
Hemolyticko-uremický syndrom – nejčastější příčina akutního renálního selhání u dětí
Komplexnost patofyziologie a nové možnosti diagnostiky a terapie atypických forem
Hemolytic Uremic Syndrome (HUS) – One of the Most Common Causes of Acute Renal Failure in Childhood.
Complexity of the Pathophysiology and Novel Diagnostic and Therapeutic Options for Atypical Forms
Hemolytic uremic syndrome (HUS) is a severe life-threatening disease, a thrombotic microangiopathy like thrombocytopenic purpura and HELLP syndrome, characterized by the clinical triad of hemolytic anemia, thrombocytopenia and acute loss of renal function. It is the most frequent cause of acute renal failure in children. The classical form of HUS (D+ HUS) typically develops after a diarrheal prodrome which in 75% of patients is triggered by Shiga-like toxin producing enterohemorrhagic Escherichia coli (EHEC). In 90% of cases, the condition is reversible, with complete recovery and no recurrence. The second form of HUS called D - HUS or atypical HUS (aHUS) affects a heterogeneous group of about 10% patients in whom infection is not confirmed. The two forms of HUS differ in symptoms and response to therapy. Severe cases and relapses are much more common in atypical HUS. Recently, the association between mutations in genes for complement cascade regulatory proteins and aHUS has been reported. Six genes whose mutations are involved in the development of about 60% of aHUS cases have been identified. Every endeavour is made to find new therapeutic options for aHUS patients. The recently reported promising therapeutic potential of the monoclonal antibody eculizumab paves the way for the treatment of aHUS.
Key words:
atypical hemolytic uremic syndrome, molecular genetics, eculizumab, kidney and liver transplantation
Authors:
M. Malina; J. Janda; T. Seeman
Authors‘ workplace:
Pediatrická klinika UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. J. Lebl, CSc.
Published in:
Čes-slov Pediat 2010; 65 (11): 648-653.
Category:
Review
Overview
Hemolyticko-uremický syndrom je závažnou chorobou ze skupiny trombotických mikroangiopatií, kam se dále řadí trombotická trombocytopenická purpura a HELLP syndrom (zkratka anglického Hemolysis, Elevated Liver enzymes, Low Platelets, syndrom kombinující jaterní postižení a hematologické změny spojovaný s těhotenstvím). Projevuje se triádou anémie, trombocytopenie a akutního renálního selhání. U dětí je HUS jednou z nejčastějších příčin akutního selhání ledvin.
Pro klasickou formu HUS, tzv. D+ HUS, je charakteristické infekční průjmové předchorobí, kdy zhruba v 75 % případů jde o infekci enterohemoragickými kmeny Escherichia coli (EHEC) produkujícími Shiga-like toxin. V 90 % případů D+ HUS se funkce ledvin po překonání akutního stavu i zcela upravují a nemoc nemá relabující charakter. V řadě případů se ale po delší době objevují poruchy glomerulární filtrace včetně chronické renální insuficience.
Existuje cca 10 % pacientů, u nichž dochází k relapsům onemocnění nebo k remisi ani nedojde. Projevy a reakce na léčbu bývají u těchto atypických forem (aHUS) značně odlišné a mají většinou i velmi těžký průběh. V poslední době byla prokázána spojitost mezi mutacemi v genech pro proteiny regulující komplementovou kaskádu a rozvojem aHUS. Do současné doby se podařilo prokázat 6 genů, jejichž mutace jsou spojené s rozvojem aHUS a vysvětlují cca 60 % těchto onemocnění.
Klíčová slova:
atypický hemolyticko-uremický syndrom, molekulární genetika, eculizumab, transplantace ledvin a jater
Úvod
Historie hemolyticko-uremického syndromu (HUS) se píše od roku 1955, kdy Gasser se svými kolegy popsal pět malých dětí s příznaky akutního renálního selhání, trombocytopenie a neimunní hemolytické anémie [1]. V originální práci bylo použito názvu v plurálu – Hämolytisch-urämische Syndrome, jako kdyby autoři tušili, že nepůjde o jednoduchou patofyziologickou a anatomickou entitu, ale spíše o různé syndromy. Nicméně popsaná triáda příznaků definuje hemolyticko-uremický syndrom doposud. Publikační priorita je poněkud komplikovaná překryvem příznaků HUS s trombotickou trombocytopenickou purpurou (TTP) popsanou Moschcowitzem již v roce 1925 ve formě kazuistiky u mladé dívky [2].
