
Kongenitálny hyperinzulinizmus – najčastejšia príčina perzistujúcich hypoglykémií u novorodencov a dojčiat
Congenital Hyperinsulinism – Most Frequent Cause of Persistent Hypoglycemia in Newborns and Infants
Congenital hyperinsulinism (CHI) is the most frequent cause of severe, persistent hypoglycemia in newborns and infants. The majority of cases refer to inherited disorders of insulin secretion. The most prevalent are the inactivating mutations of genes for Kir6.2 and SUR1 subunits of potassium channel of the B-cells. The true etiology of CHI can be identified only by methods of DNA analysis, which could also distinguish the focal and diffuse form. The treatment is intended to normalize blood glucose and to prevent the neurological consequences.
Key words:
persistent hypoglycemia, congenital hyperinsulinism, DNA analysis, 18F-DOPA PET CT
:
Monika Rosoľanková; E. Franková
:
Oddelenie patologických novorodencov a JIRS I. DK, DFNsP, Bratislava
zástupca prednostu MUDr. E. Franková
:
Čes-slov Pediat 2010; 65 (9): 516-522.
:
Review
Kongenitálny hyperinzulinizmus je najčastejšou príčinou závažnej perzistujúcej hypoglykémie u novorodencov a malých detí. Je podmienený vrodenými poruchami regulácie sekrécie inzulínu. Najčastejšie býva spôsobený inaktivačnými mutáciami génov pre Kir6.2 a SUR1 podjednotky draslíkového kanála B-buniek pankreasu. Presný typ kongenitálneho hyperinzulinizmu je možné diagnostikovať pomocou DNA analýzy, ktorá zároveň odlíši fokálnu formu od difúznej a tým usmerní ďalšiu diagnostiku a liečbu. Pri neskorej alebo neefektívnej liečbe hypoglykémie hrozí ireverzibilné poškodenie mozgových buniek s trvalými následkami.
Kľúčové slová:
perzistujúca hypoglykémia, kongenitálny hyperinzulinizmus, DNA analýza, 18F-DOPA PET CT
Úvod
Kongenitálny hyperinzulinizmus (Congenital Hyperinsulinism, CHI) známy aj pod názvom Perzistujúca hyperinzulinemická hypoglykémia novorodencov a dojčiat, je najčastejšou príčinou závažnej perzistujúcej hypoglykémie u novorodencov a malých detí [1, 2]. Frekvencia výskytu CHI v Európe je 1/40–50 tisíc živonarodených detí, vyššia incidencia je v krajinách s vyšším výskytom konsanguinity, ako napr. Saudská Arábia (1 : 3000) [1]. U 60 % detí s hyperinzulinizmom sa objavuje hypoglykémia už v novorodeneckom veku, ďalších 30 % sa diagnostikuje počas prvého roku života, zvyšok neskôr [2].
Kongenitálny hyperinzulinizmus je podmienený vrodenými poruchami regulácie sekrécie inzulínu B bunkami celého pankreasu (difúzna forma) alebo jeho určitej časti (fokálna forma) [1]. Pankreas sa stáva akoby „slepým“ voči glykémii a nadmerne produkuje inzulín bez ohľadu na koncentráciu glukózy v krvi. Hyperinzulinizmus spôsobuje vtok glukózy do inzulín-dependentných tkanív a bráni využitiu alternatívnych energetických substrátov (najmä ketolátok) potlačením lipolýzy, beta-oxidácie mastných kyselín a ketogenézy. Týmto spôsobom oberá mozog o primárny a sekundárny zdroj energie. Nedostatok energie vedie k strate funkcie neurónov, čoho prejavom môžu byť kŕče alebo kóma [2]. Pri neskorej alebo neefektívnej liečbe a prevencii hypoglykémie môže dôjsť k ireverzibilnému poškodeniu mozgových buniek a vzniku trvalých následkov (ako psychomotorická retardácia, detská mozgová obrna) alebo až k smrti dieťaťa [1, 2, 3].
Etiológia
Vo všeobecnosti rozoznávame dva typy hyperinzulinizmu – tranzientný a perzistentný.
Tranzientný hyperinzulinizmus sa môže vyskytnúť u prematúrnych novorodencov, hypotrofických novorodencov, detí diabetických matiek, pri novorodeneckej sepse, u novorodencov s fetálnym distresom a i. [2]. Tento typ hyperinzulinizmu sa vo všeobecnosti darí zvládnuť častejším kŕmením, infúziou glukózy, prípadne Diazoxidom. Spontánne vymizne v priebehu niekoľkých dní a nemá tendenciu k rekurencii [2].
Druhý typ je perzistentný hyperinzulinizmus, podmienený geneticky (synonymom je kongenitálny hyperinzulinizmus – CHI). K dnešnému dňu boli s CHI asociované mutácie 7 typov génov.
Mutácie génov podmieňujúcich CHI zasahujú do tvorby energie a vylučovania inzulínu. Miesta účinku proteínov, kódovaných génmi asociovanými s kongenitálnym hyperinzulinizmom, sú schematicky vyznačené na obrázku 1.
