
Úloha hepcidinu v regulaci metabolismu železa
The role of hepcidin in iron metabolism
Iron is essential for proper function of the cells in human body. Iron is involved in tissue oxygenation, antioxidant defence, cell proliferation and in other important metabolic processes as a part of haemoproteins (haemoglobin, myoglobin) and different iron-containing enzymes. It is crucial to keep iron homeostasis balanced, because of the potential iron toxicity. Hepcidin plays a key role in this process by controlling iron absorption from the diet, iron recycling from senescent erythrocytes and iron release from stores. Many diseases are accompanied by abnormal hepcidin level. Inappropriately low hepcidin leads to iron overload and is typical for hereditary haemochromatosis. Increased hepcidin level contributes to the development of anaemia caused by insufficient iron supply for erythropoiesis.
Molecular mechanisms affecting hepcidin production are still subject of intensive research. More detailed knowledge about these mechanisms may contribute to the identification of new molecules involved in the regulation of iron metabolism and to the development of new drugs potentially useful in the treatment of disorders associated with disrupted iron homeostasis.
Key words:
iron metabolism, hepcidin, ferroportin, hereditary haemochromatosis, anaemia of chronic diseases
:
J. Houda 1; D. Pospíšilová 1; M. Horváthová 2
:
Dětská klinika při LF UP a FN, Olomoucpřednosta prof. MUDr. V. Mihál, CSc.
1; Ústav biologie při LF UP, Olomoucpřednosta doc. RNDr. V. Divoký, Ph. D.
2
:
Čes-slov Pediat 2014; 69 (5): 301-312.
:
Review
Železo je nezbytné pro správnou funkci buněk lidského organismu. Jako součást hemoproteinů (hemoglobinu, myoglobinu) a řady enzymů se podílí na oxygenaci tkání, na obraně proti oxidativnímu stresu, na proliferaci buněk a na dalších důležitých metabolických pochodech. Protože je však nadbytek železa pro buňky a organismus potenciálně toxický, udržení homeostázy železa je pro organismus zásadní. Klíčovou roli v tomto procesu hraje hepcidin, který kontroluje vstřebávání železa ze stravy, recyklaci železa ze zaniklých erytrocytů a jeho uvolňování ze zásob. Mnoho onemocnění se vyznačuje abnormální hladinou hepcidinu. Neadekvátní snížení hepcidinu vede k přetížení organismu železem, které je typické u hereditární hemochromatózy, naopak zvýšená hladina hepcidinu se podílí na rozvoji anemie v důsledku nedostatečného přísunu železa pro erytropoézu.
Molekulární mechanismy ovlivňující produkci hepcidinu jsou dosud předmětem intenzivního výzkumu. Podrobnější znalosti o těchto mechanismech mohou sloužit k vývoji nových látek zasahujících do regulace metabolismu železa, které mohou být využity k diagnostickým a terapeutickým účelům u stavů s narušenou homeostázou železa.
Klíčová slova:
metabolismus železa, hepcidin, feroportin, hereditární hemochromatóza, anemie chronických chorob
Úvod
Železo (Fe) je esenciální prvek, který se hojně vyskytuje v živé i neživé přírodě. Nejčastěji se nachází v nerozpustné formě s minimální využitelností pro člověka. V živé přírodě se železo vyskytuje ve vazbě na hem – hemové železo a vázané na jiné proteiny – nehemové železo, přičemž v těchto sloučeninách se železo nachází jako dvojmocné Fe2+ nebo trojmocné Fe3+. Nedostatek železa vede k rozvoji sideropenie a sideropenické anemie. Jeho nadbytek naopak vede ke zvýšené produkci volných kyslíkových radikálů a zvýšení oxidačního stresu, který může být příčinou poškození buněk a tkání. Vzhledem ke své potenciální toxicitě se železo v organismu nachází ve formě vázané, a to na různé transportní a zásobní proteiny (např. transferin, feritin atd.). Přesná regulace metabolismu železa je proto nezbytná pro správnou funkci buněk lidského organismu. Udržení správné hladiny železa v organismu je zajištěno citlivým regulačním mechanismem, který je schopný reagovat na aktuální stav železa a jeho spotřebu a udržet tím nezbytnou rovnováhu. Objev několika klíčových molekul v posledním desetiletí významně přispěl k pochopení dosud nejasných aspektů metabolismu železa.
Cirkulace železa u člověka
Celkové množství železa v organismu se u novorozence pohybuje kolem 75 mg/kg hmotnosti. U dospělého člověka se liší v závislosti na pohlaví a věku. U dospělého muže je přibližně 50 mg/kg hmotnosti a v průběhu dospělosti hladina železa více nekolísá. U žen ve fertilním věku dochází postupně v důsledku přirozených ztrát při menstruaci ke snížení obsahu železa až na hladinu asi 35 mg/kg hmotnosti. Ve srovnání s celkovým množstvím železa v organismu jsou jeho denní ztráty minimální: 1 mg/den u dospělého muže a u žen mimo reprodukční věk. U menstruujících žen jsou ztráty od 0,006–0,025 mg/kg/den, a to především v důsledku menstruačního krvácení. Železo se vylučuje stolicí při odlučování starých enterocytů, močí při odlučování epitelu močového ústrojí a malá část je vylučována potem; ani jeden z těchto procesů však není regulován. Většina železa v organismu je vázána na hemoglobin erytrocytů (cca 65 %), další významná část je uložena v zásobní formě (feritin a hemosiderin) v játrech a slezině (cca 25 %) (tab. 1). Železo z erytrocytů je recyklované a uvolněné zpět do krevního oběhu, přičemž tento proces je nejvýznamnějším zdrojem železa (20–25 mg/den). Starší nebo poškozené erytrocyty jsou degradovány v monocyto-makrofágovém systému sleziny. Po fagocytóze erytrocytu makrofágy dochází k rozpadu hemoglobinu a železo je uvolněno do cytoplazmy makrofágu. Podle potřeby je železo buďto aktivně uvolňováno zpět do krevního oběhu, nebo uskladněno navázáním na feritin nebo hemosiderin (tab. 1).