V současnosti se obě klinické diagnózy společně s HELLP syndromem (zkratka anglického Hemolysis, Elevated Liver enzymes, Low Platelets, syndrom kombinující jaterní postižení a hematologické změny spojovaný s těhotenstvím) slučují pod patofyziologicky shrnující název trombotické mikroangiopatie. Příčinou klinických projevů je totiž značné poškození endotelu drobných arteriol a kapilár. Histologicky jsou léze endotelu shodné u všech těchto tří syndromů. Dochází k otoku endoteliálních buněk, ztrátě jejich krycí funkce, vzniku intravaskulárních trombů s konzumpcí značného množství trombocytů a rozvoji anémie z mechanické destrukce erytrocytů a narušení jejich morfologie při snaze proniknout tromby obturovanými cévami. HUS je pak v krevním obraze charakterizován hemolytickou anémií s anisocytózou, schistocyty až fragmentocyty a trombocytopenií. Při HUS je nejvíce postižen endotel ledvin. U HUS způsobeného shiga toxinem je tato náchylnost endotelu ledviny patofyziologicky osvětlena (viz dále), u atypických forem je stále tématem rozsáhlého výzkumu. Projevy poškození endotelu se však neomezují jen na ledviny a často jsou stejně poškozeny další orgány s bohatou mikrocirkulací (nejčastěji mozek, srdce a gastrointestinální trakt).
Incidence a klasifikace
Incidence HUS se pohybuje u dětí kolem 1–2 případů na 100 000 dětí. HUS je nejčastější příčinou akutního selhání ledvin u kojenců a batolat (0,3–0,4/100 000 dětí)[3]. HUS se klinicky dělí na takzvaný D+ HUS a D - HUS. Tato klasifikace vychází z popisu příznaku prodromálního stadia onemocnění. Typické formě D+ HUS předchází kolitida s výskytem krvavého průjmu (z anglického diarrhoea = D), po které následují příznaky hemolytické anémie, trombocytopenie a akutního selhání ledvin.
Kromě těchto primárních HUS, jejichž patofyziologie bude rozvedena níže, je známo sekundární poškození s příznaky HUS/TTP asociované s mnoha bakteriálními (pneumokoky [4]) či virovými infekcemi. Projevy HUS mohou být též nežádoucími účinky některých léků, jako cytostatika mitomycinu C či imunosupresiva cyklosporinu. HUS může též být paraneoplastickým projevem maligního tumoru [5]. V letošním roce též byla potvrzena spekulovaná asociace HUS s těhotenstvím [6].
D+ HUS
D+ HUS tvoří 90 % všech HUS u dětí. Vyskytuje se nejčastěji ve věkové skupině dětí od 3 do 6 let. Patofyziologicky zpočátku nebylo jasné, co přímo tuto formu HUS způsobuje, obviňováno bylo mnoho patogenů a byla nalezena řada asociací HUS s různým bakteriálními a hlavně virovými infekcemi. Opravdového viníka – určité kmeny E. coli se podařilo odhalit až po delší době. Dnes je za původce typického HUS považována infekce kmeny E. coli produkujícími Shiga-like toxin (dříve vero-cytotoxin), nejčastěji jde o sérotyp O157:H7, ale producentem toxinu jsou i jiné kmeny (např. některé řazené dříve k enteropatogenním E. coli vyvolávajícím závažné dětské průjmy) [7]. Zdrojem infekce je většinou hovězí skot, jehož střevo je u 15 % kolonizováno těmito bakteriemi. Vektorem jsou poté většinou živočišné produkty těmito kmeny kontaminované, např. nedostatečně tepelně upravené hovězí maso, nepasterizované mléko a mléčné produkty, ale někdy i rostlinné produkty, např. kontaminovaná zelenina (jednu z největších epidemií vyvolal sterilovaný špenát [8]).