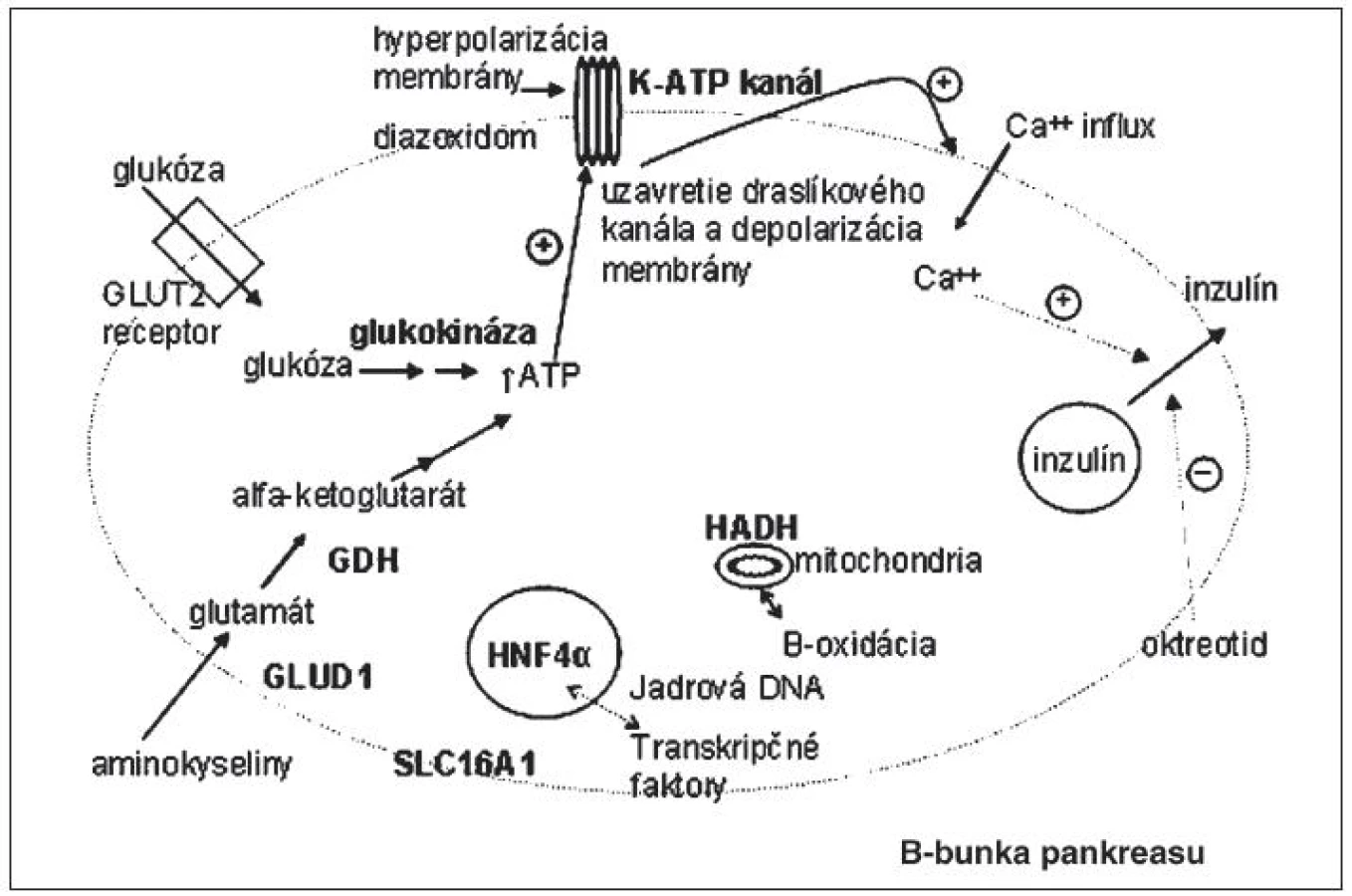
Kongenitálny hyperinzulinizmus na podklade inaktivačných mutácií génov ABCC8 a KCNJ11
Je to najčastejší typ CHI. Doteraz bolo popísaných viac ako 100 mutácií génu ABCC8 a 20 mutácií génu KCNJ11 [1]. Inaktivačné mutácie génov pre Kir6.2 a SUR1 podjednotky draslíkového kanála B-buniek pankreasu (obr. 2) podmieňujú difúznu alebo fokálnu formu hyperinzulinizmu, ktoré sú klinicky prakticky neodlíšiteľné.