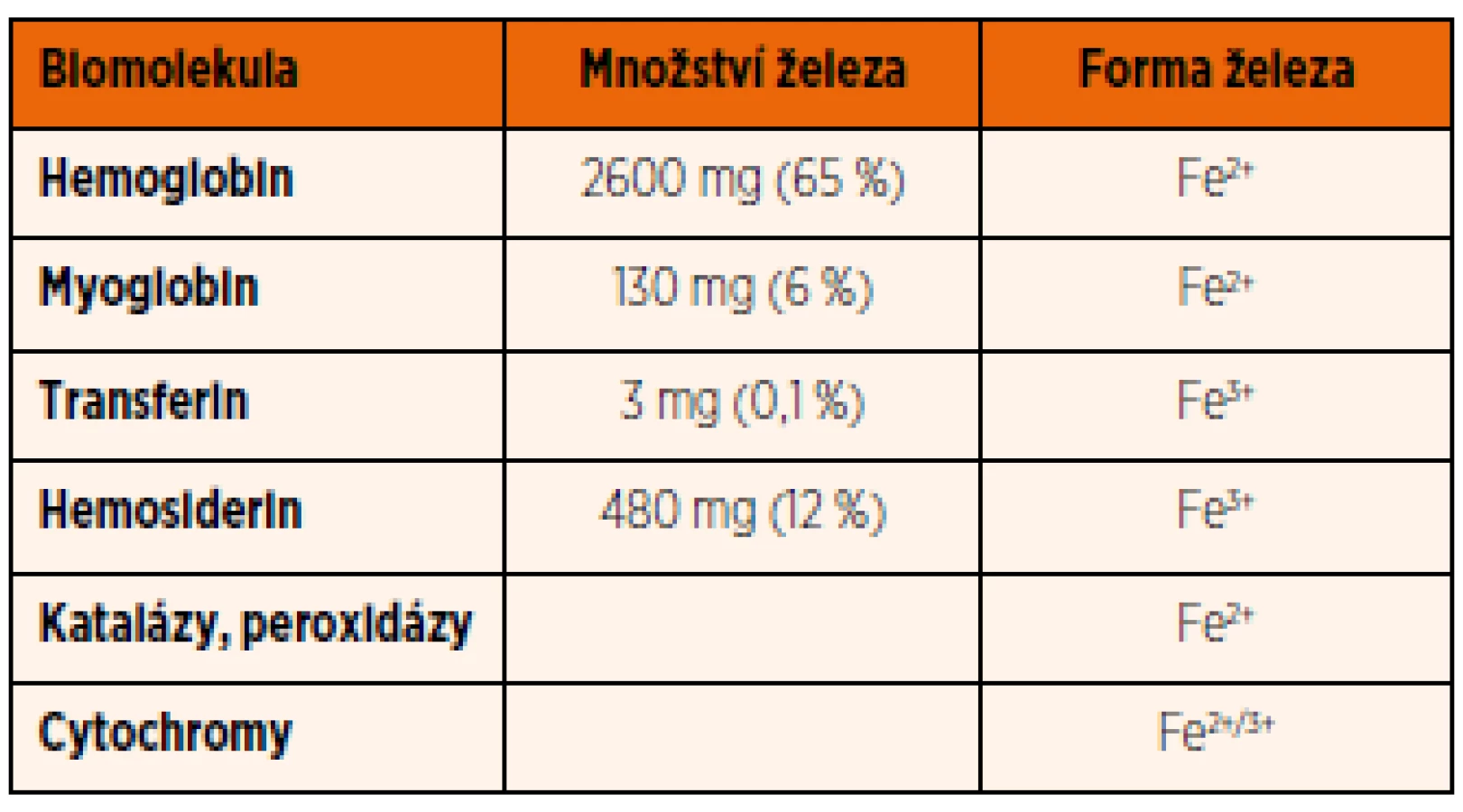
Relativně malá část železa (1–2 mg/den) je vstřebávaná ze stravy. Stejně jako uvolňování železa ze zásob, tak i jeho vstřebávání z gastrointestinálního traktu (GIT) je přísně regulováno a řízeno na úrovni buněčných membrán. Naopak u člověka a vyšších obratlovců chybí aktivní a regulovaný systém pro vylučování nadbytečného železa.
Vstřebávání železa
Železo se vstřebává jednak ve formě hemu, který pochází z hemoglobinu a myoglobinu, a jednak ve formě volné. Hemové železo v potravinách živočišného původu je vstřebáváno v zažívacím traktu mnohem efektivněji než železo volné, molekulární mechanismus však zatím není zcela objasněn. Transmembránový přenašeč SLC46A1 neboli HCP1 (heme carrier protein 1), který byl původně identifikován jako transportér hemu [1], se podílí především na transportu kyseliny listové [2].
Ve volné formě je dvojmocné železo (Fe2+) vstřebáváno na apikální straně enterocytů pomocí transmembránového přenašeče DMT1 (divalent metal transporter 1) [3] (obr. 1A). Fe2+ vzniká redukční přeměnou z Fe3+, která je na apikální straně enterocytu v GIT katalyzována enzymem DCytB (duodenal cytochrome B).
Poté, co je v enterocytu část železa spotřebována na základní biochemické procesy a část je uložena do zásob po vazbě na feritin, je zbývající část železa aktivně uvolněna do krevního řečiště (obr. 1B). K uvolňování železa z enterocytů dochází na jejich bazolaterální straně za účasti feroportinu (FPN), zatím jediného známého exportéru železa [4]. Po vstupu Fe2+ iontů do krevního řečiště je Fe2+ oxidováno na trojmocnou formu Fe3+ pomocí feroxidázy hephaestinu [5]. Následně se trojmocné železo váže na transferin (Tf), čímž se snižuje jeho reaktivita a pohotovost k produkci toxických radikálů a v této vazbě je transportováno k cílovým buňkám exprimujícím transferinový receptor 1 (TfR1). Saturace transferinu železem je důležitým ukazatelem celkové hladiny železa v organismu; za fyziologických podmínek je transferin saturován železem na 30 %, při jeho nadbytku saturace Tf stoupá.