Shiga-like toxin překvapivě bakterie sama neprodukuje do vnějšího prostředí a k jeho uvolnění dochází až po usmrcení bakterie. Z toho též vycházelo doporučení nepodávat pacientům s HUS v akutní fázi antibiotika [9]. Podrobná metanalýza však toto podezření nepotvrdila a aktivace lýzy bakterie a uvolnění toxinu je stále předmětem výzkumu [10]. Toxin z usmrcených bakterií poté prochází povrchem enterocytů a prostupuje dále do krevního řečiště, kde se váže na endotel bohatý na specifický glykoproteinový receptor globotriaosylceramidu (Gb3). Tento receptor je exprimován především na endotelu glomerulů, následkem je glomerulopatie s projevy akutního selhání ledvin.
Prognóza pacientů s D+ HUS je v akutním stadiu příznivá, mortalita se pohybuje pouze okolo 5 % [3]. Tato forma nerelabuje, ale při dlouhodobém sledování se v řadě případů objevuje postupný pokles glomerulární filtrace a stav může progredovat do chronické renální insuficience [3b]. Základem terapie je zvládnutí akutního stavu renálního selhání, dialýza, symptomatická terapie a překlenutí akutní doby, než dojde k obnovení funkce ledvin.
Atypický D - HUS
Atypický D - HUS (= aHUS, nebo též D - HUS) tvoří cca 10 % případů dětí s HUS [3]. Nemá specifický věk manifestace. Vyskytuje se u novorozenců i dospělých. Nepředchází mu většinou prodromální stadium hemoragické kolitidy (i když její přítomnost nevylučuje aHUS). Má většinou výrazně těžší průběh s extrarenálními komplikacemi a jeho dlouhodobá prognóza je také významně horší. Až u 90 % dětí první ataka D - HUS vede k reziduálnímu poškození ledvin a až u 50 % případů progreduje postižení do obrazu chronické renální insuficience a terminálního selhání ledvin, které vyžaduje dlouhodobou léčbu dialýzou nebo transplantaci ledviny. Navíc 25 % případů atypického D - HUS končí fatálně již při první atace. Některé formy D - HUS často rekurují, a to bohužel i po transplantaci ledviny [3, 11].
Již od sedmdesátých let se tušilo, že atypický HUS je vyvolán aktivací alternativní cesty komplementu [12]. Konzumpce C3 složky komplementu a normální hladiny C4 složky v séru pacientů s aHUS pro to jasně svědčily. Též histologické nálezy biopsií ledvin ukazovaly lokální aktivace komplementu až k jeho lytické složce [13] (depozice C3 v arteriolách a glomerulech a zvýšené množství finální lytické složky C5b–9 v místech intravaskulárních trombů).
Přesné důkazy, proč je alternativní složka komplementu takto neregulovatelně stimulována, se objevily až v devadesátých letech díky rozvoji metod molekulární genetiky. S ní přišel přímo skokový nárůst objevů na poli atypického HUS. Do současnosti bylo identifikováno několik genů, jejichž mutace jsou zodpovědné za familiární a sporadické formy atypického HUS. Prvním objeveným mutovaným genem asociovaným s aHUS byl gen pro regulátor komplementu faktor H [14]. Úplný nedostatek či funkční porucha tohoto proteinu vede k nekontrolované aktivaci alternativní cesty komplementu. V rychlém sledu následovaly objevy dalších asociovaných genů. V současné době je známo šest genů, jejichž změny jsou zodpovědné za vznik D - HUS (mutace v genech pro faktor H, faktor I, faktor B, MCP/CD46, C3 a trombomodulin) [14–18]. Přehled příčin D - HUS je uveden v tabulce 1.
