instability a malignitami, 4. neurofibromatózami a malignitami, 5. novým syndrómom konštitučného deficitu „DNA reparačného systému“ (CMMR-D). Prehľadné údaje o charaktere CAL, typických znakoch a druhu malignít u jednotlivých syndrómov slúži k zlepšeniu diferenciálnej diagnostiky nádorových ochorení u detí. Molekulová diagnostika zárodočných mutácií môže veľmi efektívne určiť vysokorizikové rodiny pre vývoj malignít a pomôcť zabezpečiť primárnu prevenciu.
Kľúčové slová:
café-au-lait, neurofibromatóza 1, Watsonov syndróm, NF like, CMMR-D, malignity detského veku
Úvod
V bežnej populácii sú solitárne „café-au-lait“ (CAL) ploché kávové škvrny na koži pomerne časté, kým mnohopočetné CAL sú indikátorom geneticky podmienených ochorení. CAL sú hyperpigmentované lézie, ktoré varírujú od svetlo k tmavohnedému zafarbeniu. Ich okraje môžu byť hladké, pravidelné alebo nepravidelné s obrovskými melanozómami, ktoré obsahujú zvýšené množstvo melanínu. CAL u pacientov s neurofibromatózou 1 (NF1) vykazuje v porovnaní s pacientami bez asociácie s NF1 signifikantne vyššiu melanocytovú denzitu.
Sofie De Schepper et al. [1] dokázali na zvýšení melanocytovej denzity u NF1 pacientov v porovnaní s non-NF1 populáciou účasť faktora kmeňových buniek (SCF-stem cell factor). Napriek tomu, že SCF je dôležitý cytokín, v koži pacientov s NF1 sú pre indukciu NF1-špecifických hyperpigmentácií potrebné aj iné rastové faktory alebo genetické mechanizmy. Medzi najčastejšie geneticky podmienené ochorenie s manifestáciou CAL až u 99 % pacientov do 12. roku života patrí neurofibromatóza 1, ktorá má presné diagnostické kritériá. CAL makuly sa okrem NF1 môžu vyskytovať aj pri iných ochoreniach.