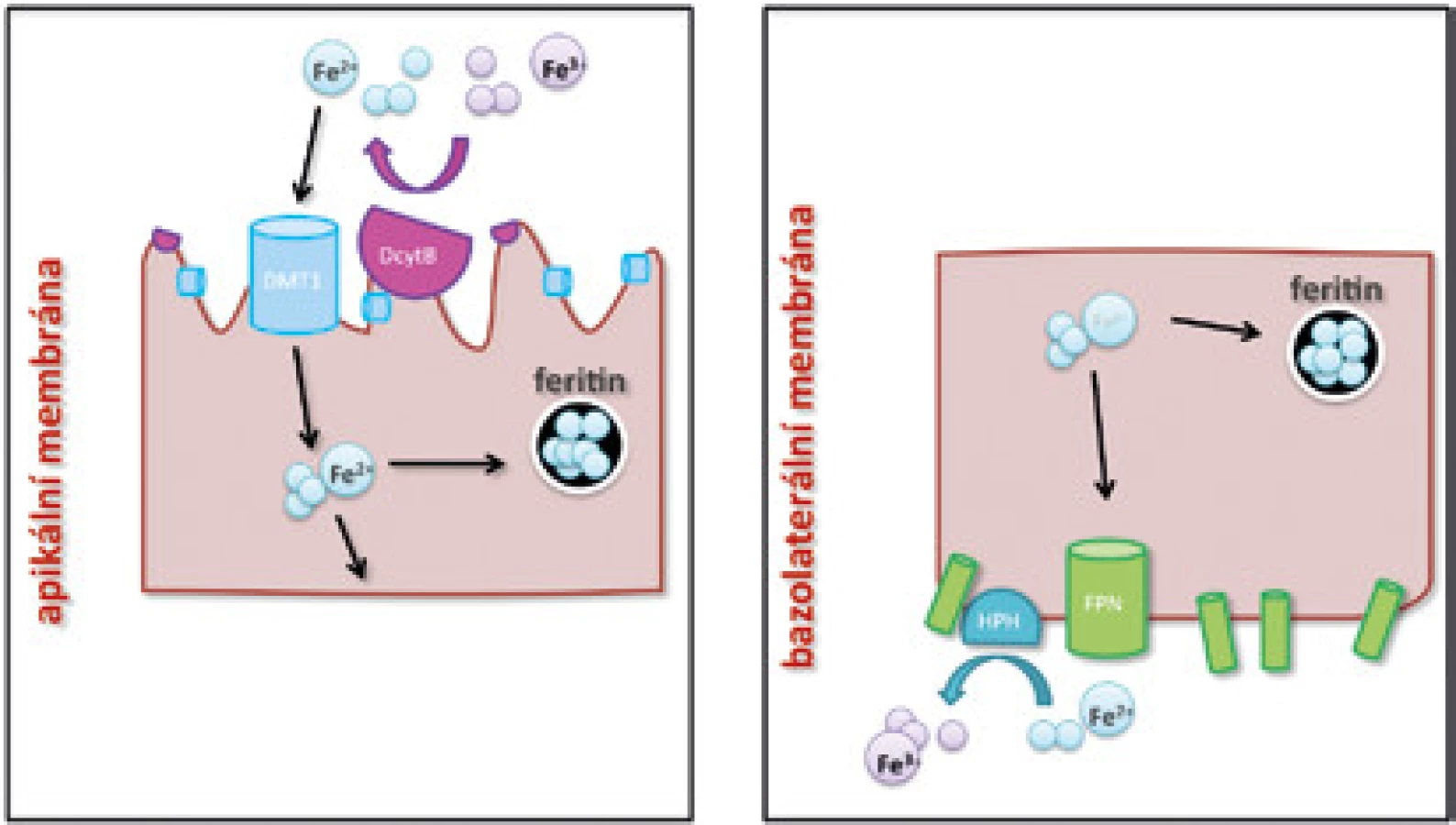
Transferin s Fe3+ (holotransferin) se na povrchu buněk naváže na membránový TfR1 a celý komplex se do intracelulárního prostředí dostává endocytózou. Z vytvořených endozomů je Fe2+ po redukci transportováno již zmíněným proteinem DMT1, klíčovou roli pro efektivitu přenosu zde hraje kyselé prostředí obsahující vodíkové protony [6]. V erytroidních buňkách je Fe2+ inkorporované do hemu a následným spojením s globinovými řetězci se vytváří molekula hemoglobinu.
Recyklace železa v makrofázích
Jak jsme již zmínili, staré nebo poškozené erytrocyty jsou pohlceny makrofágy a degradací hemoglobinu v lysozomech dochází k uvolnění železa, které je podle potřeb vráceno do cirkulace nebo uskladněno ve formě feritinu. I z makrofágů je export Fe2+ zabezpečen feroportinem [4]. Tento proces recyklace železa je klíčový pro udržení homeostázy železa a jeho porucha se podílí např. na patofyziologii hereditární hemochromatózy a anemie chronických chorob (nyní označované jako anemie zánětu).
Hepcidin, feroportin a jejich role v metabolismu železa
Hepcidin
Již před více jak deseti lety (2001) dva nezávisle pracující výzkumné týmy [7, 8] poprvé identifikovaly hepcidin, který je dnes považovaný za klíčový peptid v metabolismu železa. Hepcidin je vylučován močí a syntetizován převážně v játrech. V menším množství je exprimován i v makrofázích, adipocytech [9], β-buňkách pankreatu [10] a řadě dalších tkání včetně srdce a mozku, avšak role zde produkovaného hepcidinu je zatím nejasná. Hepcidin je peptid složený z 25 aminokyselin a 4 disulfidických můstků. Je kódován genem HAMP (hepcidin antimicrobial peptide) lokalizovaném na dlouhém raménku chromozomu 19 (19q13.1) [11]. Translací mRNA vzniká prekurzor hepcidinu pre-prohepcidin složený z 84 aminokyselin. Následně je z něj enzymaticky odštěpen prohepcidin složený z 64 aminokyselin. Poté je prohepcidin uvolněn do lumen endoplazmatického retikula, kde dochází k odštěpení dalších 39 aminokyselin enzymem furinu podobné proproteinové konvertázy a tím vzniká peptid složený z 25 aminokyselin – hepcidin-25 pocházející z C konce prekurzoru [12]. Byly identifikovány i další formy hepcidinu složené z menšího počtu aminokyselin: hepcidin-22 a hepcidin-20 [13]. Biologický význam pre-prohepcidinu nebyl prokázán [14]. U hepcidinu-20 byl in vitro prokázán antibakteriální a antifungální účinek v koncentracích 10násobně vyšších, než je běžná hladina zdravých kontrol [8]. Struktura hepcidinu je mezidruhově poměrně jednotná. Lze tedy usuzovat, že mechanismus řízení metabolismu železa, na němž se hepcidin podílí, je efektivní a fylogeneticky výhodný [15].
Původně oba objevitelské týmy určily hepcidinu roli v nespecifické imunitní odpovědi organismu při obraně proti bakteriální infekci. Vzápětí byl na myším modelu objeven vztah hepcidinu a homeostázy železa [16] a ještě téhož roku byla blíže popsána jeho role hlavního regulátoru metabolismu železa [17]. Proteinem, který se spolu s hepcidinem podílí na tomto procesu, je feroportin.
