
Neonatální hemochromatóza asociovaná s renální tubulární dysgenezí
Neonatal hemochromatosis associated with renal tubular dysgenesis
Neonatal hemochromatosis (NH) is a clinical syndrome consisting of severe liver disease accompanied by pathologic siderosis in various extrahepatic tissues. Gestational alloimmune liver disease (GALD) has been established as the cause of fetal liver injury resulting in nearly all cases of NH. The presenting findings of NH are fetal demise during late 2nd and 3rd trimester or, after delivery, signs of liver failure and typically multiple organ dysfunction syndrome with high mortality. Renal tubular dysgenesis (RTD) represents a developmental disorder of fetal kidneys differentiation as a consequence of angiotensinogen deficiency because of evolving fetal liver injury. Oligohydramnion, congenital oligoanuria and refractory hypotension are the leading symptoms of RTD. The coincidence between RTD and NH is well known as the most common renal pathology seen in NH. GALD can be prevented by repetitive intravenous immunoglobulin application during the next pregnancy, starting usually between 14th to 16th postmenstrual weeks.
The authors present a case of GALD manifested as severe neonatal hemochromatosis and renal tubular dysgenesis followed by successful treatment of the mother in the next pregnancy resulting in delivery of the healthy offspring.
Key words:
neonatal hemochromatosis, liver failure, extrahepatic siderosis, gestational alloimmune liver disease, renal tubular dysgenesis, anuria, hypotension, intravenous immunoglobulin
:
M. Navratilova 1; E. Šimáková 2; M. Podholová 3; P. Eliáš 4; P. Dědek 1; Z. Kokštein 1; J. Malý 1
:
Dětská klinika FN a LF UK, Hradec Královépřednosta prof. MUDr. M. Bayer, CSc.
1; Fingerlandův ústav patologie FN a LF UK, Hradec Královépřednosta prof. MUDr. A. Ryška, Ph. D.
2; Porodnická a gynekologická klinika FN a LF UK, Hradec Královépřednosta doc. MUDr. J. Špaček, Ph. D.
3; Radiologická klinika FN a LF UK, Hradec Královépřednosta prof. MUDr. A. Krajina, CSc.
4
:
Čes-slov Pediat 2014; 69 (6): 342-349.
:
Case Report
Neonatální hemochromatóza (NH) je klinickým syndromem, jehož součástí je onemocnění jater plodu (resp. novorozence) a patologické přetížení extrahepatálních tkání železem. Příčinou neonatální hemochromatózy je v drtivé většině případů gestační aloimunní onemocnění jater (GALD). V plně rozvinuté formě se NH manifestuje úmrtím plodu koncem druhého nebo v průběhu třetího trimestru těhotenství. Při porodu novorozence s NH jsou typickými projevy známky nitroděložní růstové restrikce a časně nastupující šok s hypoglykemií, koagulopatií, oligurií, edémy a vysokou mortalitou. Renální tubulární dysgeneze (RTD) je porucha vývoje fetálních ledvin vznikající v důsledku nedostatečné produkce angiotenzinogenu v játrech plodu. U všech pacientů s GALD se objevuje snížení počtu proximálních tubulů a jaterní exprese angiotenzinogenu inverzně koreluje s jejich denzitou. Klinickým důsledkem je různý stupeň oligohydramnia, vrozená oligoanurie a refrakterní hypotenze. Rozvoji GALD v následujícím těhotenství lze předcházet opakovanou aplikací intravenózních imunoglobulinů těhotné od 14.–16. týdne těhotenství.
Autoři prezentují případ GALD s klinickým obrazem neonatální hemochromatózy a renální tubulární dysgeneze a následnou úspěšnou léčbu matky v další graviditě a porod zdravého sourozence.
Klíčová slova:
neonatální hemochromatóza, jaterní selhání, extrahepatální sideróza, gestační aloimunní jaterní onemocnění, renální tubulární dysgeneze, anurie, hypotenze, intravenózní imunoglobuliny
Úvod
Neonatální hemochromatóza (NH) je vzácným klinickým syndromem, patřícím na čelní místo diferenciální diagnostiky závažných hepatopatií novorozeneckého období. Jedná se o soubor příznaků rozvíjejících se ve druhém trimestru gravidity v důsledku postižení jater plodu a extrahepatální siderózy, která je typickým nálezem. Ač původně byla NH popsána jako pravděpodobná porucha metabolismu železa, poměrně záhy bylo z klinických pozorování patrné, že fenotypické projevy jsou manifestací těžkého jaterního postižení plodu. Zásadní zlom v pochopení patofyziologie stavu přinesl poznatek, že téměř všechny případy NH vznikají jako následek gestační aloimunní jaterní nemoci (GALD). Tento fakt vedl ke změně názvosloví a z neonatální hemochromatózy se stal symptom, který je nejčastějším následkem fetálního poškození jater v důsledku GALD. V literatuře se nicméně nadále setkáváme s oběma termíny [1, 2, 3, 14]. GALD vzniká jako důsledek mateřské senzitizace a produkce transplacentárně pronikajících protilátek IgG3 proti dosud neidentifikovanému povrchovému antigenu hepatocytu plodu s navazující aktivací komplementu a formací komplexu atakujícího membránu hepatocytu. K aloimunní teorii jaterního poškození dále přispívá fakt, že gravidita ženy s anamnézou porodu novorozence postiženého GALD je zatížena až 90% rizikem opakování této nemoci u dalšího potomka. Zároveň byl jednoznačně prokázán protektivní efekt podávání intravenózních imunoglobulinů (IVIG) těhotným s potenciálním rizikem rozvoje NH plodu [4]. Sideróza jater a ostatních tkání je výsledkem poruchy regulace transplacentárního transportu železa řízeného selhávajícími játry a jeho pokračujícím ukládáním do mimojaterních tkání [1, 14]. Kromě GALD může k NH (tzv. non-GALD NH) vést deficit mitochondriální deoxyguanozin kinázy nebo porucha syntézy žlučových kyselin (deficience delta 4-3-oxosteroid 5 beta-reduktázy) [5, 15].