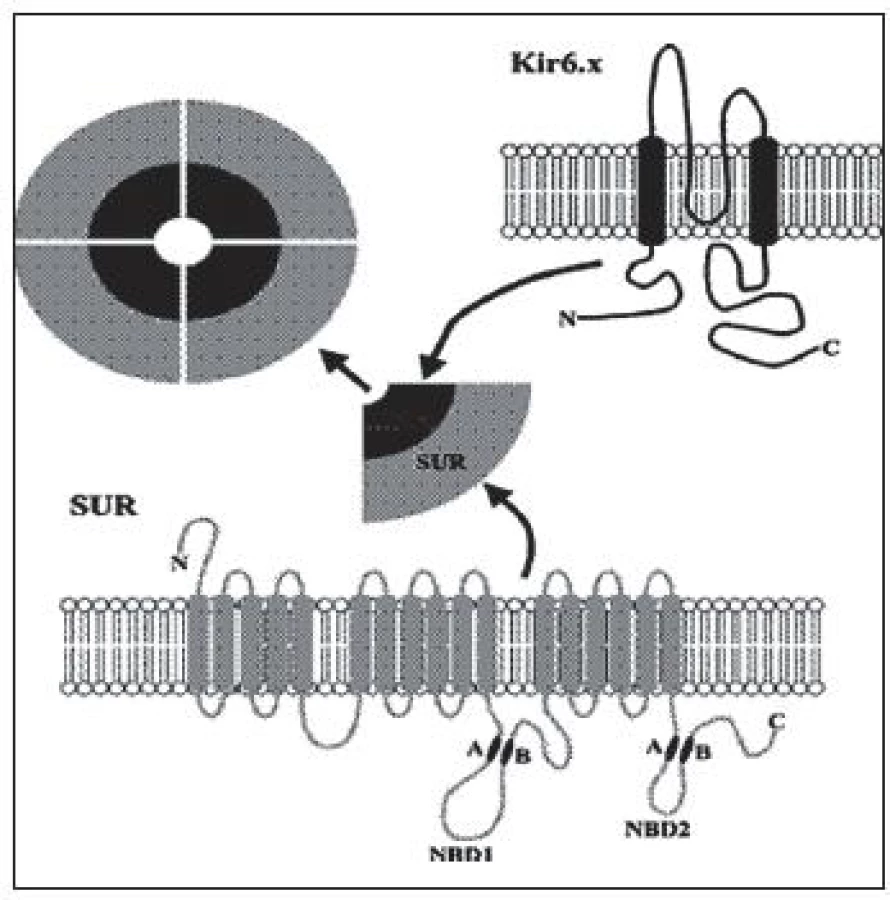
Difúzna forma, pri ktorej majú všetky B-bunky pankreasu defektný káliový kanál, sa dedí autozómovo recesívne, menej často autozómovo dominantne. Rozsah dysfunkcie K-ATP kanála závisí od typu genetického defektu. Autozómovo recesívne mutácie vedú k úplnej strate funkcie draslíkového kanála s ťažkým klinickým priebehom, zlou odpoveďou na lieky, neskoršou a zriedkavejšou spontánnou remisiou. Nakoľko jeden typ podjednotiek (všetky Kir6.2 alebo všetky SUR1) je poškodený, zvyšková funkcia kanála je nulová, čo sa prejavuje aj rezistenciou na Diazoxid (znižuje sekréciu inzulínu väzbou na SUR1 podjednotku s následným otvorením draslíkového kanála, s hyperpolarizáciou membrány, čo zamedzí prestup sekrečných granúl s inzulínom bunkovou membránou). Pre závažné hypoglykémie s rizikom trvalého poškodenia mozgu väčšinou vyžadujú chirurgický zákrok – subtotálnu pankreatektómiu. Po operácii a v priebehu detstva je vysoké riziko vzniku diabetu v dôsledku odstránenia veľkého počtu B-buniek. Zriedkavejšie sú autozómovo dominantné mutácie (opísané boli pre gén ABCC8), ktoré sa klinicky prejavujú u jedinca v heterozygotnej forme s jednou zdravou alelou. Mávajú miernejší priebeh, čo súvisí len s parciálnou stratou funkcie K-ATP kanála (polovica podjednotiek je funkčných). Vo väčšine prípadov pacienti reagujú na vyššie dávky Diazoxidu [7, 8, 9, 10].
Fokálna forma je podmienená autozómovo recesívnou inaktivačnou mutáciou génu pre Kir6.2 alebo SUR1, v heterozygotnej forme (mutácia sa dedí takmer výlučne od otca). Mutácia sa za normálnych okolností neprejaví, pretože na normálnu fukciu postačuje jedna zdravá alela – od matky. Až počas intrauterinného vývoja pankreasu dochádza u postihnutého jedinca k strate heterozygozity – špecifickej strate funkcie materskej alely. Gén sa nachádza v blízkosti oblasti chromozómu 11p15, ktorá podlieha imprintingu. Imprinting cielene vyraďuje niektoré materské gény v tejto oblasti z funkcie. Poruchou imprintingu dochádza k inaktivácii materských génov pre Kir6.2 a SUR1, čo spôsobuje expresiu len abnormálnej otcovskej alely v tejto oblasti pankreasu. Porucha imprintingu zároveň spôsobuje aktiváciu priľahlých génov stimulujúcich rast. Vzniká tak fokálna hyperplázia abnormálnych buniek [1, 7, 11, 12, 13]. Keďže tieto lézie vznikajú počas fetálneho obdobia, neporušujú normálnu architektúru pankreasu a nie sú obalené kapsulou, čo je naopak typickou vlastnosťou inzulinómu [1].