Feroportin
Feroportin byl objeven v roce 2000 třemi nezávislými týmy, které jej popsaly jako transmembránový přenašeč železa nacházející se ve zvýšené míře na bazolaterální membráně enterocytů duodena a na membráně makrofágů monocyto-makrofágového systému a Kupfferových buněk jater. V menší míře se pak nachází v plicních buňkách, buňkách renálních tubulů a v erytroidních prekurzorech. Feroportin je kódován genem SLC40A1 (SLC11A3, 2q32). Je to jedinečný peptid s konzervativní strukturou společnou pro savce a jiné obratlovce a jediný doposud objevený specifický transmembránový exportér železa [4]. Prostřednictvím feroportinu je intracelulární železo přenášeno do krevního oběhu, avšak přesný mechanismus přenosu není doposud objasněn. Byly popsány dvě formy feroportinu [18]. FPN1A (Feroportin 1A) obsahuje ve své struktuře 5’ element senzitivní na hladinu železa, který v buňkách s nedostatkem železa tlumí jeho syntézu. FPN1B (Feroportin 1B) tento 5’ element neobsahuje a nachází se především v duodenálních enterocytech, kde zajišťuje přenos železa z GIT do krevního oběhu a jeho syntéza není tlumena nedostatkem železa.
Mechanismus účinku hepcidinu a feroportinu
Po navázání hepcidinu na feroportin dochází k internalizaci komplexu a jeho degradaci v lysozomech buňky [19]. Snížené množství feroportinu na membráně buňky způsobuje snížený výdej železa z intracelulárního prostoru do prostoru extracelulárního. Stejným mechanismem snižuje hepcidin v enterocytech vstřebávání železa ze zažívacího traktu a výdej železa do krevní cirkulace. V případě nedostatku železa dojde naopak k utlumení produkce hepcidinu a ke zvýšenému uvolňování železa do krevního oběhu prostřednictvím feroportinu. Tímto mechanismem je udržována relativně stabilní rovnováha železa v organismu (obr. 2).
![Homeostáza železa v organismu a řízení jeho cirkulace hepcidinem. Hepcidin je produkován v hepatocytech jater v odpovědi na stav železa v organismu (stimulační signál, +) a aktivitu erytropoézy (tlumící signál, -). Hepcidin (černá čára) inhibuje transport železa z enterocytů duodena, makrofágu sleziny a hepatocytů jater (x) a snižuje tak hladinu železa v plazmě. Patologické zvýšení produkce hepcidinu (v důsledku zánětu a inaktivačních mutací matriptázy 2, MT-2) vede k utlumení uvolňování železa z výše zmíněných buněčných kompartmentů a ke snížení jeho hladiny v plazmě, což vede k rozvoji anemie. Naopak při patologickém snížení produkce hepcidinu (v důsledku zvýšených hladin erytroidních faktorů nebo při mutacích genů asociovaných s hemochromatózou) dochází ke zvýšení hladiny železa v plazmě a eventuálnímu rozvoji přetížení organismu železem. (Upraveno dle: Ganz, 2011 [20])](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/848bc88ac818fd1ea801324a50f13dec.jpg)
Narušení této rovnováhy v důsledku mutací genů pro feroportin a hepcidin vede k rozvoji primárního přetížení organismu železem, neboli hereditární hemochromatóze. Při dominantních mutacích genu pro feroportin (hemochromatóza typ 4b), které způsobují jeho rezistenci na hepcidin, a při recesivních mutacích HAMP genu, které vedou k produkci neúčinného hepcidinu, dochází k nadměrnému uvolňování železa z makrofágů a enterocytů a rozvoji onemocnění podobnému klasické hemochromatóze (asociované s mutacemi v HFE, viz níže) s hyperferitinemií a přetížením parenchymatózních tkání železem (obr. 2). V důsledku časného nástupu klinických příznaků u pacientů s mutací v HAMP genu se tomuto typu hemochromatózy říká juvenilní (typ 2b). Byly však popsány i dominantní mutace feroportinu, které vedou k jeho inaktivaci a způsobují tak hromadění železa v makrofázích a fenotyp odlišný od klasické hemochromatózy, kdy se u pacienta v důsledku nízké saturace transferinu může eventuálně rozvinout anemie. Tento typ hemochromatózy (4a) nebývá obvykle asociován s orgánovým poškozením železem a označuje se někdy i jako feroportinová nemoc [21].
Faktory ovlivňující produkci hepcidinu
Hepcidin efektivně řídí distribuci železa v lidském organismu. Regulací produkce tohoto peptidu organismus vyrovnává výkyvy v homeostáze železa. Mezi faktory, které stimulují produkci hepcidinu, patří zvýšená hladina železa a zánětlivé procesy, naopak deficit železa, akcelerovaná erytropoéza a hypoxie působí na produkci hepcidinu supresivně (obr. 2).
Produkce hepcidinu ovlivněná hladinou železa v krevním oběhu
Exprese hepcidinu je zpětně regulována hladinou železa v krevním oběhu, přičemž BMP/SMAD signální dráha je považována za hlavní regulační osu transkripce hepcidinu [22] v hepatocytech jater (obr. 3). BMP (bone morphogenetic proteins) je skupina molekul regulujících několik důležitých procesů jako embryogenezi, tvorbu a remodelaci kostí a hojení tkání. Zdrojem BMP jsou pravděpodobně intersticiální a stelární jaterní buňky. Ze skupiny BMP molekul se na stimulaci hepcidinu závislé na železe podílí BMP6 [23]. Interakce BMP6 s příslušnými receptory (BMP receptor I a II, BMPRI-II) [24] vede k aktivaci komplexu SMAD1/5/8-SMAD4 (small mothers against decapentaplegic), který je translokován do jádra, kde aktivuje expresi HAMP genu. Důležitou roli v tomto procesu hraje hemojuvelin (HJV), glykosylfosfastidylinositolem ukotvený membránový protein, který funguje jako koreceptor pro BMP6 [22, 25]. Recesivní mutace genu pro HJV způsobují deficit hepcidinu a vedou k rozvoji dalšího typu hemochromatózy označované i jako juvenilní hemochromatóza typu 2a.