Nejčastější (a nejméně často rozpoznanou) manifestací NH je nitroděložní úmrtí plodu koncem druhého nebo v průběhu třetího trimestru v důsledku intrauterinního jaterního selhání. Většina živě narozených dětí přichází na svět předčasně a má známky nitroděložní růstové restrikce. Jaterní postižení se projeví obvykle krátce po narození, nicméně existuje malá část novorozenců, která může mít jen diskrétní projevy či dokonce být zcela bez klinických symptomů. V rozvinuté (a nejčastější) formě NH připomíná septický šok s rychle nastupujícími edémy a oligurií. Koagulopatie, hypofibrinogenémie, hypoglykemie, hypoalbuminémie, trombocytopenie a anemie jsou hlavními laboratorními odchylkami [1, 3]. Žloutenka s elevací konjugovaného i nekonjugovaného bilirubinu se vyvíjí s odstupem několika dní. Hladina sérových aminotransferáz neodráží závažnost jaterního inzultu (norma – mírné zvýšení), typickým nálezem bývá vysoká hladina alfa-fetoproteinu (AFP). Laboratorní vyšetření metabolismu železa vykazují hypersaturaci transferinu, hypotransferinémii a hyperferritinémii. Nízká hladina sérového transferinu vypovídá o závažnosti jaterního poškození [1]. U NH bývá prokazována vyšší hladina sérového tyrosinu, která by mohla vést k podezření na tyrosinémii, ve skutečnosti ale odráží nedostatečnou metabolickou funkci jater [1, 6].
Neonatální hemochromatóza může být provázena poruchou vývoje proximálních tubulů, tzv. renální tubulární dysgenezí (RTD). Jedná se o vzácnou poruchu diferenciace fetálních ledvin spojenou s vrozenou oligoanurií, refrakterní hypotenzí, oligohydramniem, event. i hypoplazií plic [7, 14]. Samostatně se vyskytující RTD je autozomálně recesivní porucha s mutacemi v genech kódujících složky renin-angiotenzinového systému [8]. V rámci NH vzniká sekundárně v důsledku těžkého jaterního poškození a je vysvětlena chyběním vlivu jaterního angiotenzinogenu na nitroděložní vývoj tubulů a navazujících glomerulů [7]. U všech pacientů s GALD se objevuje určité snížení počtu proximálních tubulů a periferních glomerulů a jaterní exprese angiotenzinogenu inverzně koreluje s jejich následnou denzitou. Porucha vývoje ledvin spadá do druhé poloviny druhého trimestru těhotenství (období kolem 24. gestačního týdne), a proto, v závislosti na době vzniku, může či nemusí dojít k rozvoji plicní hypoplazie. V novější literatuře se setkáváme s označením renální dysgeneze asociovaná s deficitem angiotenzinogenu [14].
Kazuistika
Na jednotku intenzivní a resuscitační péče (JIRP) pro novorozence Dětské kliniky FN a LF UK Hradec Králové byl v březnu 2011 přijat chlapec z první gravidity ženy léčené pro inzulin dependentní diabetes mellitus, který se narodil ve 30. gestačním týdnu (GT) s porodní hmotností 1200 g. V rámci II. ultrazvukového screeningu ve 30. GT byly odhaleny oligohydramnion a růstová restrikce plodu (IUGR), avšak s normálními poměry v placentární a fetální cirkulaci. Pro tyto nálezy byla žena hospitalizována, nicméně ještě týž den pro známky hrozící hypoxie plodu (dle kardiotokografického záznamu) bylo těhotenství ukončeno akutním císařským řezem, při kterém odteklo malé množství čiré plodové vody. Byl vybaven vitální novorozenec, skóre dle Apgarové bylo hodnoceno 8, 8, 9 body. Pro známky dechové tísně byl chlapec překládán na JIRP pro novorozence s distenční dechovou podporou aplikací kontinuálního přetlaku v dýchacích cestách (NCPAP), bez potřeby oxygenoterapie. Při přijetí na JIRP byl novorozenec čilý, bledý, dominoval fenotyp IUGR. Překvapujícím nálezem byla hluboká hypotenze kontrastující s dobrým klinickým stavem, invazivní střední arteriální tlak se pohyboval v rozmezí 12–15 mmHg. Volumoterapie krystaloidy nevedla ke zlepšení, proto byl dále ordinován dopamin a současně byl zaveden centrální žilní katétr cestou vena umbilicalis. Při její kanylaci se objevilo velmi obtížně stavitelné krvácení z pupečníkových cév, laboratorně byla zjištěna anemie a trombocytopenie (tab. 1). Dostupný výsledek v kontextu s klinickým stavem vedl k pracovní diagnóze diseminované intravaskulární koagulopatie při šoku neznámé etiologie a byla zahájena substituce krevních derivátů (antitrombin III, antihemofilní plazma, erymasa, trombonáplav) s následným laboratorním potvrzením závažné koagulopatie (tab. 1). Přes negativní zánětlivé parametry byla ordinována antibiotická léčba (ampicilin, amikacin). Pro trvající hypotenzi po adekvátní tekutinové resuscitaci oběhu a terapii dopaminem byla léčba rozšířena o noradrenalin a dobutamin, dále byl podán hydrokortizon.