Niekoľko prác publikovalo kazuistiky detí s mnohopočetnými CAL makulami v rámci multisystémového ochorenia. Korf [2] referuje o vyšetrení 41 detských pacientov s kožným nálezom 6 a viac CAL vo veku od 1 mesiaca do 14. roku života, z ktorých 24 detí malo známky charakteristické pre NF1. U 6 sa na základe ich distribúcie potvrdil segmentálny typ NF1. U troch detí diagnostikoval Leopardov syndróm s početnými lentiginóznymi morfami, u ďalších troch McCune Albrightov syndróm s veľkými pruhovitými hyperpigmentáciami. Ostatné diagnózy zahŕňali iné zriedkavé nozologické jednotky ako napríklad Russelov-Silver a Chediakov-Hadachiho syndrómy.
Prehľad monogénovo podmienených genetických ochorení s výskytom hyperpigmentácií bez malignity uvádzame v tabuľke 1.
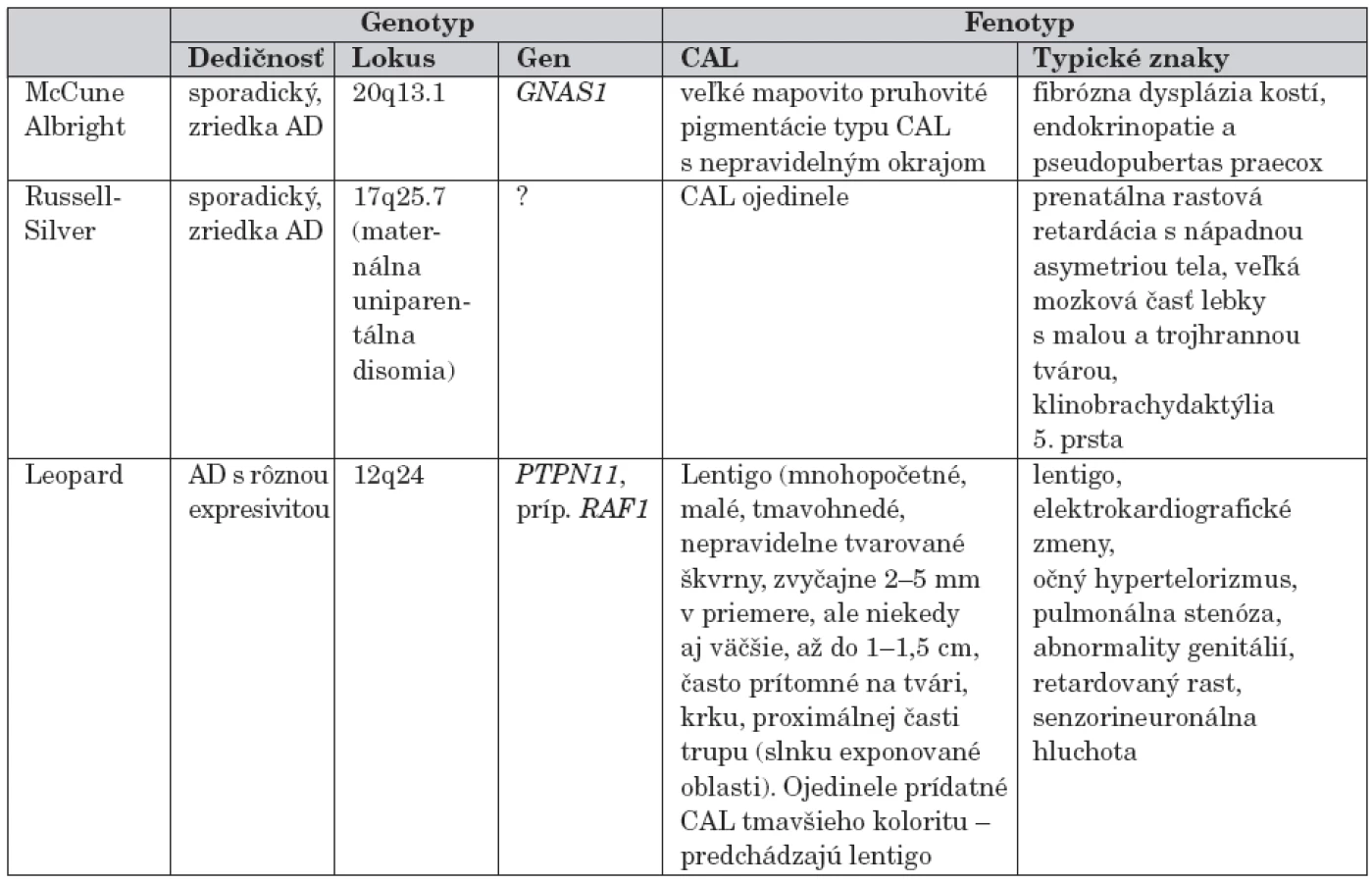
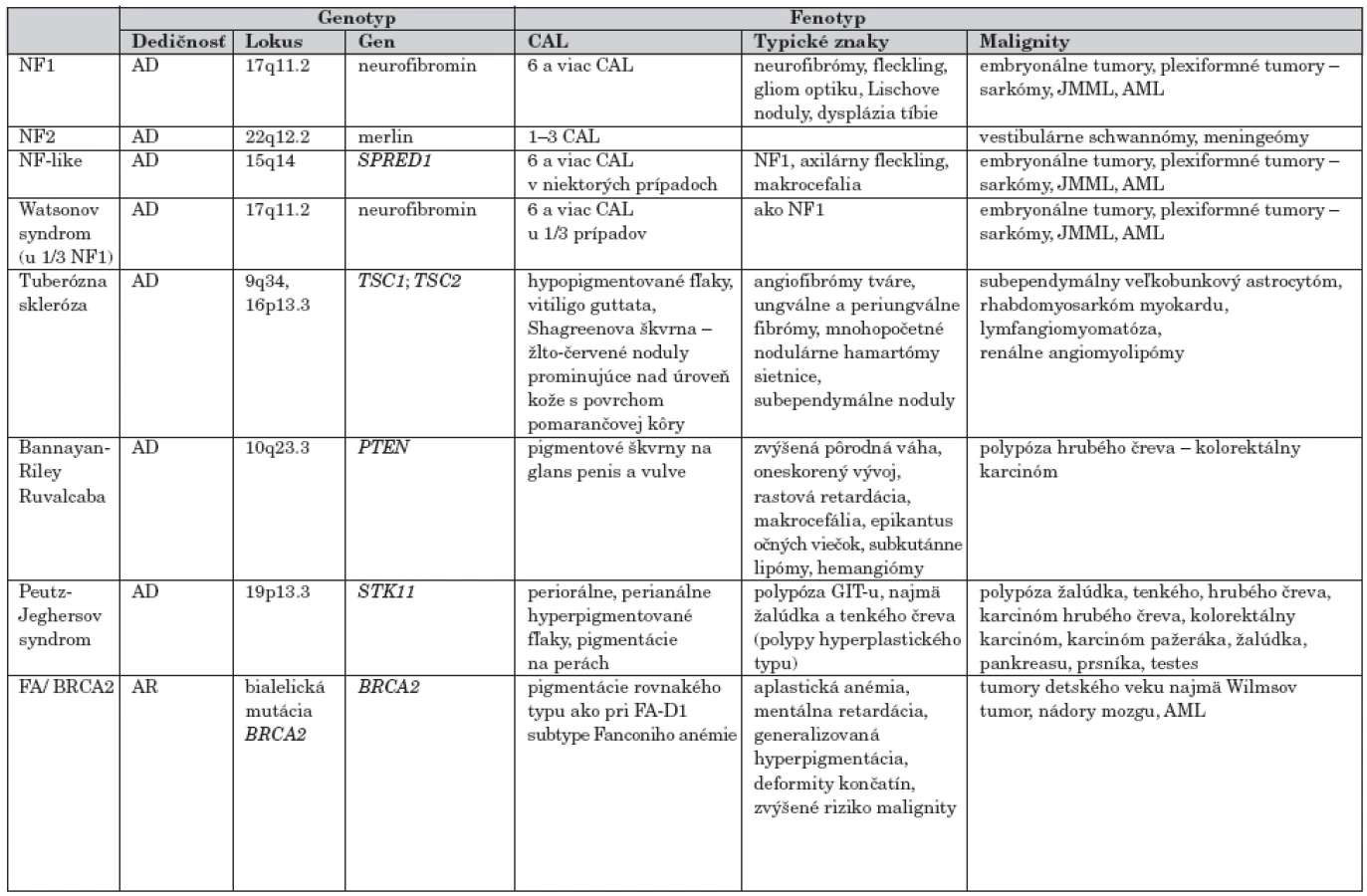
Podľa frekvencie výskytu je ďalším ochorením s hyperpigmentovými makulami tuberózna skleróza. Pigmentové škvrny s lokalizáciou na glans penis a vulve sú charakteristické pre Bannayan-Riley Ruvalcaba, na perách, periorálne a perianálne ich pozorujeme u pacientov s Peutz-Jeghersovým syndrómom. U pacientov s uvedenými dedičnými ochoreniami sa okrem hyperpigmentácií môžu vyskytovať rôzne typy malignít (tab. 2).