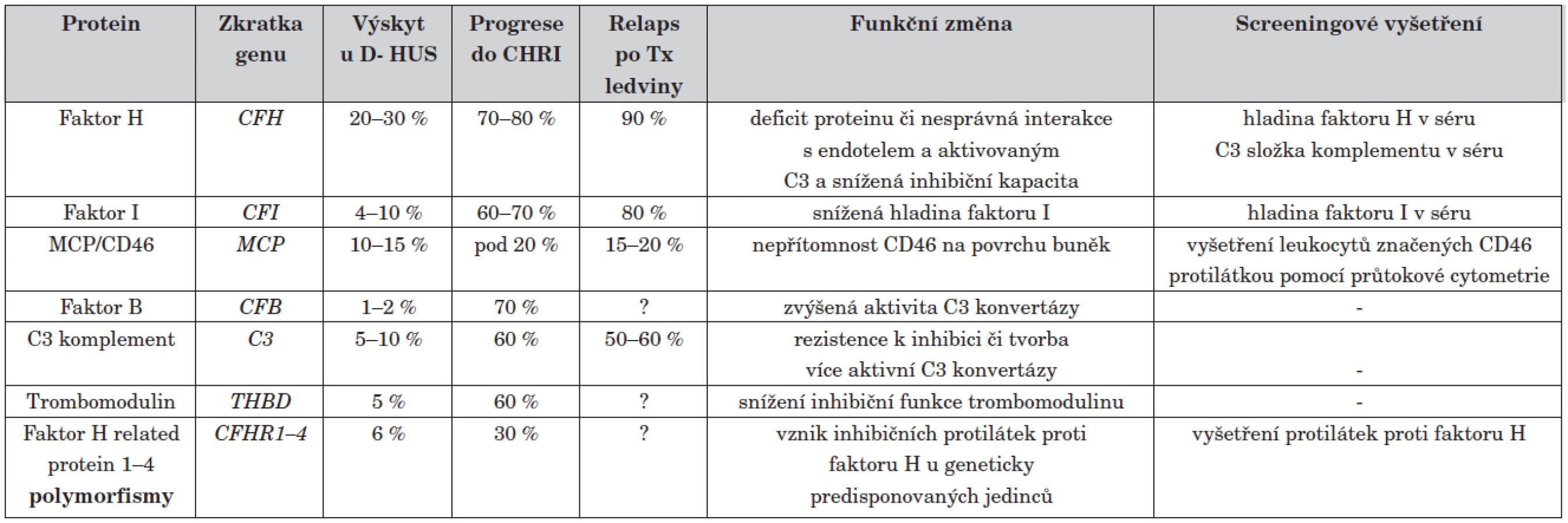
Jednotlivé formy se od sebe značně liší svým průběhem. Mutace v některých genech způsobují velmi závažné vrozené formy HUS s velmi špatnou prognózou (geny pro faktor H, faktor B, C3), jiné mutace, např. v genu MCP mají lepší prognózu s minimem relapsů po transplantaci ledviny.
Situace etiologie aHUS se ještě zkomplikovala po nálezu inhibičních protilátek proti faktoru H [19]. Tyto autoprotilátky pravděpodobně vznikají u geneticky predisponovaných jedinců s určitými polymorfismy genů jako následek prolomení imunitní tolerance (tab. 1) [20].
Diagnostika D - HUS
Jak vyplývá z tabulky 1, znalost genetického pozadí onemocnění je nezbytná nejen pro přesné zařazení pacienta do definované etiologické skupiny, ale hlavně pro upřesnění jeho prognózy. S objevem nových možností terapie pro určité genetické formy začíná být toto vyšetření zcela zásadní, zvláště při plánování zařazení pacienta do transplantačního programu. Tato vyšetření jsou velmi náročná časově i finančně a dosud nebyla v ČR dostupná. Pediatrická klinika v Motole v poslední době spolupracuje s ÚHKT (Ústav hematologie a krevní transfuze) ve snaze zavádět některé metody v ČR.