Dalším patologickým příznakem byla kompletní anurie, která byla v prvních hodinách přičítána hypoperfuzi ledvin při šokovém stavu. Diuréza ale nenastoupila ani při dosažení krevního tlaku na dolní hranici normy a rychle došlo k rozvoji povšechných otoků. Po dechové stránce byl pacient v prvních 6 hodinách života stabilní, při podpoře NCPAP s fyziologickými hodnotami acidobazické rovnováhy. Poté se začaly navyšovat nároky na oxygenoterapii a progredovala tachy-dyspnoe, což bylo indikací k zahájení umělé plicní ventilace. I nadále trval závažný stav s refrakterní hypotenzí, anurií a prohlubující se metabolickou acidózou. V průběhu prvního dne života byla doplněna ultrazvuková vyšetření – UZ ledvin a močového měchýře s normálním nálezem, na UZ mozku bylo zjištěno subependymální krvácení I. stupně, echokardiografie při vysokých dávkách katecholaminů (noradrenalinu, dobutaminu, dopaminu) vyloučila vrozenou srdeční vadu, byla zjištěna dysfunkce atrioventrikulárních chlopní (trikuspidální a mitrální insuficience) s normální kontraktilitou myokardu, otevřené foramen ovale s levopravým zkratem a perzistující Botallova dučej s bidirekčními zkraty. Během 1,5 dne trvající intenzivní péče s cílem udržení stabilního vnitřního prostředí a normálního systémového krevního tlaku nedošlo k nástupu diurézy, progredovala anasarka a stav