Klinický priebeh u typickej fokálnej formy je stredne ťažký, máva častejšiu spontánnu remisiu (80 % do 8. roku života). Novorodenci s mutáciami Kir6.2 a SUR1 mávajú väčšinou vysokú pôrodnú hmotnosť vzhľadom ku gestačnému veku (obr. 3). Závažná hypoglykémia sa objavuje už počas prvých dní života a vyžaduje agresívnu liečbu s vysokým parenterálnym príjmom cukrov na prevenciu rekurentných hypoglykemických epizód. V klinickom obraze dominujú príznaky hypoglykémie ako letargia, dráždivosť, poruchy vedomia až kóma, kŕčová aktivita a tiež apnoické pauzy, hypotonický syndróm. Postupne sa pridružujú komplikácie spojené s medikamentóznou liečbou a s vysokým príjmom cukrov, napr. výrazné prírastky na hmotnosti, problémy s perorálnym príjmom, hypertrofická kardiomyopatia, cholecystolitiáza a i. [2, 3, 15].

Terapia je medikamentózna a chirurgická (enukleácia fokusu). Ak sú účinné lieky, môžeme dosiahnuť rovnaký výsledok ako chirurgický zákrok. Napriek liečbe asi u 40 % detí vzniká ľahká tranzientná psychomotorická retardácia, riziko neskoršieho vzniku diabetu je nízke [1, 2, 7, 14].
Ďalšie príčiny CHI sú menej časté a prejavujú sa ako klinicky mierne difúzne formy hyperplázie B-buniek.
Druhou najčastejšou príčinou CHI (po mutáciách Kir6.2 a SUR1) sú aktivačné mutácie génu GLUD1 (AD dedičnosť), kódujúcom glutamát dehydrogenázu. Tento enzým má dôležitú úlohu v regulácii sekrécie inzulínu stimulovanej aminokyselinami (hlavne Leucínom), preto sa u nosičov mutácií GLUD1 objavuje hypoglykémia po požití jedla bohatého na proteíny. Hypoglykémie sú menej frekventné a vznikajú neskôr ako u pacientov s mutáciami podjednotiek draslíkového kanála. Často sa diagnostikujú vo veku 3–4 mesiacov, keď dieťa vynechá nočnú dávku mlieka. Ochorenie sa asociuje s hyperamonémiou (cca 100–200 µmol/l), preto je známe aj pod názvami syndróm Hyperinzulinizmu/Hyperamonémie (HI/HA syndróm), či Leucín-senzitívna hypoglykémia. Vzniká difúzna forma hyperplázie B-buniek, dobre reagujúca na liečbu Diazoxidom. V prevencii hypoglykémie je potrebná reštrikcia proteínov v diéte, prípadne konzumácia bielkovinových pokrmov v kombinácii so sacharidmi.
Kongenitálny hyperinzulinizmus vzniká aj na podklade aktivačných mutácií génu pre glukokinázu (AD dedičnosť). Glukokináza fosforyluje glukózu v B-bunkách a umožnuje jej vstup do metabolizmu a tvorbu ATP. Hladina ATP ovplyvňuje vylučovanie inzulínu väzbou na ATP-dependentný draslíkový kanál B-bunky. Predstavuje tzv. „glukosenzor“ v B-bunkách pankreasu. Diazoxid je liekom voľby.
Kongenitálny hyperinzulinizmus pri SCHAD (Short Chain Acyl Coenzyme A Dehydrogenase Deficiency) je podmienený inaktivačnou mutáciou HADH (AR dedičnosť), génu, ktorý kóduje L-3-hydroxy-acylkoenzým A dehydrogenázu. Ide o enzým, zúčastňujúci sa na beta-oxidácii mastných kyselín. V klinickom obraze dominujú intermitentné nepredvídateľné hypoglykémie s kŕčovou aktivitou. V diagnostike pomáha pozitivita vyšetrenia profilu acylkarnitínov a organických kyselín – zvýšený 3-OH-butyryl-karnitín v plazme a 3-OH-glutarát v moči. Diazoxid je účinnou liečbou.
Ostatné príčiny CHI sú veľmi zriedkavé. Patria medzi ne aktivačné mutácie SLC16A1 (AD dedičnosť), kóduje monokarboxylový transportér 1. Inaktivačné mutácie HNF4A (AD dedičnosť) kóduje hepatálny nukleárny faktor 4α [1, 2, 7, 14].
Diagnostika
Ak sa u dieťaťa objavia epizódy spontánnych ťažkých hypoglykémií trvajúcich viac ako 48 hodín, musíme myslieť na CHI. Glykémia menej ako 3,3 mmol/l (podľa niektorých autorov 2,5 mmol/l u donosených novorodencov a u prematúrnych novorodencov až menej ako 1,7 mmol/l) a súčasne koncentrácia inzulínu nad 3 μU/ml robí diagnózu CHI veľmi pravdepodobnou. Avšak hladina inzulínu u detí s CHI počas dňa výrazne kolíše. Preto nález normálnej hladiny inzulínu počas hypoglykemickej epizódy nevylučuje hyperinzulinizmus.
Diagnózu kongenitálneho hyperinzulinizmu podporuje zachytenie ďalších biochemických markerov excesívneho účinku inzulínu, ako napr. nízka hladina mastných kyselín a ketolátok v krvi, neprítomnosť ketonúrie počas hypoglykemickej epizódy a extrémna nálož cukrov v infúzii (viac ako 10 mg/kg/min.), potrebná na prevenciu hypoglykemických epizód.