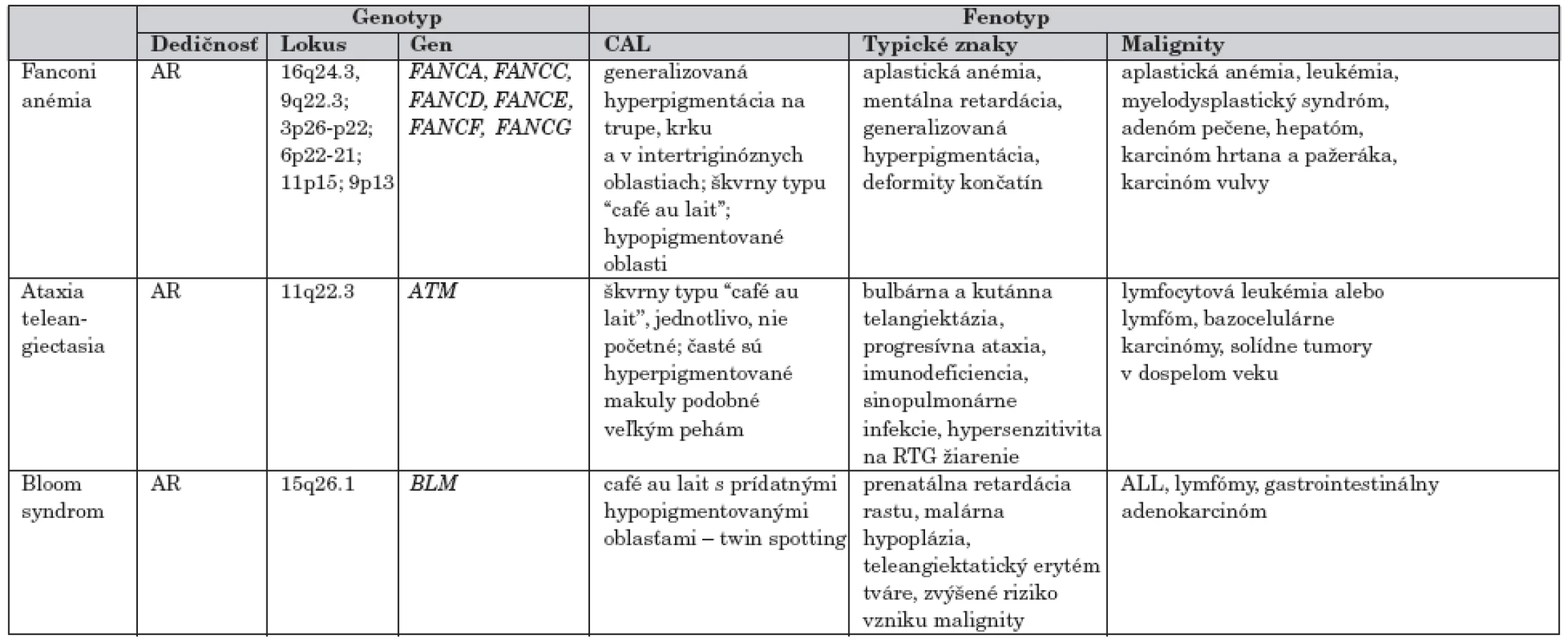
Hypo - hyperpigmentované makuly v rôznych lokalitách na koži krku, trupu a intertriginózne nachádzame tiež u pacientov zo skupiny syndrómov chromozómovej instability, ktoré predisponujú k vývoju hematologických malignít ako aj rôznych typov solídnych tumorov (tab. 3).
Výskyt mnohopočetných CAL makúl pozorovaný v rodinách viacgeneračne bez prítomnosti ďalších významných príznakov svedčí pre samostatnú autozómovo dominantnú nozologickú jednotku. CAL makuly teda tvoria skupinu geneticky heterogénnych chorôb.
Podľa dermatologických vyšetrení bývajú hyperpigmentácie podobné CAL označované pod názvom névus pilus a lentigo. Névus pilus predstavuje mnohopočetné malé, tmavé névy, ktoré sú často rozosiate na slabo hyperpigmentovanom teréne kože a lentigá sú tmavé silne pigmentované mnohopočetné makuly v priemere niekoľko milimetrov.
Klinický význam detekcie CAL u detí umožňuje včas rozpoznať potenciálne rizika multiorgánového poškodenia a riziko vývoja malignity.
V ďalšom texte uvádzame prehľadnú charakteristiku zriedkavých CAL syndrómov, ktorá môže napomôcť k odhaleniu horeuvedeného rizika a ďalšiemu manažmentu zdravotnej starostlivosti.
1. CAL makuly v rámci ochorení s multiorgánovými zmenami
Táto skupina ochorení asociuje veľmi zriedkavo s malignitou.
McCune Albrightov syndróm (MCS)
Patrí k autozómovo-dominantným (AD) ochoreniam s častým sporadickým výskytom, ktoré okrem CAL charakterizujú iné významnejšie znaky (endokrinopatie, polyostotická fibrózna dysplázia kostí). Najčastejšou kauzálnou príčinou je mutácia génu kódujúceho alfa podjednotku G proteínu (GNAS1). CAL majú zvyčajne nepravidelné okraje a sú väčšie než u ostatných syndrómov. Najčastejšie sa objavujú v sakrálnej, gluteálnej oblasti a u 50 % pacientov sú unilaterálne.
Leopardov syndróm (LS)
Leopardov syndróm býva nazývaný aj Gorlinov syndróm II alebo syndróm mnohopočetného lentiga a patrí k zriedkavému, AD multisystémovému ochoreniu. Jeho názov je odvodený od komplexu znakov: Lentigo, Elektrokardiografické zmeny, Okulárny hypertelorizmus, Pulmonálna stenóza, Abnormality genitálií, Rastová retardácia, Deafness.