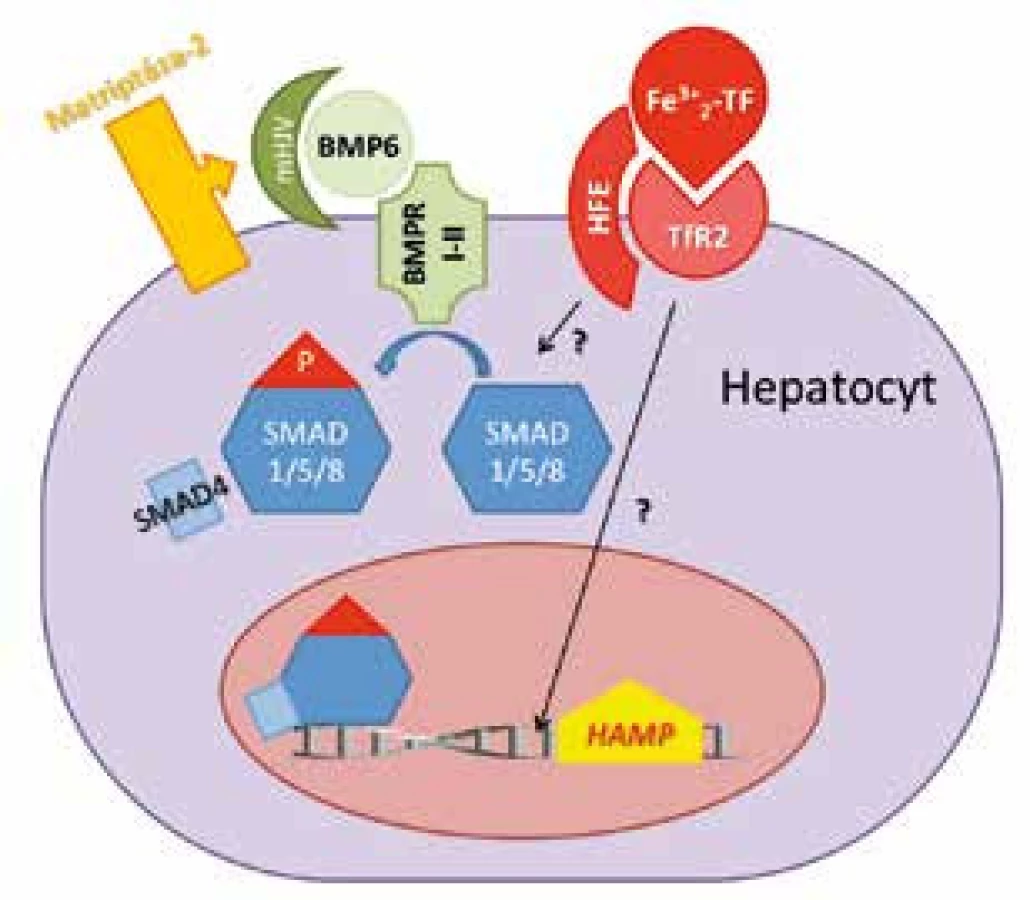
Přesná role dalších dvou proteinů, HFE (human hemochromatosis protein) a transferinového receptoru 2 (TfR2), na regulaci hepcidinu v závislosti na hladině železa je méně objasněna (obr. 3). Nicméně mutace obou proteinů narušují expresi hepcidinu a vedou k rozvoji hemochromatózy typu 1 (HFE) a 3 (TfR2) (obr. 2), co podporuje jejich podíl na regulaci exprese hepcidinu [26, 27]. HFE je membránový protein strukturálně podobný molekulám MHC třídy I, který kompetitivně váže TfR1 [27, 28]. Předpokládá se, že vazba HFE na TfR1 je inhibována při nadbytku holotransferinu, který obsazuje vazebné místo pro HFE. Následně se HFE nenavázaný na TfR1 váže na TfR2, kde je jeho vazba méně silná. V kombinaci s holotransferinem je vazba HFE na TfR2 zvýšena [29]. Celý komplex poté stimuluje expresi hepcidinu aktivací signální dráhy, která dosud zůstává předmětem dalšího výzkumu. Některé studie [27, 30] popisují ovlivnění BMP kaskády a/nebo MAPK (mitogen-activated protein kinase) kaskády komplexem holotransferin-HFE-TfR2. Výsledkem je zvýšení hladiny hepcidinu a následný útlum vstřebávání a uvolňování dalšího železa do krevního oběhu z enterocytů a buněk monocyto-makrofágového systému.
Dalšími významnými molekulami, které však naopak tlumí produkci hepcidinu, jsou matriptáza-2 (MT-2) kódována genem TMPRSS6 (transmembrane protease serine 6) a neogenin. Oba proteiny štěpí membránový hemojuvelin a snižují tak jeho množství na povrchu buněk[31–33], což vede k utlumení BMP6/SMAD signální dráhy a supresi hepcidinu (obr. 3). Děje se tak při nedostatku železa [31]. V roce 2008 byla popsána bodová mutace Ala736Val v genu TMPRSS6 na chromozomu 22q12-13. Mutace vedla k inaktivaci MT-2 a v in vitro modelu ke zvýšení exprese hepcidinu. Posléze byly různé mutace TMPRSS6 nalezeny i u dalších pacientů s hypochromní mikrocytární anemií, která je rezistentní na perorální podávání železa a parenterální aplikace železa vede jen k jejímu malému zlepšení [34]. Jedná se o autozomálně recesivně dědičnou formu sideropenické anemie rezistentní na železo, která se označuje jako IRIDA (iron refractory iron deficiency anaemia) [35]. Zvýšená exprese TMPRSS6 byla kromě akutního alimentárního nedostatku železa [36] zjištěna i u hypoxie [37, 38] a po injekční aplikaci BMP6 [39].
Erytropoéza potlačuje produkci hepcidinu
Největší část železa je v organismu spotřebovávána při produkci erytrocytů, a to jeho vazbou v molekule hemoglobinu, která probíhá v erytroidních prekurzorových buňkách. Erytropoéza je téměř výhradně závislá na hladině železa v krevním oběhu vázaného na holotransferin [40]. Předpokládá se, že zvýšený obrat erytropoézy při krvácení nebo při zvýšeném působení erytropoetinu tlumí produkci hepcidinu v játrech pomocí signálu, který vychází z kostní dřeně [41]. Tento tlumivý efekt erytropoézy se vyskytuje i u onemocnění charakterizovaných zvýšenou, avšak neefektivní erytropoézou, kde často dochází k apoptóze erytroidních prekurzorů a nedostatečné tvorbě zralých erytrocytů. Důsledkem je patologická suprese hepcidinu a následná akumulace železa ve tkáních (obr. 2). Prototypem toho typu onemocnění je β-thalassemia intermedia [42, 43], u které dochází k přetížení železem i bez transfuzí. Z toho vyplývá, že signály přicházející z kostní dřeně jsou nadřazené signálům, které informují o hladině železa v organismu. Nicméně funkční erytropoéza ve dřeni je esenciální pro utlumení exprese hepcidinu, protože pacienti s kongenitální Diamondovou-Blackfanovou anemií (DBA), která se vyznačuje těžkou anemií a erytroidní aplazií v kostní dřeni, nejsou schopni suprimovat hladinu hepcidinu, ta zůstává zvýšená [44]. Za potenciální mediátory supresivního signálu z kostní dřeně jsou u β-thalasemie považovány GDF15 [45] a TWSG1 [46], jejich podíl na fyziologické regulaci hepcidinu zatím nebyl prokázán [47].