Glukagón ako kontraregulačný hormón stimuluje uvoľnenie glukózy zo zásob glykogénu v pečeni. Preto vzostup glykémie po aplikácii glukagónu počas hypoglykémie je senzitívnym markerom hyperinzulinizmu.
Odber biologického materiálu počas hypoglykemickej ataky má z diferenciálno-diagnostických príčin zahŕňať aj odber na hormonálny profil a selektívny metabolický skríning. Hypoglykémia, elevovaný inzulín a amoniak, pozitivita vyšetrenia organických kyselín v moči – elevácia 3-OH-glutarátu v moči a profilu acylkarnitínov – elevácia 3-OH-butyryl-karnitínu v plazme suponujú diagnózy HI/HA syndrómu, respektíve SCHAD.
Keďže klinické odlíšenie fokálnej a difúznej formy CHI na poklade mutácií Kir6.2 a SUR1 nie je možné, v praxi využívame pomocné vyšetrovacie metódy, ako je DNA analýza a 18F-DOPA PET CT.
DNA analýza umožňuje určiť presný typ CHI a suponovať fokálnu formu a tým urýchliť ďalšiu diagnostiku a usmerniť liečbu. Potvrdenie, či ide o difúznu alebo fokálnu formu CHI, dokáže DNA analýza presnejšie ako zobrazovacie metódy, nakoľko veľmi malé ložiská sa nemusia zobraziť vôbec a veľmi veľké imitujú difúzne formy.
Vyšetrenie 18F-DOPA PET CT sa najčastejšie používa na lokalizáciu ložiska v prípade potvrdenia fokálnej formy DNA analýzou. Keďže B-bunky metabolizujú L-DOPA na dopamín účinkom dekarboxylázy, ich zvýšená aktivita sa dá identifikovať pomocou rádiofarmaka 18F(fluoro)-DOPA. Difúzna akumulácia rádiofarmaka nad celou žľazou potvrdzuje difúznu formu a tzv. „horúci uzol“ fokálnu formu (obr. 4).
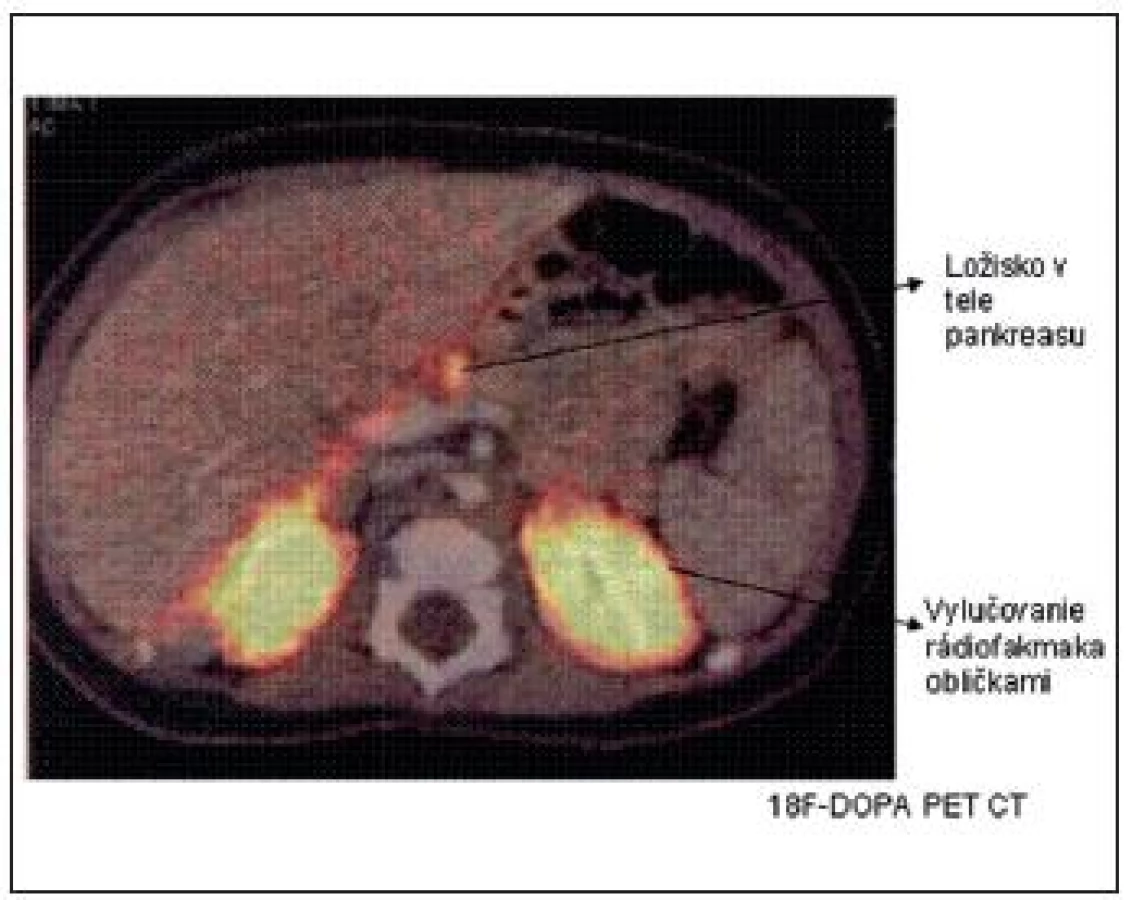
Synchrónne získavanie metabolických a anatomických dát pomocou PET a CT umožní presne lokalizovať ložisko v pankrease, ktoré je peroperačne makroskopicky vzhľadom na zachovanú normálnu lobulárnu štruktúru pri fokálnej forme ťažko identifikovateľné. Vyšetrenie 18F-DOPA PET CT je teda pre úspešnú chirurgickú liečbu (enukleáciu ložiska, ev. parciálnu resekciu pankreasu) nevyhnutné. Senzitivitu tohto vyšetrenia pri CHI naznačil dôkaz ektopickej fokálnej lézie v stene duodena a jejuna. Určité problémy s interpretáciou snímok nastali podľa publikovaných prác v minulosti pri atypických formách CHI, pri viacpočetných ložiskách v pankrease alebo pri interferencii s exkréciou rádiofarmaka obličkami, pečeňou a biliárnym traktom.