V současnosti diagnostika stojí na molekulárně genetickém vyšetření přímým sekvenováním všech genů asociovaných s aHUS. Je známo, že existují pacienti, kteří mají mutaci ve více než jednom genu a tudíž nález mutace v jednom genu není důvodem ukončení diagnostického procesu. V současné době se daří prokázat genetickou anomálii v asociovaných genech až u 60 % pacientů. Většina mutací je heterozygotních a penetrance se zdá být relativně nízká, kolem 50 %. Často se najde mutace u jednoho z rodičů či sourozence, který je kompletně zdravý. Tato nízká penetrance nemoci ji vyčleňuje ze standardního konceptu monogenní choroby. Zdá se, že mutace je pouze predisponujícím faktorem a ke spuštění nemoci je třeba více genetických a externích faktorů [21]. Pro nízkou penetranci je i eticky obtížné určit indikaci k prenatální diagnostice. Po spouštěcích faktorech se nyní intenzivně pátrá.
Vzhledem k velké časové náročnosti vyšetření se též pracuje na rychlých screeningových vyšetřeních pro předběžné určení etiologie. Tato vyšetření jsou shrnuta v tabulce 1.
Základem vyšetření by mělo být získání vzorku séra, plazmy, EDTA krve a DNA od pacienta v akutním stavu před započetím plazmaferézy. Po vyslovení podezření ze základních diagnostických parametrů (klasické příznaky HUS, vyloučení EHEC infekce a přítomnosti shiga toxinu a sekundárního HUS, nízké C3, normální C4) se započíná s vyšetřením sekvenováním od nejčetnějších známých genů po méně četné. Zároveň se testuje sérum pacienta na deficit faktoru H, provádí se screening protilátek proti faktoru H a vyšetření hladin ostatních regulátorů komplementu (faktor I, faktor B). Rychlým diagnostickým testem pro vyšetření mutací MCP je průtoková cytometrie mononukleárů a granulocytů z periferní krve označených protilátkou proti CD46 [3].
Terapie D - HUS
Dlouhá léta se ví o příznivém efektu plazmaferézy, jejíž použití je v akutní fázi doporučeno co nejdříve od manifestace nemoci a opakované plazmaferézy se doporučují s cílem dosáhnout klinické i laboratorní remise. Předpokládá se, že plazmaferéza odstraní mutované faktory a protilátky proti faktorům komplementu a nahradí je dostatečným množstvím normálních proteinů komplementové kaskády.
Plazmaferéza je výrazně efektivnější než pouhé infuze plazmy a navíc nezatěžuje pacienta excesivním přívodem tekutin, který je u pacientů v renálním selhání nevhodný. Přes svou nespornou účinnost je ale extrémně zatěžující a není trvalým řešením. Navíc u některých forem aHUS není dostatečně účinná. MCP/CD46 je povrchová molekula endotelu, která se plazmaferézou nedá odstranit či nahradit. Též odstranění v cirkulaci abundantních a mutací hyperaktivovaných molekul, jako C3 nebo faktor B, je i při masivní plazmaferéze neefektivní [22, 23].
Nové možnosti léčby D - HUS
Dalším zkoušeným terapeutickým postupem s dlouhodobým efektem by mohla být kombinovaná transplantace jater a ledvin. Jedná se o zajímavé řešení pro faktory produkované v játrech, jako například faktor H. První dva pokusy s touto terapií skončily bohužel fatálně pro rychlý rozvoj akutní rejekce jaterního transplantátu [24]. Vybuzený komplement reagoval přehnaně na transplantát, který se velmi rychle odhojil. Po této zkušenosti další kohorta osmi pacientů byla před transplantací připravena masivní opakovanou plazmaferézou, která pokračovala i po transplantaci. Výsledky byly po této úpravě velmi dobré a sedm z osmi pacientů dlouhodobě přežívá bez relapsu onemocnění [25, 26].
V současné době jsou ve fázi testování látky, které by mohly působit přímo na „místě činu“, tedy v komplementové kaskádě a její regulaci. Dvě nezávislé skupiny testují rekombinantní a purifikovaný faktor H pro pacienty s jeho deficiencí [27]. Další skupiny se pokouší o léky, které se cíleně směřují do míst s vysokou aktivací komplementu a působí inhibičně na jeho alternativní cestu.