Hypoxie potlačuje produkci hepcidinu
Hypoxie snižuje hladinu hepcidinu, ale mechanismus účinku dosud není přesně známý. Ve studii, ve které byli dobrovolníci vystaveni vysoké nadmořské výšce, došlo k rychlému poklesu hladiny hepcidinu, a to před ostatními biochemickými změnami [48]. Regulační osa pravděpodobně zahrnuje HIF-1α (hypoxií indukovaný faktor 1 alfa) transkripční faktor, který je syntetizován játry. Snížení hladiny hepcidinu v důsledku hypoxie je výsledkem jak nepřímé [49], tak i přímé suprese exprese hepcidinu [50]. Podíl samotného erytropoetinu, produkovaného při hypoxii, na regulaci hepcidinu je nepřímý a vyžaduje aktivní erytropoézu ve dřeni. Již zmínění pacienti s DBA a erytroidní aplazií mají vysokou hladinu hepcidinu i při abnormálně vysokých koncentracích erytropoetinu v plazmě [44].
Infekce a zánět zvyšují produkci hepcidinu
Produkce zánětlivých mediátorů při infekcích a systémových zánětlivých onemocněních rovněž ovlivňuje syntézu hepcidinu na úrovni transkripce. IL-6 (interleukin 6), působící přes STAT-3 (signal transducer and activator of transcription 3) signální dráhu, je hlavním cytokinem ovlivňujícím produkci hepcidinu [19]. STAT-3 obsahuje vazebnou část, která aktivuje promotor genu pro hepcidin [51]. U dobrovolníků bylo po aplikaci IL-6 sledováno několikanásobné zvýšení vylučované hladiny hepcidinu během 2 hodin po podání [19]. V in vitro podmínkách byly objeveny další cytokiny (např. IL-1, TNF-α), které mají vliv na produkci hepcidinu, jejich účinky u člověka však nebyly dosud potvrzeny [52, 53]. Tento mechanismus působí jako obrana proti infekci. Snížením dostupnosti železa je narušena látková výměna mikroorganismu a snižuje se jeho schopnost dělení. Význam této nespecifické obrany organismu proti patogenním mikroorganismům je předmětem dalšího studia. Důsledkem zvýšené hladiny hepcidinu a snížené hladiny železa v krevní cirkulaci je i jeho snížená dostupnost pro erytropoézu (obr. 2). Při dlouhodobém působení zánětlivé aktivity nebo třeba u septických stavů se proto rozvíjí tzv. anemie chronických chorob.
Produkce hepcidinu ovlivněná množstvím železa uloženého v zásobách
Jak jsme detailně popsali, produkce hepcidinu v hepatocytech je ovlivněna hladinou železa v krevním oběhu. Předpokládá se však, že svůj podíl na expresi hepcidinu má i množství železa uložené v zásobní formě. Přesný mechanismus však nebyl doposud plně objasněn. Určitou roli v tomto procesu mohou hrát Fe-dependentní ligázy, prolyl hydroxylázy, hypoxie a BMP6 [54].
Závěr
Objev hepcidinu a feroportinu a jejich vzájemného propojení vedl k hlubšímu porozumění metabolismu železa u člověka. Byla upřesněna molekulární podstata onemocnění, jako je hereditární hemochromatóza, anemie chronických chorob [55], či IRIDA a objasněná příčina přetížení železem u anemií s neefektivní erytropoézou (obr. 2).
V poslední době se objevují studie popisující další patologické stavy asociované s narušením produkce nebo funkce hepcidinu a feroportinu [21]. Některá chronická zánětlivá onemocnění (např. nespecifické střevní záněty) spojená se zvýšenou hladinou cytokinů ovlivňují produkci hepcidinu, což vede k narušení metabolismu železa [56]. Souhrnný přehled onemocnění s abnormální hladinou hepcidinu, která se podílí na patofyziologii dané nemoci, je uveden v tabulce 2.
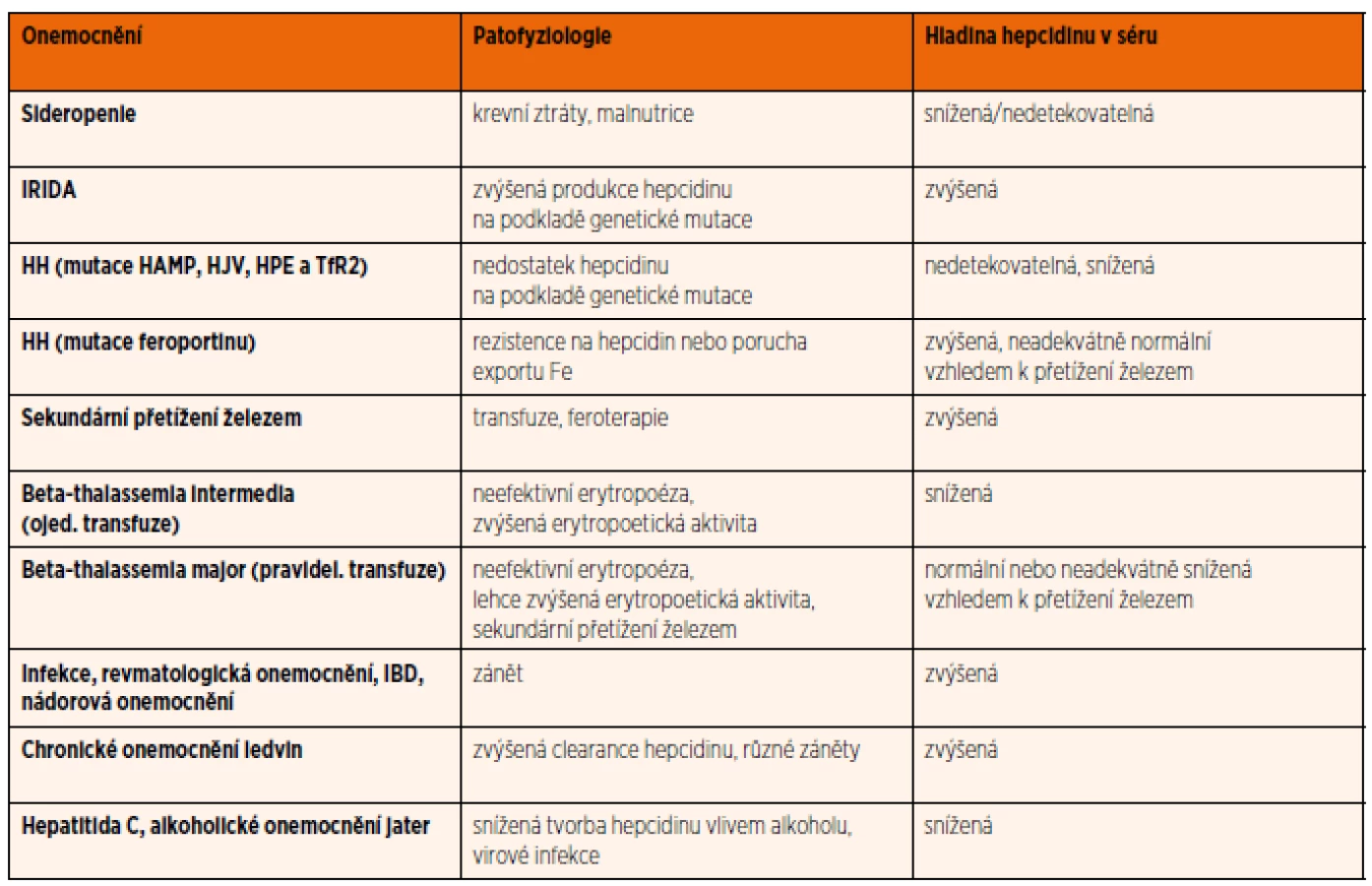
Hladina hepcidinu se zatím stanovuje pouze experimentálně imunochemickými a proteomickými metodami. Na mezinárodní úrovni se stále pracuje na standardizaci měření hladin hepcidinu [57]. Stanovení hladin hepcidinu by mohlo pomoci při nastavení léčby anemií železem, erytropoetinem, a to především u anemií chronických chorob doprovázejících různá onemocnění nebo naopak nutnosti chelatace u anemií se sekundárním přetížením železem. Výpočet indexu hepcidin/feritin, který určuje, zdali produkce hepcidinu odpovídá množství železa, by mohl být podobně diagnosticky přínosný jako výpočet indexu sTfR/log feritinu v diferenciálně diagnostické rozvaze právě u anemie chronických chorob, ale i u sideropenické anemie [58].