Ďalším problémom, ktorý je potrebné prekonať, je interpretácia peroperačných bioptických vzoriek. Následné pooperačné imunohistochemické vyšetrenie vzorky tkaniva s protilátkami proti inzulínu a proinzulínu dokáže správnosť peroperačnej diagnózy [3, 16–21].
V súčasnosti sa upustilo od používania invazívnych stimulačných metód, ako sú PASVS (Pancreatic Arterial Stimulation Venous Sampling) a THPVS (Transhepatic Portal Venous Sampling), ktoré boli technicky náročné, nízko senzitívne a špecifické a ohrozovali pacienta hypoglykémiou počas vyšetrenia [2, 3].
Liečba
Vzhľadom na fakt, že pri hyperinzulinizme nie je mozog chránený alternatívnymi zdrojmi energie (ketolátky, laktát), na prevenciu poškodenia mozgu je nevyhnutná bezodkladná a intenzívna liečba hypoglykémie.
Difúzne formy CHI (okrem recesívnych mutácií Kir6.2 a SUR1) vo väčšine prípadov odpovedajú dobre na liečbu Diazoxidom, ktorý inhibuje sekréciu inzulínu otvorením K-ATP kanála. Perorálna dávka je 15 mg/kg/deň (5–20 mg/kg/deň) rozdelená do 3 dávok. Medzi nežiadúce účinky patrí retencia tekutín (problém hlavne u novorodencov – kardiálne zlyhanie), excesívne ochlpenie na čele, chrbte, obočí (ústup o pár mesiacov po ukončení liečby Diazoxidom).
Naopak väčšina nosičov heterozygotných Kir6.2 a SUR1 mutácií v heterozygotnej (fokálne CHI) alebo homozygotnej, či zloženej heterozygotnej forme (difúzne CHI) nereaguje na liečbu Diazoxidom, nakoľko B-bunky obsahujú len defektné K-ATP kanály. V týchto prípadoch sa odporúča intravenózna infúzia glukózy (rýchlosť často viac ako 10 mg/kg/min.). V prípade, že dieťa toleruje perorálny príjem, môžeme doplniť intravenóznu liečbu perorálnym podaním materského mlieka, prípadne ho môžeme aj fortifikovať pridaním sacharidov.
Oktreotid (somatostatínový analóg) inhibuje sekréciu inzulínu. Používa sa dočasne na preklenutie obdobia do pankreatektómie. Aplikuje sa parenterálne (1–4 µg/kg/dávku každých 6 – 8 – 12 – 24 hodín subkutánne alebo v kontinuálnej intravenóznej infúzii rýchlosťou 1–5 µg/kg/hod., v niektorých centrách aj subkutánnou infúziou). V úvode liečby je dobre účinný, avšak časom stráca efektivitu. Niektorí autori uvádzajú nepriamo úmernú závislosť efektivity oktreotidu so zvyšujúcou sa dávkou (viac ako 20–40 µg/kg/deň). Z nežiadúcich účinkov sa môže objaviť cholecystolitiáza, alterácia črevnej motility s následným nechutenstvom, hypotyroidizmus, riziká spojené s parenterálnou aplikáciou (bolesť, infekcia a i.).
Glukagón stimuluje sekréciu glukózy z pečene. V terapii sa využíva v subkutánnej, intramuskulárnej aplikácii alebo ako kontinuálna intravenózna infúzia počas prípravy na operáciu. Oktreotid a glukagón sú účinné u väčšiny typov hyperinzulinizmu.
Niektoré centrá používajú aj nifedipín, kalciový blokátor používaný na liečbu hypertenzie. Jeho účinok je pri hyperinzulinizme diskutabilný.
Chirurgická liečba – selektívna resekcia ložiska, eventuálne enukleácia ložiska, subtotálna pankreatektómia (80–94 %) alebo takmer totálna pankreatektómia (viac ako 95 % tkaniva) s rizikom iatrogénneho diabetu a exokrinnej insuficiencie pankreasu. Je dôležité si uvedomiť, že úspešná chirurgická liečba umožňuje úplné vyliečenie pacienta s fokálnou formou CHI a zmiernenie príznakov u difúznej a atypickej formy s následnou medikamentóznou liečbou [2, 3, 14].