nezadržitelně směřoval do multiorgánového selhání. Kontrolní UZ mozku zobrazil intraventrikulární krvácení IV. stupně s časně nastupujícím hydrocefalem a nitrolební hypertenzí. Po diskusi s rodiči byla zadržena resuscitační péče. Chlapec zemřel ve stáří 41 hodin. Kultivační vyšetření byla sterilní.
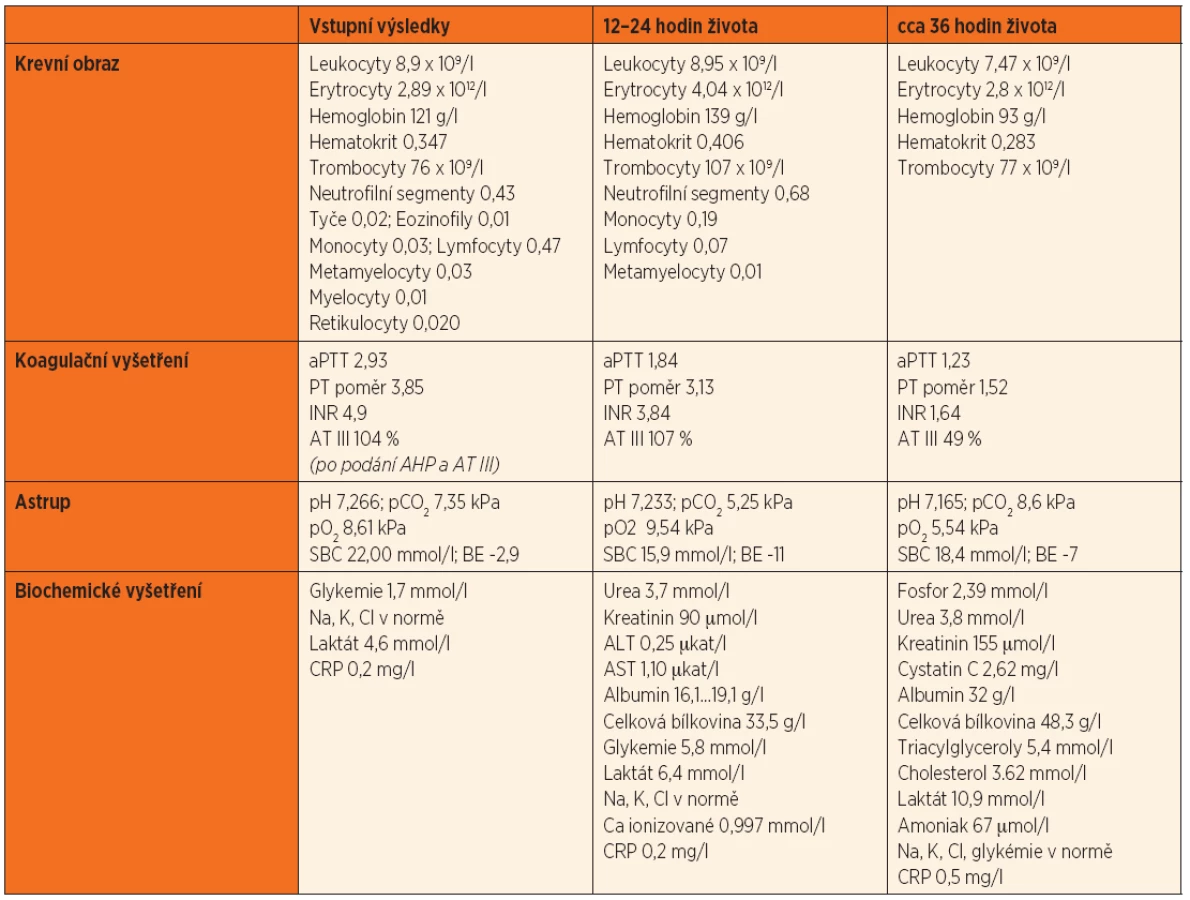
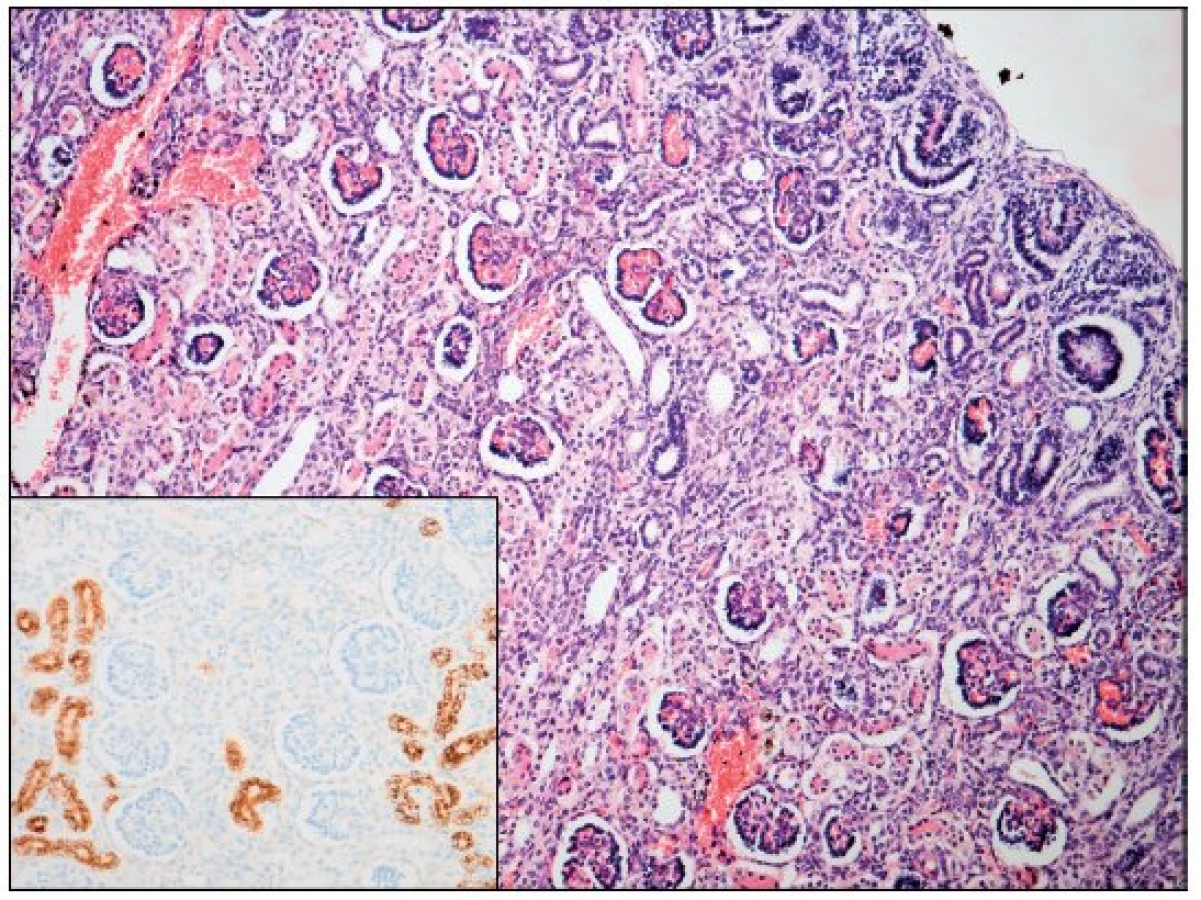
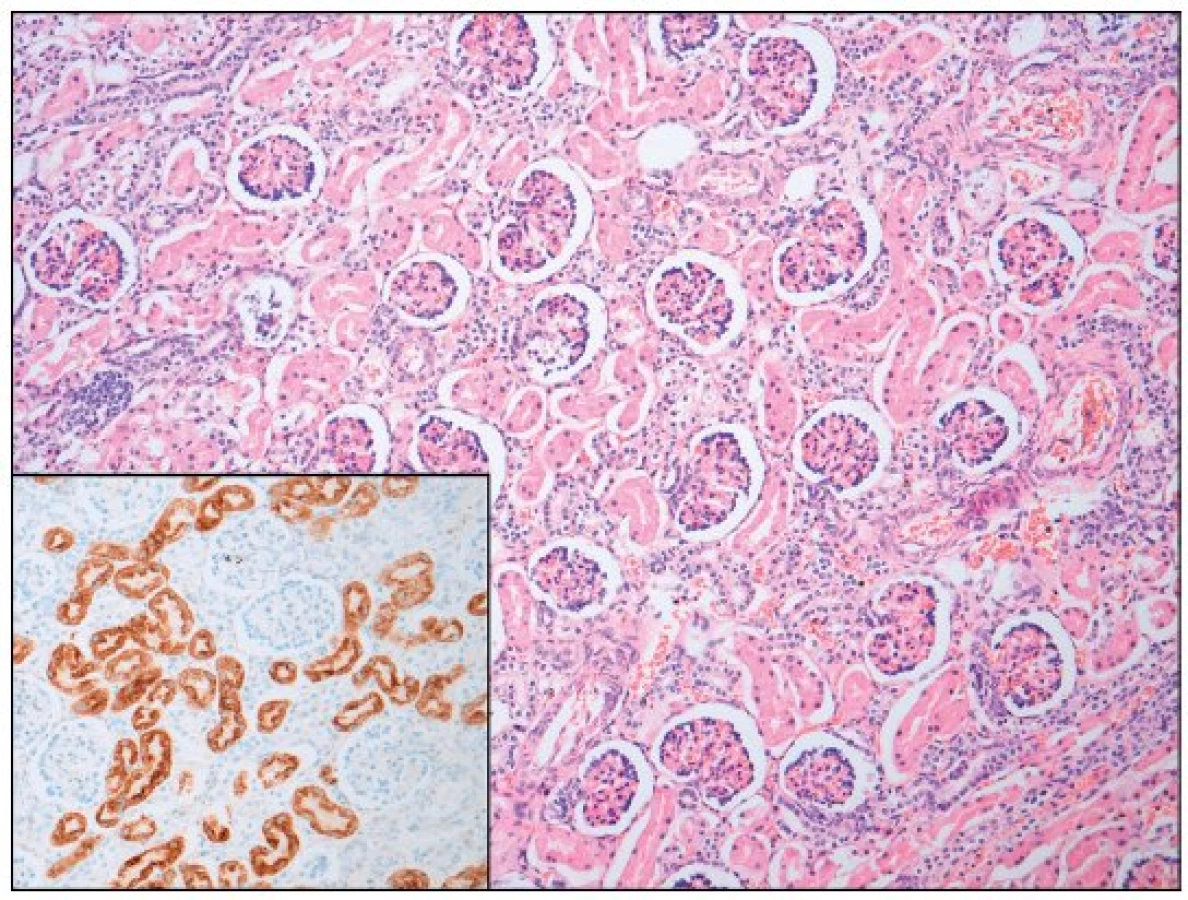
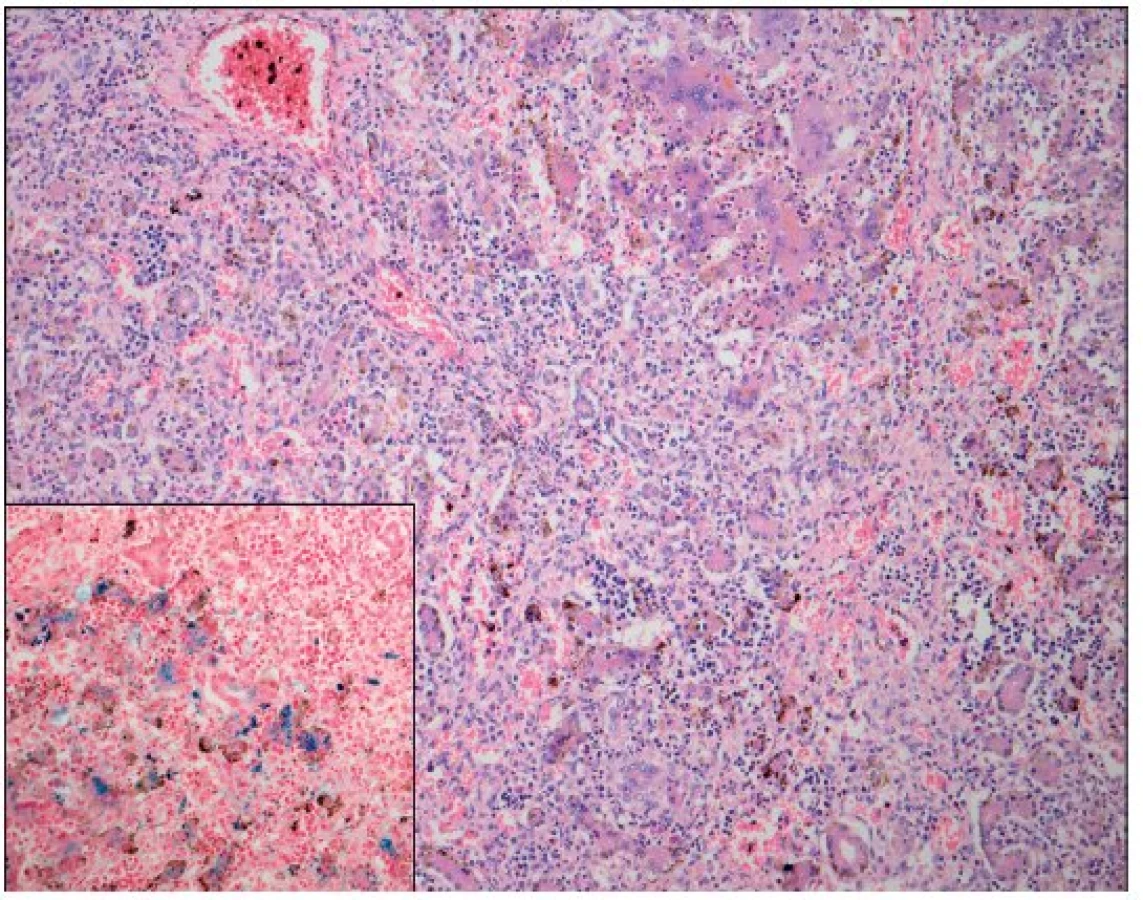
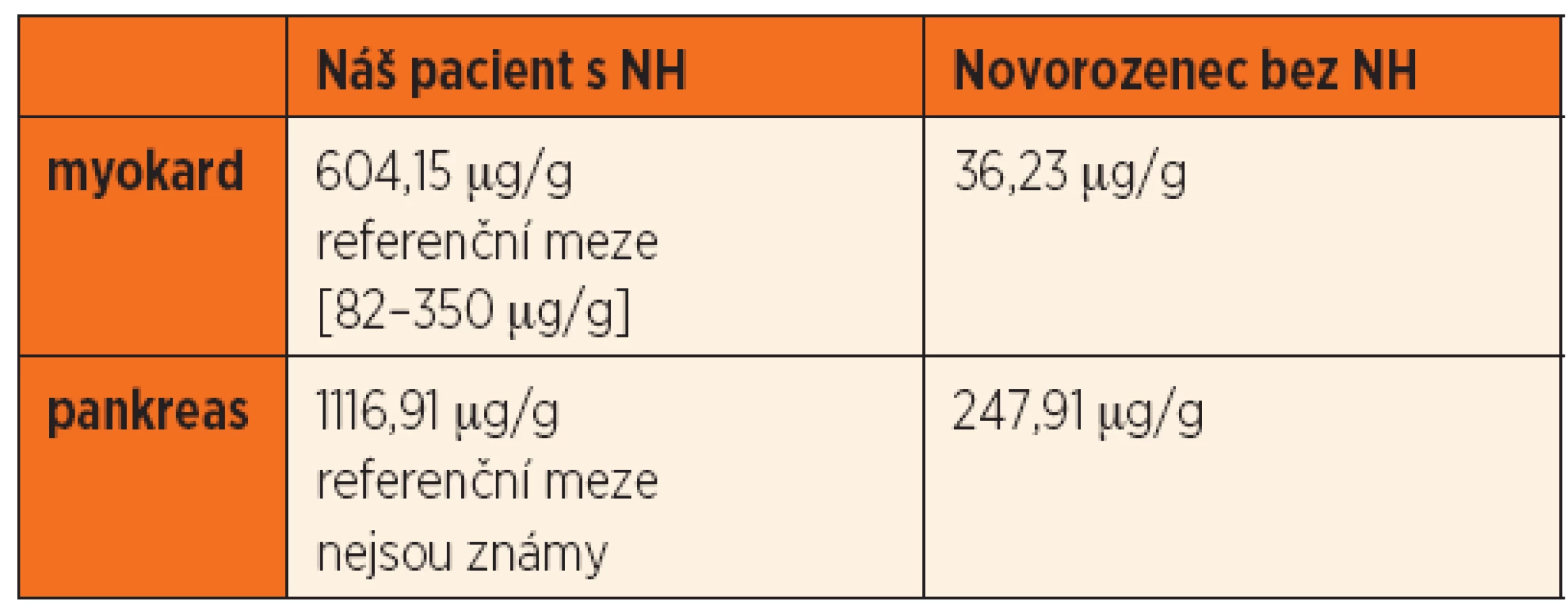
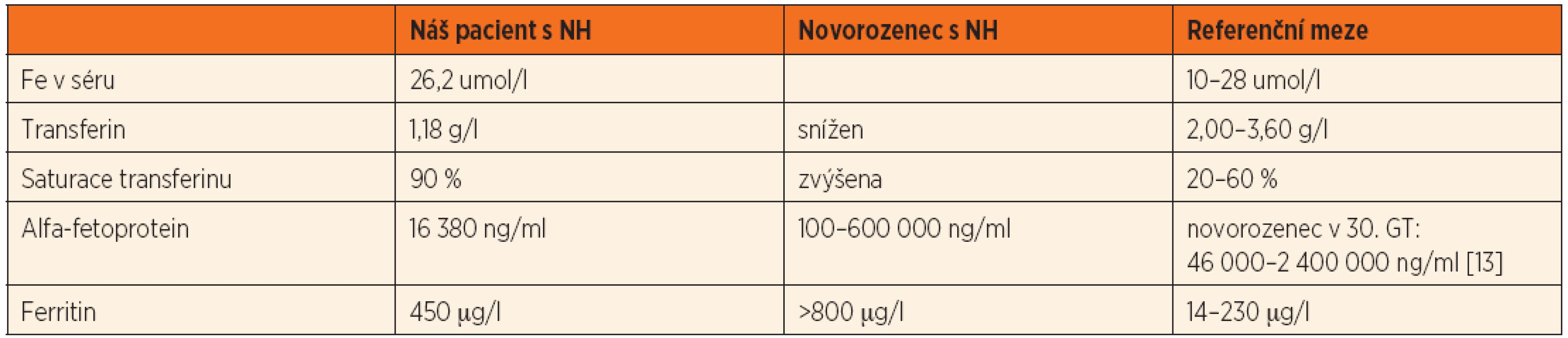
Po smrti byla provedena četná vyšetření k objasnění její příčiny. Dle klinického průběhu bylo vysloveno podezření na renální tubulární dysgenezi (RTD), která byla potvrzena imunohistochemickým vyšetřením mikroskopických preparátů ledvin (obr. 1, 2). Nález fibroticky změněných malých jater vedl v kontextu s klinickým průběhem a uvažovanou renální patologií k podezření na neonatální hemochromatózu (NH), pro kterou svědčilo histologické vyšetření jaterní tkáně (obr. 3). Druhé čtení histologických preparátů provedl pan profesor M. Elleder z Ústavu dědičných metabolických poruch UK 1. LF a VFN v Praze. Kvantitativní průkaz extrahepatální siderózy v sušině tkání myokardu a pankreatu (tab. 2) definitivně potvrdil diagnózu, kterou podpořily i laboratorní odchylky v metabolismu železa (tab. 3).