Až 70 % prípadov má familiárny výskyt a sú asociované s mutáciou v géne kódujúcom proteín tyrosínovej fosfatázy (PTPN11). Lentigo a lentiginy sa vyskytujú až u 90 % prípadov. Lentigo je prítomné pri narodení alebo sa vyvinie v detstve, časom škvrny tmavnú a ich počet sa zvyšuje (10 000 a viac, pokrývajú veľkú časť tela). Lentiginy sú malé, tmavohnedé, polygonálne, nepravidelné tvarované makuly, zvyčajne 2–5 mm v priemere, často prítomné na tvári, krku, proximálnej časti trupu, ale tiež na dlaniach, plantách a sklérach a genitáliách. Okrem týchto škvŕn sa popisujú iné zmeny kože, pehy v axilách (freckling) a lokalizované hypopigmentácie podobné CAL pri NF1 [3].
2. CAL makuly asociované s ochoreniami so zvýšeným rizikom malignity
Neurofibromatóza 1 (NF1)
Ide o najfrekventovanejšie autozómovo dominantné ochorenie (1 : 3000), kde CAL makuly sú hlavným príznakom klinického spektra je NF1. Vzniká v dôsledku mutácie NF1 tumor-supresorového génu, ktorého produktom je neurofibromín. Neurofibromín inhibuje proliferáciu buniek moduláciou cesty mitogénnej aktivity prostredníctvom inaktivácie p21ras protoonkogénu. Stratou funkcie génu dochádza k aktivácii RAS, čoho dôsledkom je zvýšená proliferácia buniek spojená s rastom tumorov. U približne 30–50 % pacientov vzniká NF1 ako mutácia de novo [4].
Diagnostika ochorenia si vyžaduje splnenie aspoň dvoch z nasledujúcích kritérií: prítomnosť CAL, kožných/podkožných neurofibrómov alebo plexiformného neurofibrómu, hyperpigmentovaných škvŕn v axilárnej a inguinálnej oblasti, Lischových uzlíkov (hamartómov dúhovky), gliómov optického nervu, charakteristických kostných dysplázií a/alebo pseudoartritídy a existencia priameho príbuzného s NF1 [4]. U jedincov s NF1 je zvýšené riziko hematologických a rôznych nádorových malignít, tak ako uvádza tabuľka 5.
Watsonov syndróm (WS)
Prítomnosť CAL makúl asociované s pulmonálnou valvulárnou stenózou, nízkym vzrastom a makrocefaliou sa klasifikuje ako Watsonov syndróm. Keďže sa u niektorých pacientov zistila mutácia NFl génu, vznikol predpoklad, že Watsonov syndróm a neurofibromatóza 1 sú alelické ochorenia. V oblasti 17q však existuje séria susediacich génov pre pulmonálnu stenózu, neurokutánne anomálie, nízky vzrast a mentálnu retardáciu, ktoré podporujú ďalšiu etiológiu ako syndrómu naliehajúcich génov [5].
NF1-like syndróm (NF1-like)
V ostatnom čase Brems H. et al. [6] identifikovali ako prví u pacientov s fenotypom podobným NF1 mutáciu v inom géne (SPRED1) nachádzajúcom sa v lokuse 15q25. Niektorí z nich mali dysmorfiu tváre podobnú Noonanovej syndrómu, kongenitálne VVCH srdca a problémy s učením. Doterajšie publikované prípady vykazovali autozómovo dominantný typ dedičnosti [6].
Neurofibromatóza 2 (NF2)
Neurofibromatóza 2 predtým nazývaná ako centrálna forma neurofibromatózy je autozómovo dominantné ochorenie s podstatne nižšou frekvenciou výskytu 1 : 40 000 živonarodených. Asi polovicu prípadov tvoria nové mutácie. CAL makuly, ktoré sa vyskytujú v malom počte, nie sú hlavným rozpoznávajúcim znakom ochorenia.
Ochorenie charakterizuje výskyt bilaterálnych vestibulárnych schwanómov a častokrát mnohopočetných nádorov mozgu a miechy ako napríklad meningeómy a gliómy. Symptomatológia v detstve je zriedkavá. Nádory sa zvyčajne diagnostikujú u mladých ľudí po 20. roku života, nezriedka s komplikáciami. Gén pre NF2 je lokalizovaný na chromozóme 22. Jeho produktom je merlín, ktorý patrí do skupiny proteínov asociovaných s cytoskeletom bunky. Má dôležitú úlohu pre integritu a stabilitu bunkovej membrány.
Tuberózna skleróza (TS)
Tuberózna skleróza je AD ochorenie charakteristické rastom početných nenádorových tumorov. Symptomatológia varíruje v závislosti od lokalizácie a samotnej veľkosti tumorov. Najčastejšie sú lokalizované na koži, mozgu, obličkách, ale aj iných orgánoch. Postihnutí pacienti majú okrem hyperpigmentácií typu café-au-lait prítomné aj iné kožné zmeny – hypopigmentované, hypomelanotické makuly, elipsovitého tvaru, rôznej veľkosti, ktoré jemne prominujú nad úroveň kože. Ďalej sú to tvárové angiofibrómy v nosovolícnej lokalite a benígne fibrómy [7]. Pacienti môžu mať rôznu neurologickú symptomatológiu.
Ochorenie je geneticky heterogénne v kauzalite s mutáciami v géne TSC1, TSC2. Medzi malignity asociované s tuberóznou sklerózou patrí subependymálny veľkobunkový astrocytóm, rhabdomyosarkóm myokardu, lymfangiomyomatóza, prípadne renálne angiomyolipómy.