V literatúre publikované zlyhanie chirurgickej liečby súviselo so zriedkavým nálezom viacpočetných ložísk, s inkompletnou resekciou ložiska alebo s nerovnomerným rozložením gradientu aktivity B-buniek pri 18F-DOPA PET CT vyšetrení a tým podmieneným nesprávnym označením atypickej alebo difúznej formy za fokálnu formu CHI [3, 20–22].
Úspešnosť chirurgickej liečby závisí od miery resekcie. V jednotlivých centrách sa pohybuje od 92–98 % pri parciálnej resekcii pri fokálnom CHI, až po 7–11% úspešnosť pri difúznej forme. Miera resekcie pankreasu je dlhodobo diskutovanou témou. V mnohých prípadoch CHI je medikamentózna liečba dostačujúcim prostriedkom na prevenciu poškodenia mozgu hypoglykémiou. V literatúre boli popísané aj raritné prípady spontánnej remisie podmienené pravdepodobne apoptózou alebo maturáciou buniek pankreasu [23, 24]. Preto musíme striktne predchádzať ireverzibilnej rozsiahlej resekcii pankreasu (nad 95 %) v novorodeneckom veku, ktorá je spojená s vysokým rizikom vzniku diabetu (30–40 %) a exokrinnej insuficiencie pankreasu. Medzi popisované komplikácie operačného riešenia patria perzistujúce hypoglykémie (40 %), poranenie ductus choledochus (17 %), prolongovaná sekrécia z drénu v rane, vznik pseudocysty pankresu, chylózny ascites a i. [3, 25–30].
Prognóza
Riziko poškodenia mozgu závisí od včasnosti objavenia sa hypoglykémií, od ich závažnosti a trvania. Preto je nevyhnutná včasná agresívna liečba s cieľom udržať normoglykémiu.
Podľa literárnych údajov bola v súbore sledovaných pacientov s CHI hypoglykémia vznikajúca v novorodeneckom období hlavným prognosticky nepriaznivým faktorom pre vznik ťažkej psychomotorickej retardácie a epilepsie. Ak dieťa dobre reaguje na medikamentóznu liečbu, býva prognóza podobná ako pri chirurgickej liečbe. Avšak dôležité je uvedomiť si, že pri fokálnej forme CHI je chirurgická liečba kuratívna [3, 15, 30].
Záver
Kongenitálny hyperinzulinizmus je relatívne starou nozologickou jednotkou, s ktorou sa môžeme stretnúť v neonatológii, ako aj v pediatrii ako takej. Avšak až metódy DNA analýzy umožnili odhalenie jej presnej príčiny. DNA analýza totiž dokáže identifikovať príčinu až 85 % najťažších prípadov CHI a zároveň rozlíšiť difúznu a fokálnu formu s odlišným terapeutickým prístupom aj prognózou [1, 2, 3, 7].
Počas roku 2009 boli na našom pracovisku hospitalizovaní dvaja pacienti s perzistujúcimi hypoglykémiami, u ktorých bolo možné (vďaka novej možnosti vyšetrenia pacientov pomocou DNA analýzy už aj na Slovensku) potvrdiť a následne liečiť fokálnu formu kongenitálneho hyperinzulinizmu. Podrobnejší rozbor oboch kazuistík bude nasledovať v ďalšom čísle.
Poďakovanie
Autori vyslovujú poďakovanie MUDr. J. Staníkovi za odborné usmernenie pri tvorbe článku.
MUDr. Monika Rosoľanková
Oddelenie patologických novorodencov
a JIS I. DK
DFNsP
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: mnkrsnkv@gmail.com
Sources
1. Kelly P, Stanley CHA, Sarafoglou K, et al. Pediatric Endocrinology and Inborn Errors of Metabolism. McGraw-Hill, 2009 : 39–50.
2. Thornton PS. Congenital Hyperinsulinism. Children´s Hospital of Philadelphia, http://www. sur1.org/hyperinsulism.htm.
3. Barthlen W, Blankenstein O, Mau H, et al. Evaluation of 18F-DOPA PET CT for surgery in focal congenital hyperinsulinism. JCEM 2008;;93(3): 869–875.
4. Sweet IR, Najafi H, Matschinsky FM, et al. Annotated questions and answers about glucose metabolism and insulin secretion of B cells. Diabetes Rev. 1996;;4 : 130–144.
5. Kelly A, Li CH, Matter A, et al. Amino acid-stimulated insulin secretion: lessons learned from congenital hyperinsulinism. Pediatric Academic Society´s Annual Meeting, 2004.
6. Fourtner SH, Stanley CHA, Kelly A, et al. Protein sensitivity not synonymous with leucine sensitivity. The Endocrine Society´s, 85th Annual Meeting, 2003.
7. Staník J, Rosoľanková M, Gašperíková D, et al. Prínos DNA analýzy pre diagnostiku a liečbu perzistujúcich hypoglykémií u detí. Čes.-slov. Pediat. 2009;;64(11): 559–560.