Diskuse
Stanovení diagnózy GALD s klinickou symp-tomatologií neonatální hemochromatózy spočívá v podstatě v identifikaci a diagnostice NH, neboť téměř všechny případy neonatální hemochromatózy vznikají v důsledku GALD. Diagnostika NH je u fulminantně probíhajících forem obtížná. Přestože ne všechny případy probíhají tak perakutně jako v popisované kazuistice, bývá v mnoha případech NH prokázána až z autopsie mikroskopickým vyšetřením jaterní tkáně a průkazem extrahepatální siderózy (nejčastěji v exokrinní části pankreatu, myokardu, folikulech štítné žlázy a malých slinných žlázách). Nález siderózy v játrech není pro NH diagnostický a objevuje se u řady jiných hepatopatií [1]. Rovněž je nutné zdůraznit, že nepřítomnost barvitelného železa v játrech nevylučuje NH, neboť v důsledku jaterního postižení dochází k masivnímu úbytku hepatocytů, které jediné obsahují železo v případě intrahepatální siderózy v rámci NH. Zásadní podmínkou pro histopatologickou diagnostiku je provedení cíleného barvení specifických tkání Pruskou (Berlínskou) modří, které by mělo být realizováno v případě každé pitvy novorozence zemřelého na jaterní selhání, ev. suspektní hepatopatii i u nejasných novorozeneckých úmrtí a potratů. Nenaplnění této podmínky vede k přehlédnutí řady případů NH s děsivým dopadem na další těhotenství postižených žen [14].
K podezření na NH u našeho pacienta vedlklinický průběh a sekční nález malých, fibroticky změněných jater. Mikroskopický obraz těžké dysplazie jaterní tkáně s neuspořádanými trabekulami, cholestázou, dilatací žlučovodů a mnohojadernými hepatocyty se siderózou [6] naše podezření prohloubil a vedl k doplnění dalších vyšetření. Odchylky v metabolismu železa – hypersaturace transferinu, hypotransferinémie a vysoké hladiny ferritinu diagnózu podpořily. Zásadním nálezem pro konečnou diagnózu byl průkaz železa v sušině myokardu a pankreatu. Vzhledem k absenci referenčních mezí obsahu železa v sušině tkání (vyjma myokardu a jater) byly vzorky tkání porovnány se vzorky tkání gestačně stejně starého zemřelého novorozence bez NH. Extrahepatální siderózu u žijícího novorozence je možno prokázat z bioptického vzorku (např. ze slinných žláz dutiny ústní) nebo vyšetřením magnetickou rezonancí [1].
V některých (velmi závažných) případech je NH provázena renální hypoplazií s dysgenezí proximálních tubulů a úbytkem periferních glomerulů. Ve světové literatuře je popsáno jen velmi málo takových případů [8, 9, 10], přestože GALD vede v důsledku ztráty funkční jaterní tkáně ke snížení produkce angiotenzinogenu. Ten je řídicí molekulou z hlediska normálního vývoje proximálních tubulů a navazujících glomerulů ve druhém a na začátku třetího trimestru těhotenství a v podstatě každý případ GALD je spjat s rozvojem určitého stupně renální dysgeneze asociované s deficitem angiotenzinogenu [7]. Diagnostika onemocnění je opět komplikovaná a zakládá se na průkazu sníženého počtu („paucity“) proximálních tubulů [7]. V prezentovaném případě byl nález RTD potvrzen imunohistochemickým vyšetřením ledvin a opět porovnáním histologického vzorku ledvin s gestačně stejně starým novorozencem (obr. 1, 2).