Velká naděje se nyní vkládá do monoklonální protilátky Eculizumab vyvinuté pro léčbu paroxysmální noční hemoglobinurie, která blokuje komplement na úrovni faktoru C5 a netlumí odstraňování imunokomplexů ani opsonizaci, ale pouze lytickou schopnost komplementu. První výsledky u dospělých a větších dětí se zdají být slibné. Eculizumab byl u D - HUS poprvé použit u pacientky s C3 mutací s opakovanými relapsy, která odmítla pokračování pravidelných plazmaferéz. Po podání jedné dávky došlo k zastavení aktivace komplementu a pacientčin stav se stabilizoval [28]. Existují i dvě recentní publikace o použití Eculizumabu u dětských aHUS pacientů s dobrým výsledkem [29, 30]. V letošním roce též vyšla krátká zpráva o prvním profylaktickém použití Eculizumabu při transplantaci ledviny u dítěte s mutací ve faktoru H [31]. Problémem této terapie je vysoká cena preparátů a pravděpodobně i nutnost podávat lék u některých pacientů celoživotně. Tomuto by se alespoň u některých mutací dalo předejít kombinovanou transplantací jater a ledvin s přípravnou terapií Eculizumabem.
Závěr
Je možné konstatovat, že molekulárně genetická diagnostika těchto vzácných onemocnění může být modelovým případem, jak odhalit jejich podstatu a pokusit se je cíleně léčit. Navíc upřesněné poznání systému komplementu a jeho regulace umožňuje pochopení jeho role v dalších pochodech. Komplement totiž hraje významnou roli například v mnoha imunitních mechanismech boje se závažnými infekcemi a též má zásadní roli v transplantační medicíně. Je možné, že patologické mechanismy popsané u pacientů s atypickým HUS přispějí k lepšímu pochopení těchto pochodů a možná otevřou i terapeutické možnosti pro další léčbu častějších onemocnění a umožní efektivnější transplantace (experimentálně se uvažuje např. o užití Eculizumabu při transplantacích ledvin k zastavení rejekce) [32].
Jelikož se jedná o velmi vzácné onemocnění, jeho studium je limitováno dosažením dostatečně reprezentativního vzorku pacientů. Mezinárodní spoluprací se však v posledních letech podařilo odhalit některé patofyziologické mechanismy zodpovědné za vznik tohoto závažného a obávaného onemocnění a zdá se, že terapeutické možnosti jsou již jen krůček od standardního použití. Dobrou zprávou je fakt, že pracoviště dětské nefrologie v ČR jsou zapojena do multicentrických mezinárodních studií, jejichž výsledky dávají naději, že se znalosti i naše terapeutické možnosti významně posunou směrem vpřed a umožní dětem s D - HUS co nejpodrobnější diagnostiku a nejmodernější léčbu.
Podporováno grantem IGAMZČR reg. č. NT11457 a VZ MŠMT ČR reg. č. 0021620819.
Věnováno k životnímu jubileu prof. MUDr. Miroslava Šašinky, DrSc.
Došlo: 16. 8. 2010
Přijato: 14. 10. 2010
MUDr. Michal Malina
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: michal.malina@lfmotol.cuni.cz
Sources
1. Gasser C, et al. Hemolytic-uremic syndrome: bilateral necrosis of the renal cortex in acute acquired hemolytic anemia. Schweiz Med. Wochenschr. 1955; 85(38–39): 905–909.
2. Moschcowitz E. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: an undescribed disease. 1925. Mt Sinai J. Med. 2003; 70(5): 352–355.
3. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009; 361(17): 1676–1687.
3b. Bláhová K, Janda J, Kreisinger J, Matejková E, Sedivá A. Long-term follow-up of Czech children with D+ hemolytic-uremic syndrome. Pediatr. Nephrol. 2002; 17(6): 400–403.
4. Copelovitch L, Kaplan BS. Streptococcus pneumoniae associated hemolytic-uremic syndrome: classification and the emergence of serotype 19A. Pediatrics 2010; 125(1): e174–182.