8. Huopio H, Reimann F, Ashfield R, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J. Clin. Invest. 2000; 106 : 897–906.
9. Magge SN, Shyng SL, McMullen C, et al. Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the B cell sulfonylurea receptor. JCEM 2004; 89 : 4450–4456.
10. Thornton PS, McMullen C, Ganguly A, et al. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes 2003; 52 : 2403–2410.
11. Verkarre V, Fournet JCH, De Lonlay P, et al. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J. Clin. Invest. 1998; 102 : 1286–1291.
12. Pinney SE, MacMullen C, Becker S, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant K ATP channel mutations. J. Clin. Invest. 2008; 118 : 2877–2886.
13. De Lonlay P, Fournet JCH, Rahier J, et al. Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J. Clin. Invest. 1997; 100 : 802–807.
14. Zschocke J, Hoffmann GF, et al. Vademecum Metabolicum. 2nd ed. Germany, Schattauer: Milupa, 2004; 109–110.
15. Menni F, De Lonlay P, Sevin C, et al. Neurologic outcomes of 90 neonates and infants with PHH. Pediatrics 2001; 107(3): 476–478.
16. Otonkoski T, Nanto-Salonen K, Hussain K, et al. Noninvasive diagnosis of focal hyperinsulinism of infancy with 18F-DOPA PET. Diabetes 2006; 55 : 13–18.
17. Ribeiro MJ, Boddaert N, De Lonlay P, et al. The added value of 18F-DOPA PET in the diagnosis of hyperinsulinism of infancy: a retrospective study involving 49 children. Eur. J. Nucl. Med. Mol. Imaging 2007; 34 : 2120–2128.
18. Hardy OT, Hernandez-Pampaloni M, Saffer JR, et al. Accuracy of 18F - DOPA positron emission tomography for diagnosing and localizing focal congenital hyperinsulinism. JCEM 2007; 92 : 4706–4711.
19. Hussain K, Seppanen M, Nanto-Salonen K, et al. The diagnosis of ectopic focal hyperinsulinism of infancy with 18F-DOPA positron emission tomography. JCEM 2006; 91 : 2839–2842.
20. Peranteau WH, Bathaii SM, Pawel B, et al. Multiple ectopic lesions of focal islet adenomatosis identified by PET scan in an infant with congenital hypeinsulinism. J. Pediatr. Surg. 2007; 42 : 188–192.
21. Giurgea I, Sempoux CH, Bellanne - Chantelot CH, et al. The Knudson´s two-hit model and timing of somatic mutation may account for the phenotypic diversity of focal congenital hyperinsulinism. JCEM 2006; 91 : 4118–4123.
22. De Lonlay P, Simon A, Galmiche-Rolland L, et al. Neonatal hyperinsulinism: clinicopathologic correlation. Hum. Pathol. 2007; 38 : 387–399.
23. Kassem SA, Ariel I, Thornton PS, et al. B cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 2000; 49 : 1325–1333.
24. Kubota A, Yonekura T, Usui N, et al. Two cases of persistent hyperinsulinemic hypoglycemia that showed spontaneous regression and maturation of the Langerhans islets. J. Pediatr. Surg. 2000; 35 : 1661–1662.
25. Stanley CHA, Adzick NS, Thornton PS, et al. A multidisciplinary approach to the focal form of congenital hyperinsulinism leads to successful treatment by partial pancreatectomy. J. Pediatr. Surg. 2004; 39 : 270–275.
26. Suchi M, Courtney M, Thornton PS, et al. Congenital hyperinsulinism – intraoperative biopsy interpretation can direct the extent of pancreatectomy. Am. J. Surg. Pathol. 2004; 28 : 1326–1335.
27. McAndrew HF, Smith V, Spitz L, et al. Surgical complications of pancreatectomy for persistent hyperinsulinaemic hypoglycaemia of infancy. J. Pediatr. Surg. 2003; 38 : 13–16.
28. De Lonlay-Debeney P, Poggi-Trvert F, Fournet JCH, et al. Clinical features of 52 neonates with hyperinsulinism. N. Engl. J. Med. 1999; 340 : 1169–1175.
29. Crétolle C, Fekete CN, Jan D, et al. Partial elective pancreatectomy is curative in focal form of PHHI. J. Pediatr. Surg. 2002; 37 : 155–158.
30. Meissner T, Wendel U, Burgard P, et al. Long-term follow up of 114 patients with congenital hyperinsulinism. Eur. J. Endocrinol. 2003; 149 : 43–51.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2010 Issue 9
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Congenital Hyperinsulinism – Most Frequent Cause of Persistent Hypoglycemia in Newborns and Infants
- Neonatal Hydrocephalus – The Value of Evaluation of the Brain by Means of Sonography
- The Effect of Initial Management of Ventilation on the Incidence of Bronchopulmonary Dysplasia and Other Morbidities in Neonates Born at 24th–27th Weeks of Gestation at Clinic of Neonatology Slovak University Hospital Nove Zamky
- Expression Pattern of Homeodomain Genes Does Not Define the Known Subgroups of Childhood Acute Lymphoblastic Leukemias