Peutz-Jeghersov syndróm (PJS)
Je autozómovo dominantne dedičný syndróm, inak označovaný aj ako syndróm mukokutánnej pigmentácie a intestinálnej polypózy. Ochorenie je podmienené mutáciou v géne STK11. CAL nie sú prítomné. Tmavé hnedé alebo šedomodročierne pigmentované škvrny vyskytujúce sa periorálne sú sprievodným znakom ochorenia. Ďalším významným znakom je hamartomová polypóza tráviaceho traktu – zvlášť jejuna a ilea, ochorenie býva diagnostikované okolo 22. roku života.
Viac ako polovica pacientov je v priebehu života postihnutá gastrointestinálnymi, ale aj extragastrointestinálnymi malígnymi tumormi ako nádor pankreasu, duktálny karcinóm prsníka, adenokarcinóm pľúc a nádor ovária.
Bannayan Riley Ruvalcaba syndróm (BRRS)
Patrí k zriedkavým autozómovo dominantným syndrómom nadmerného vzrastu s ojedinelými CAL makulami, hamartomatóznymi léziami a pigmentáciami na glans penis u mužov a vulve u žien. Kauzálnou príčinou je mutácia v géne PTEN. Z malignít sa u pacientov najčastejšie vyskytuje kolorektálny karcinóm na podklade polypózy (hamartómového typu).
FA/BRCA2 syndróm (FABRS)
Je spôsobený bialelickou recesívnou mutáciou génu BRCA, ktorého fenotypový prejav je identický s niektorými subtypmi Fanconiho anémie (FA-D1).
Ochorenie je sprevádzané kožnými prejavmi hyperpigmentácií ako pri Fanconiho anémii, teda charakteristickými CAL. Malignity asociované s bialelickou mutáciou BRCA2 sa vyskytujú najmä v detskom veku. Jedná sa o Wilmsov tumor, nádory mozgu a akútnu myeloidnú leukémiu [8]. Karcinóm prsníka ani ovária sa doposiaľ pri bialelickej recesívnej mutácii génu BRCA2 nepopísal.
3. CAL makuly v rámci syndrómov chromozómovej instability s vysokým rizikom malignity
Syndrómy chromozómovej instability sú známe endogénnou precitlivelosťou na rôzne faktory prostredia (žiarenia, chemické látky) a tým aj spontánnou poruchou integrity chromozómov (tvorba trhlín, zlomov a prestavbou chromozómov). V dôsledku tejto poruchy sú ochoreniami, ktoré najčastejšie asociujú s výskytom malignít.
Fanconiho anémia (FA)
Je charakterizovaná generalizovanou hyperpigmentáciou na trupe a krku s typickými škvrnami typu “café-au-lait” (>50 %) a niekedy aj s hypopigmentovanými oblasťami. Ak sa objavia aj pehy, alebo iné hyperpigmentácie v ingvinálnej, resp. intertriginóznej oblasti, môže sa pri FA a málo výraznej hypoplázii radiálneho lúča chybne diagnostikovať ako neurofibromatóza. Pri FA je však ďalším dominujúcim a vysoko sugestívnym diagnostickým znakom pancytopénia. Obyčajne sa manifestuje najprv aplastickou anémiou, s prechodom do myelodysplastického syndrómu a leukémie. Zo solidných tumorov sa zriedkavo môže vyskytnúť adenóm pečene, hepatóm, karcinóm hrtana, pažeráka, či karcinóm vulvy [9].
Ochorenie je autozómovo recesívne s lokusovou heterogenitou. V kauzálnej súvislosti sa potvrdili mutácie niekoľkých génov FANCA, FANCC, FANCD, FANCE, FANCF, FANCG.
Ataxia teleangiektázia (Louis Barr syndróm) (AT)
Hlavným príznakmi sú ataxia a telengiektázie. Asociácia s malým počtom jednotlivo sa vyskytujúcich CAL je častá. Pacienti s AT majú približne 100-násobne vyššie riziko vzniku malígneho ochorenia ako bežná populácia. Viac ako 85 % zo všetkých malignít v detskom veku tvorí lymfocytová leukémia alebo lymfóm. U dospelých s AT sa častejšie vyskytujú rôzne solídne tumory, vrátane karcinómu prsníka u žien [10].
Jediný známy gén asociovaný s touto chorobou je tumorsupresorový gén ATM, v ktorom bolo popísaných viac ako 270 mutácií. Produktom génu je proteín ATM zo skupiny fosfatidylinositol 3-kináz, hrajúci dôležitú úlohu v kontrole nádorového rastu, účasti na kontrole bunkového cyklu a reparačných pochodov [11].
Bloomov syndróm (BS)
Incidencia ochorenia je 1/10 000 a predominantne postihuje populáciu Židov rodu Aškenázi. Na koži sa po expozícii slnečným žiarením objavujú rôzne pigmentové zmeny a dilatované cievy v podkoží. Na tvári býva typický motýľovitý exantém. Ďalšie príznaky sú v dôsledku imunodeficiencie, mentálny deficit býva zriedkavý. Pri cytogenetickom vyšetrení zistíme vyššiu frekvenciu chromozómových zlomov a prestavieb.
Uvedené autozómovo recesívne ochorenie spôsobujú mutácie v géne BLM, v dôsledku čoho alterujú alebo redukujú BLM proteínovú DNA helikázovú aktivitu ovplyvňujúci replikačný proces [12].
4. CAL Syndróm konštitučného deficitu „Mismeč reparačného systému“ (CMMR-D) – syndróm s veľmi vysokým rizikom vývoja malignít v detskom veku
Toto autozómovo-recesívne ochorenie je známe aj pod názvom Lynchov syndrom III. Novšie sa uvádza ako Syndróm deficitu konštitučného „Mismeč reparačného systému“ (CMMR-D), ktoré charakterizujú hematologické malignity, atypické nádory mozgu a nádory hrubého čreva so včasným nástupom [13]. Kožné makuly s difúznymi okrajmi a nerovnomernou pigmentáciou sú podobné CAL typu neurofibromatózy 1 a môžu sa vyskytovať spolu s hypopigmentovanými ložiskami (obr. 1).