Znalost regulace metabolismu železa rovněž umožní vývoj nových léčebných postupů a léků pro stavy spojené s přetížením železa nebo stavy s nedostatkem železa v organismu. Byly vyvinuty látky působící jako agonisté hepcidinu [59] i látky stimulující produkci hepcidinu [60], které se mohou uplatnit v léčbě nebo prevenci akumulace železa u hemochromatózy a β-thalasemie. Schmidt a kol. [28] prokázali účinek inhibice produkce matriptázy-2 (pomocí siRNA proti TMPRSS6) na zvýšení tvorby hepcidinu u myšího modelu s β-thalassemia intermedia. U těchto myší došlo po podávání látky ke snížení jaterního přetížení železem a ke zvýšení erytropoézy. Naopak antagonistů hepcidinu [61] či inhibitorů produkce hepcidinu [62–64] by bylo možno léčebně využít u pacientů s anemií chronických chorob nebo IRIDA. U těchto onemocnění by byla vhodná kombinace s látkami stimulujícími krvetvorbu.
I navzdory obrovskému pokroku v pochopení fungování regulace metabolismu železa, který přispěl ke zlepšení diferenciální diagnostiky nemocí asociovaných s tímto procesem a k rozvoji nových terapeutických přístupů, mnoho aspektů regulace homeostázy železa zůstává nejasných. Z klinického hlediska bude především významná identifikace erytroidních signálů suprimujících hepcidin i zhodnocení možnosti a bezpečnosti léčby patologických stavů s narušenou homeostázou železa látkami ovlivňujícími produkci anebo funkci hepcidinu.
Tento článek byl podpořen grantem Ministerstva zdravotnictví ČR IGA NT/13587 a Interním grantem Univerzity Palackého LF_2014_011.
Došlo: 7. 5. 2014
Přijato: 9. 6. 2014
MUDr. Jiří Houda
Dětská klinika FN Olomouc
Puškinova 6
779 00 Olomouc
e-mail: jiri.houda@fnol.cz
Sources
1. Shayeghi M, Latunde-Dada G, Oakhill J, et al. Identification of an intestinal heme transporter. Cell 2005; 122 (5): 789–801.
2. Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006; 127 (5): 917–928.
3. Gunshin H, Mackenzie B, Berger U, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997; 388 (6641): 482–488.
4. Donovan A, Lima C, Pinkus J, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1 (3): 191–200.
5. Vulpe C, Kuo Y, Murphy T, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21 (2): 195–199.
6. Gunshin H, Fujiwara Y, Custodio A, et al. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest 2005; 115 (5): 1258–1266.
7. Krause A, Neitz S, Mägert H, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 2000; 480 (2–3): 147–150.
8. Park C, Valore E, Waring A, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276 (11): 7806–7810.
9. Bekri S, Gual P, Anty R, et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology 2006; 131 (3): 788–796.
10. Kulaksiz H, Fein E, Redecker P, et al. Pancreatic beta-cells express hepcidin, an iron-uptake regulatory peptide. J Endocrinol 2008; 197 (2): 241–249.
11. Hunter H, Fulton D, Ganz T, et al. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem 2002; 277 (40): 37597–37603.
12. Wallace D, Jones M, Pedersen P, et al. Purification and partial characterisation of recombinant human hepcidin. Biochimie 2006; 88 (1): 31–37.
13. Kemna E, Tjalsma H, Podust V, et al. Mass spectrometry-based hepcidin measurements in serum and urine: analytical aspects and clinical implications. Clin Chem 2007; 53 (4): 620–628.
14. Nagy J, Lakner L, Poór V, et al. Serum prohepcidin levels in chronic inflammatory bowel diseases. J Crohns Colitis 2010; 4 (6): 649–653.
15. Hilton K, Lambert L. Molecular evolution and characterization of hepcidin gene products in vertebrates. Gene 2008; 415 (1–2): 40–48.
16. Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276 (11): 7811–7819.
17. Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 2001; 98 (15): 8780–8785.
18. Zhang A-S, Enns C. Iron homeostasis: recently identified proteins provide insight into novel control mechanisms. J Biol Chem 2009; 284 (2): 711–715.
19. Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113 (9): 1271–1276.
20. Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med 2011; 62 : 347–360.
21. Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis 2004; 32 (1): 131–138.
22. Babitt J, Huang F, Wrighting D, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006; 38 (5): 531–539.
23. Xia Y, Babitt J, Sidis Y, et al. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 2008; 111 (10): 5195–5204.
24. Steinbicker A, Sachidanandan C, Vonner A, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011; 117 (18): 4915–4923.
25. Andriopoulos B, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009; 41 (4): 482–487.
26. Roy C, Enns C. Iron homeostasis: new tales from the crypt. Blood 2000; 96 (13): 4020–4027.
27. Gao J, Chen J, Kramer M, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009; 9 (3): 217–227.
28. Schmidt P, Toran P, Giannetti A, et al. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 2008; 7 (3): 205–214.
29. Goswami T, Andrews N. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem 2006; 281 (39): 28494––28498.
30. Ramey G, Deschemin J-C, Vaulont S. Cross-talk between the mitogen activated protein kinase and bone morphogenetic protein/hemojuvelin pathways is required for the induction of hepcidin by holotransferrin in primary mouse hepatocytes. Haematologica 2009; 94 (6): 765–772.
31. Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab 2008; 8 (6): 502–511.
32. Lee D-H, Zhou L-J, Zhou Z, et al. Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood 2010; 115 (15): 3136–3145.
33. Cau M, Melis M, Congiu R, et al. Iron-deficiency anemia secondary to mutations in genes controlling hepcidin. Expert Rev Hematol 2010; 3 (2): 205–216.
34. Nai A, Pagani A, Silvestri L, et al. Increased susceptibility to iron deficiency of Tmprss6-haploinsufficient mice. Blood 2010; 116 (5): 851–852.
35. Yilmaz-Keskin E, Sal E, De Falco L, et al. Is the acronym IRIDA acceptable for slow responders to iron in the presence of TMPRSS6 mutations? Turk J Pediatr 2013; 55 (5): 479–484.
36. Zhang S-P, Wang Z, Wang L-X, et al. AG490: an inhibitor of hepcidin expression in vivo. World J Gastroenterol WJG 2011; 17 (45): 5032–5034.
37. Lakhal S, Schödel J, Townsend A, et al. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. The Journal of biological chemistry 2011; 286 (6): 4090–4097.
38. Maurer E, Gütschow M, Stirnberg M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol Chem 2012; 393 (6): 535–540.
39. Meynard D, Vaja V, Sun C, et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011; 118 (3): 747–756.
40. Bartnikas T, Andrews N, Fleming M. Transferrin is a major determinant of hepcidin expression in hypo-transferrinemic mice. Blood 2011; 117 (2): 630–637.
41. Pak M, Lopez M, Gabayan V, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006; 108 (12): 3730–3735.
42. Adamsky K, Weizer O, Amariglio N, et al. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol 2004; 124 (1): 123–124.
43. Papanikolaou G, Tzilianos M, Christakis J, et al. Hepcidin in iron overload disorders. Blood 2005; 105 (10): 4103–4105.
44. Pospisilova D, Holub D, Zidova Z, et al. Hepcidin levels in Diamond-Blackfan anemia reflect erythropoietic activity and transfusion dependence. Haematologica 2014.
45. Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007; 13 (9): 1096–1101.
46. Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009; 114 (1): 181–186.
47. Tanno T, Rabel A, Lee Y, et al. Expression of growth differentiation factor 15 is not elevated in individuals with iron deficiency secondary to volunteer blood donation. Transfusion (Paris) 2010; 50 (7): 1532–1535.
48. Talbot N, Lakhal S, Smith T, et al. Regulation of hepcidin expression at high altitude. Blood 2012; 119 (3): 857–860.
49. Smith T, Robbins P, Ratcliffe P. The human side of hypoxia-inducible factor. Br J Haematol 2008; 141 (3): 325–334.
50. Gordeuk V, Miasnikova G, Sergueeva A, et al. Chuvash polycythemia VHLR200W mutation is associated with down-regulation of hepcidin expression. Blood 2011; 118 (19): 5278–5282.
51. Pietrangelo A, Dierssen U, Valli L, et al. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology 2007; 132 (1): 294–300.
52. Inamura J, Ikuta K, Jimbo J, et al. Upregulation of hepcidin by interleukin-1beta in human hepatoma cell lines. Hepatology research: Official Journal of the Japan Society of Hepatology 2005; 33 (3): 198–205.
53. Lee P, Peng H, Gelbart T, et al. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci USA 2005; 102 (6): 1906–1910.
54. Salahudeen A, Bruick R. Maintaining Mammalian iron and oxygen homeostasis: sensors, regulation, and cross-talk. Ann NY Acad Sci 2009; 1177 : 30–38.
55. Goodnough L, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood 2010; 116 (23): 4754–4761.
56. Oustamanolakis P, Koutroubakis I, Messaritakis I, et al. Serum hepcidin and prohepcidin concentrations in inflammatory bowel disease. Eur J Gastroenterol Hepatol 2011; 23 (3): 262–268.
57. Kroot J, Kemna E, Bansal S, et al. Results of the first international round robin for the quantification of urinary and plasma hepcidin assays: need for standardization. Haematologica 2009; 94 (12): 1748–1752.
58. Šuláková A, Pozler O, Nováčková L. Prevalence a typ anémie v době stanovení diagnózy nespecifického střevního zánětu u dětí. Čes-slov Pediat 2012; 67 (1): 3–10.
59. Preza G, Ruchala P, Pinon R, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest 2011; 121 (12): 4880–4888.
60. Corradini E, Schmidt P, Meynard D, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010; 139 (5): 1721–1729.
61. Sasu B, Cooke K, Arvedson T, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010; 115 (17): 3616–3624.
62. Hashizume M, Uchiyama Y, Horai N, et al. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int 2010; 30 (7): 917–923.
63. Yu P, Hong C, Sachidanandan C, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 2008; 4 (1): 33–41.
64. Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011; 117 (3): 997–1004.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2014 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Beta-blockers in the treatment of hemangiomas in childhood
- Ethical concerns involving the newborns at risk
- The role of hepcidin in iron metabolism
- The importance of classical cytogenetic examination in clinical paediatric practice: analysis of 384 indications