5. Besbas N, et al. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006; 70(3): 423–431.
6. Fakhouri F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J. Am. Soc. Nephrol. 2010; 21(5): 859–867.
7. Mark Taylor C. Enterohaemorrhagic Escherichia coli and Shigella dysenteriae type 1-induced haemolytic uraemic syndrome. Pediatr. Nephrol. 2008; 23(9): 1425–1431.
7b. Zimmerhackl LB, Rosales A, Hofer J, Riedl M, Jungraithmayr T, Mellmann A, Bielaszewska M, Karch H. Enterohemorrhagic Escherichia coli O26:H11-associated hemolytic uremic syndrome: Bacteriology and clinical presentation. Semin. Thromb. Hemost. 2010 Sep; 36(6): 586–593.
8. Grant J, et al. Spinach-associated Escherichia coli O157:H7 outbreak, Utah and New Mexico, 2006. Emerg. Infect. Dis. 2008; 14(10): 1633–1636.
9. Zhang X, et al. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 2000; 181(2): 664–670.
10. Safdar N, et al. Risk of hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 enteritis: a meta-analysis. JAMA 2002; 288(8): 996–1001.
11. Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr. Transplant. 2008; 12(6): 619–629.
12. Stuhlinger W, et al. Letter: Haemolytic-uraemic syndrome: evidence for intravascular C3 activation. Lancet 1974; 2(7883): 788–789.
13. Barre P, et al. Hemolytic uremic syndrome with hypocomplementemia, serum C3NeF, and glomerular deposits of C3. Arch. Pathol. Lab. Med. 1977; 101(7): 357–361.
14. Warwicker P, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998; 53(4): 836–844.
15. Noris M, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 2003; 362(9395): 1542–1547.
16. Fremeaux-Bacchi V, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J. Med. Genet. 2004; 41(6): e84.
17. Goicoechea de Jorge E, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. U.S.A. 2007; 104(1): 240–245.
18. Delvaeye M, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009; 361(4): 345–357.
19. Dragon-Durey MA, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005; 16(2): 555–563.
20. Moore I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood 2010; 115(2): 379–387.
21. Sánchez-Corral P, Melgosa M. Advances in understanding the aetiology of atypical Haemolytic Uraemic Syndrome. Br. J. Haematol. 2010; 150(5): 529–542.
22. Taylor CM, et al. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br. J. Haematol. 2010; 148(1): 37–47.
23. Ariceta G, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr. Nephrol. 2009; 24(4): 687–696.
24. Remuzzi G, et al. Hemolytic uremic syndrome: a fatal outcome after kidney and liver transplantation performed to correct factor H gene mutation. Am. J. Transplant. 2005; 5(5): 1146–1150.
25. Jalanko H, et al. Successful liver-kidney transplantation in two children with aHUS caused by a mutation in complement factor H. Am. J. Transplant. 2008; 8(1): 216–221.
26. Saland JM, Ruggenenti P, Remuzzi G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2009; 20(5): 940–949.
27. Fakhouri F, et al. Treatment with human complement factor H rapidly reverses renal complement deposition in factor H-deficient mice. Kidney Int. 2010; 78(3): 279–286.
28. Chatelet V, et al. Safety and long-term efficacy of eculizumab in a renal transplant patient with recurrent atypical hemolytic-uremic syndrome. Am. J. Transplant. 2009; 9(11): 2644–2645.
29. Mache CJ, et al. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2009; 4(8): 1312–1316.
30. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009; 360(5): 544–546.
31. Zimmerhackl LB, et al. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2010; 362(18): 1746–1748.
32. Sheerin NS. Should complement activation be a target for therapy in renal transplantation? J. Am. Soc. Nephrol. 2008; 19(12): 2250–2251.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2010 Issue 11
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Henochova-Schönleinova purpura – diagnostika a léčba z pohledu současných poznatků
-
Hemolyticko-uremický syndrom – nejčastější příčina akutního renálního selhání u dětí
Komplexnost patofyziologie a nové možnosti diagnostiky a terapie atypických forem - Současné trendy v léčbě invaginací u dětí
- Imunoglukan P4H® v prevencii recidivujúcich infekcií dýchacích ciest v detskom veku