![Charakter CAL pigmentácií pri syndróme deficitu konštitučného „Mismeč reparačného systému“
(CMMR-D) [18].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/e501d853749bf1c9119b4e7191c51d49.jpeg)
Za vznik tohto nedávno popísaného syndrómu sú zodpovedné bialelické mutácie v génoch, ktoré sú v heterozygótnom stave asociované s ochorením Lynchov syndrom (dedičný nádor hrubého čreva bez polypózy – HNPCC). Heterozygotné mutácie v jednom z „DNA reparačných génov“ (MMR génov) MLH1, MSH2, MSH6 a PMS2 spôsobujú inaktiváciu DNA reparačných proteínov, čoho následkom je nedostatočná oprava chybne spárovaných báz pri DNA replikácii. Nádory hrubého čreva, maternice a iné solidné tumory vznikajú u jej nositeľov mutácie v dospelom veku.
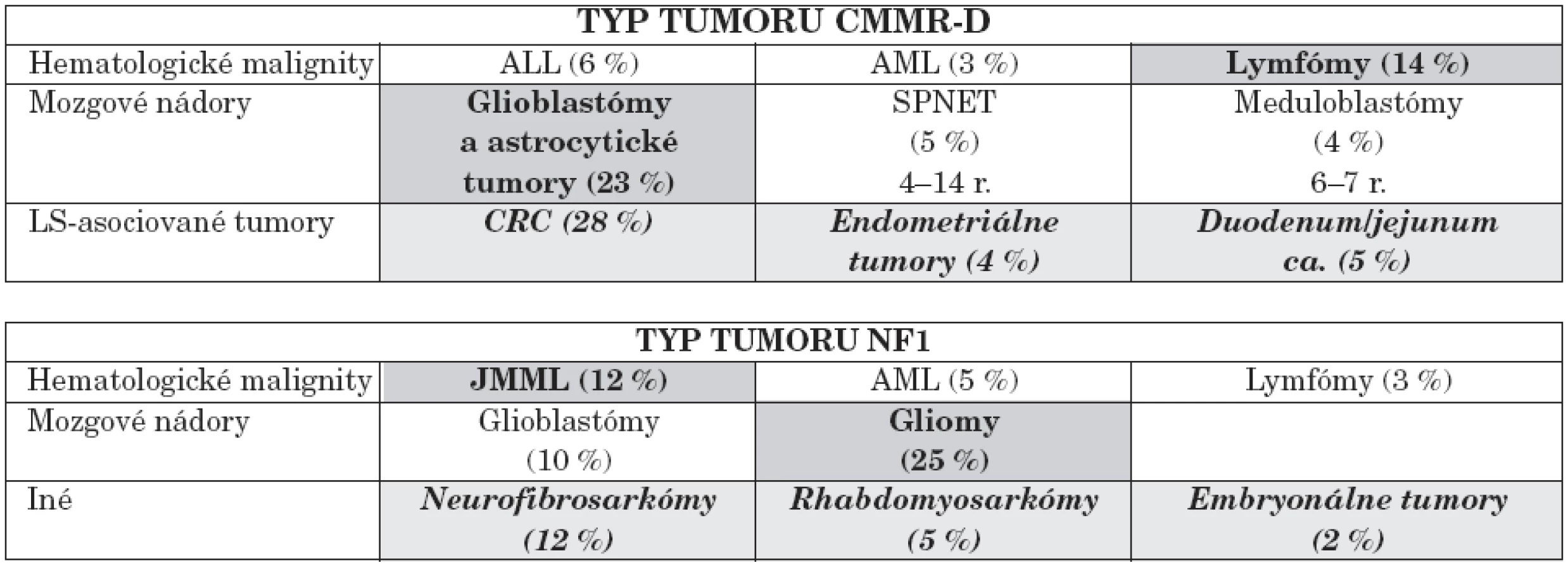
Bialelické mutácie horeuvedených génov charakterizuje výskyt prvých malignít (nádory mozgu, leukémie) už od 2. roku života a ďalších typov nádorov so zvyšujúcim sa vekom. Genotyp a fenotyp pacientov s homozygótnymi mutáciami v jednom z týchto MMR génov autori zosumovali z 35 publikácií za ostatných 10 rokov s výstupom genotypovo-fenotypových korelácií [14]. Porovnanie NF1 s CMMR-D poukázalo na rozdielne spektrum malignít. U CMMR-D z hematologických malignít prevažujú lymfómy, z nádorov astrocytómy, glioblastómy a iné solídne tumory, ako uvádzame v tabuľke 4.
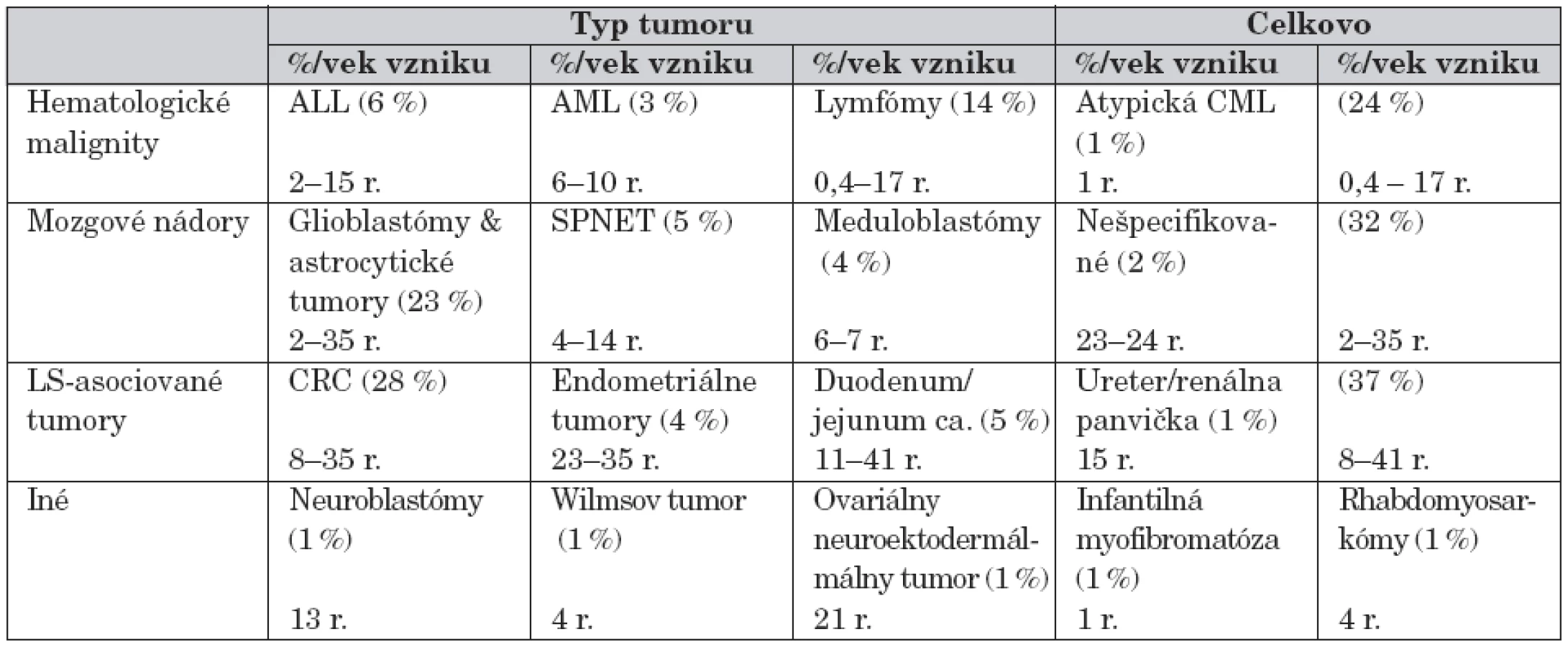
Tabuľka 5 poukazuje na percentuálny výskyt hematologických malignít a solídnych tumorov v porovnaní s CMMR-D. Rozpoznávajúcim znakom pre diferenciálnu diagnózu CMMR-D je výskyt mnohopočetných malignít u pacienta, jeho súrodenca a výskyt tumorov asociovaných s Lynchovým syndrómom v mladom veku. Za vznikom tohto AR ochorenia treba pátrať i po prípadnej konsangvinite rodičov.
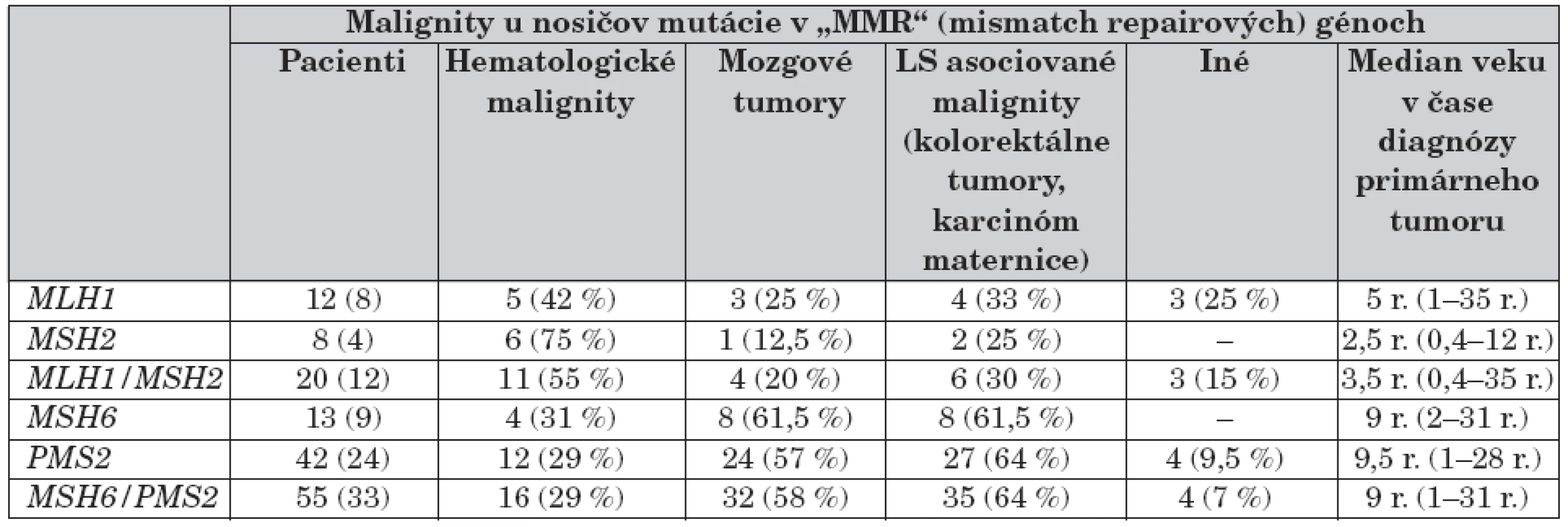
Podiel mutácií v génoch MLH1/MSH2 a MSH6/PMS2 na vzniku a spektre malígnych tumorov zosumarizovala v nedávnej publikácii K. Wimmer [12] – tabuľka 6. Na stanovenie mutácie v týchto génov sa používajú rôzne DNA techniky až po sekvenovanie, vrátane RNA-PCR stratégia so širokým rozsahom pri PMS2 [13, 15].
Záver
Klinický znak CAL skrýva za sebou mnohé syndrómy s multiorgánovým poškodením a malignitami. Precízna klinická diagnostika na základe typu kožných morf, asociovaných príznakov a iných charakteristík v spolupráci s dermatológmi a ďalšími odborníkmi umožňuje včasné rozpoznanie dedičného ochorenia. Diagnostika mnohých syndrómov je na základe typických fenotypových znakov menej náročná a umožňuje cielený manažment zdravotnej starostlivosti [16]. Záchyt niektorých z nich umožňujú rôzne laboratórne metódy vrátane klasického cytogenetického vyšetrenia s dôkazom chromozómovej instability. Klinická diagnostika pri CAL syndrómoch veľmi podobných s NF1, napr. Watsonovým syndrómom, NF1-like syndrómom a syndrómom konštitučného deficitu mismeč opravného systému si vyžaduje klinické skúsenosti [17, 18, 19].
V mnohých prípadoch môže k exaktnej diagnóze významne prispieť klinický genetik cieleným vyšetrením vrátane molekulovo-genetických metód. Laboratórna diagnostika, ktorá deteguje rôzne typy genetických mechanizmov, býva často zložitá a finančne náročná.
Včasná identifikácia heterozygotov/homozygotov jednotlivých typov dedičných nádorových ochorení pomáha optimalizovať liečbu a ďalšiu starostlivosť o pacientov a vysokorizikových jedincov pre skorý vznik malignít.
Práca bola prednesená na celoslovenskej konferencii s medzinárodnou účasťou 3.–5. september 2008, Izakovičov memoriál Vysoké
Tatry, Podbanské.
Došlo: 23. 3. 2009
Přijato: 20. 7. 2009
Prim. MUDr. Denisa Ilenčíková, PhD.
Oddelenie onkologickej genetiky
Národný onkologický ústav
Klenová 1
833 10 Bratislava
Slovenská republika
e-mail: denisa.ilencikova@nou.sk
Sources
1. Schepper S, Haeghen YV, Messiaen L, et al. Café-au-lait spots in neurofibromatosis type 1 and in healthy control individuals: hyperpigmentation of a different kind? Arch. Dermatol. Res. 2006;297 : 439–449.
2. Korf B. Statins, bone, and neurofibromatosis type 1. BMC Medicine 2008;6 : 22.
3. Digilio MC, Sarkozy A, de Zorzi A, Pacileo G, Limongelli G, Mingarelli R, Calabro R, Marino B, Dallapiccola B. LEOPARD syndrome: clinical diagnosis in the first year of life. Am. J. Med. Genet. 2006;140 : 740–746.
4. Friedman JM. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999 Mar 26;89(1): 1–6.
5. Allanson JE, Upadhyava M, Watson GH, et al. Watson syndrome: is it a subtype of type 1 neurofibromatosis? J. Med. Genet. 1991 November;28(11): 752–756.
6. Brems H, Chmara M, Sahbatou M, et al. Germline loss-of function mutations in SPRED1 cause neurofibromatosis 1-like phenotype. Nature Genetics 2007;39(9): 1120–1126.
7. Patel U, Simpson E, Kingswood JC, et al. Tuberose sclerosis complex: analysis of growth rates aids differentiation of renal cell carcinoma from atypical or minimal-fat-containing angiomyolipoma. Clin. Radiol. 2005;60(6): 663–664.
8. Hirsch B, Shimamura A, Moreau L, et al. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004;103(7): 2554–2559.
9. Taniguchi T, D’Andrea AD. The molecular pathogenesis of Fanconi anemia: recent progress. Blood 2006;107 : 4223–4233.
10. Seemanová E, Mišovicová N, Schindler D. Louis-Barové syndrom (ataxia teleangiectasia) v konsanguinní rodině. Čes.-slov. Pediat. 2006;61(11): 666–668.
11. Ball LG, Xiao W. Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacol. Sin. 2005;26(8): 897–907.
12. Amor-Guéret M. Bloom syndrome, genomic instability and cancer: the SOS-like hypothesis. Cancer Lett. 2006;236(1): 1–12.
13. Wimmer K, Etzler J. Constitutional mismatch-repair deficiency syndrom: Have we so far seen only the tip of an iceberg? Hum. Genet. 2008;124(2): 105–122.
14. De Vos M, Hayward BE, Charlton R, et al. PMS2 mutations in childhood cancer. Journal of the National Cancer Institute 2006;98(5): 358–362.
15. Tan TY, Orme LM, Lynch E, et al. Biallelic PMS2 mutations and a distinctive childhood cancer syndrome. J. Pediatr. Hematol. Oncol. 2008;30(3): 254–257.
16. Ďurovčíková D. Neurofibromatosis typ 1 ochorenie multidisciplinárneho charakteru. Revue Mediciny v Praxi 2008;6(4): 10–12.
17. Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam. Cancer 2005;4(4): 323–333.
18. Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair: is it time for Lynch III? Clin. Genet. 2007;71(6): 499–500.
19. Krüger S., Kinzel M., Walldorf C, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur. J. Hum. Genet. 2008;16(1): 62–72.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 9
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- „Café-au-lait“ makuly v diagnostike detských nádorových ochorení
- Nová klinická jednotka DSD (Disorders of Sexual Development) – poruchy sexuálního vývoje
- Antimikrobiálna rezistencia uropatogénov u detí s febrilnými infekciami močových ciest
- Význam předoperačního vyšetření hemostázy u dětí před adenotomií a tonzilektomií