Literární data dokazují, že prognóza pacientů s NH je velmi špatná, a to přesto, že NH bývá častou indikací k transplantaci jater v prvních 3 měsících života [1,14]. Přežití dětí indikovaných k transplantaci jater z důvodu NH je jen 35% [12]. Vedle symptomatické terapie jaterního selhání je používána směs chelátů a antioxidantů (deferoxamin, selen, vitamin E, N-acetylcystein, prostaglandin E1) s přežitím mezi 10–20 % případů [1, 6, 12]. Průlom v léčebném přístupu a výsledcích znamenaly práce týmu prof. Whitingtona. Kombinace výměnné transfuze s navazující aplikací intravenózních imunoglobulinů (1 g/kg) vedla k zásadnímu zlepšení péče o děti s NH v porovnání s historickými kontrolami a ve skupině 44 dětí léčených uvedeným způsobem bylo popsáno přežití 79 % [12, 14]. Vzhledem k 90% riziku opakování NH u dalších potomků bylo extrémně důležitým zjištěním, že rozvoji GALD a NH může být zabráněno profylaktickou aplikací intravenózních imunoglobulinů v druhém a třetím trimestru gravidity ženám, jejichž předchozí gravidita byla zatížena potratem, resp. porodem dítěte s NH [4]. Při dodržení tohoto přístupu lze zabránit vzniku GALD a NH téměř ve 100 % případů s normálním vývojem a normálními jaterními funkcemi těchto dětí [4, 14].
Samotná RTD je terapeuticky velmi obtížně ovlivnitelná. Ve většině případů končí úmrtím pacienta krátce po narození. Literárně jsou popisovány ojedinělé případy přeživších dětí s RTD s několikatýdenní anurií a systémovou hypotenzí [11, 16], v jejichž léčbě byla použita dočasná peritoneální dialýza a fludrokortizon [16].
Závěr
Cílem našeho sdělení bylo upozornit na vzácná onemocnění, kterými GALD s klinickým obrazem neonatální hemochromatózy a renální tubulární dysgeneze jsou. Obě diagnózy, zvláště RTD, obvykle vedou přes veškerá léčebná úsilí k úmrtí pacienta. Průkaz NH, ač u zemřelého novorozence, se jeví jako důležitý s ohledem na další graviditu ženy, která nese až 90% riziko postižení plodu NH. Pravidelné podávání intravenózních imunoglobulinů těhotným prakticky eliminuje riziko závažného postižení plodu/novorozence v důsledku gestační aloimunní jaterní nemoci. Při současných znalostech o léčbě novorozence s NH výměnnou transfuzí a intravenózními imunoglobuliny má i žena s anamnézou porodu dítěte s NH naději na zdravého potomka bez nutnosti transplantace jater.
Poděkování
Speciální poděkování patří zesnulému prof. MUDr. Milanu Ellederovi, DrSc., za pomoc při mikroskopické diagnostice neonatální hemochromatózy.
Dodatek
V lednu 2014 se ve FN Hradec Králové narodil sourozenec zemřelého pacienta, chlapec s porodní hmotností 4170 g. Gravidita ženy byla s ohledem na anamnézu pečlivě plánována a sledována. Matce byly od 16. do 38. gestačního týdne aplikovány intravenózní imunoglobuliny v dávce 1 g/kg/týden (celkem 22x), a to s dobrou tolerancí a bez komplikací. V průběhu těhotenství byly nad rámec běžné prenatální péče prováděny doplňující ultrazvukové kontroly plodu v průběhu II. trimestru gravidity (ve 23., 26. a 30. gestačním týdnu). Dále bylo realizováno vyšetření magnetickou rezonancí ve 23. a 32. gestačním týdnu cílené na oblast jater a sleziny plodu. Výsledky všech těchto vyšetření nasvědčovaly fyziologickému vývoji plodu. Porod byl veden plánovaným císařským řezem v 39. gestačním týdnu. Chlapec se po narození dobře adaptoval, hospitalizace na oddělení fyziologických novorozenců proběhla nekomplikovaně. Laboratorní vyšetření zaměřená na funkci jater včetně hodnoty alfa-fetoproteinu a parametrů metabolismu železa nezachytila odchylku od normy, UZ jater byl rovněž s normálním nálezem. Chlapec byl propuštěn do domácí péče s plánem dispenzarizace v gastroenterologické ambulanci Dětské kliniky a ve stáří 5 týdnů byl zcela bez obtíží, plně kojen, prospívající, se zcela normální funkcí jater.
Došlo: 29. 9. 2013
Přijato: 21. 3. 2014
Korespondující autor
MUDr. Jan Malý, Ph.D.
Dětská klinika FN a LF UK
Sokolská 581
500 05 Hradec Králové
e-mail: malyj@lfhk.cuni.cz
Sources
1. Whitington PF. Neonatal hemochromatosis: A congenital alloimmune hepatitis. Semin Liver Dis 2007; 27 : 243–250.
2. Magner M, Ješina P, Klement P. Význam časné diagnostiky dědičných metabolických poruch s manifestací v novorozeneckém věku. Čes-slov Pediat 2013; 68 (1): 3–11.
3. Murray KF, Kowdley KV. Neonatal hemochromatosis. Pediatrics 2001; 108 : 960–964.
4. Whitington PF, Kelly S. Outcome of pregnancies at risk for neonatal hemochromatosis is improved by treatment with high-dose intravenous immunoglobulin. Pediatrics 2008; 121 (6): e1615–e1621.
5. Hanchard NA, Shchelochkov OA, Angshumoy R, et al. Deoxyguanosine kinase deficiency presenting as neonatal hemochromatosis. Mol Genet Metab 2011; 103 : 262–267.
6. Elleder M, Chlumská A, Hadravská Š, et al. Neonatal (perinatal) hemochromatosis. Čes-slov Patol 2001; 37 (4): 146–153.
7. Bonilla SF, Melin-Aldana H, Whitington PF. Relationship of proximal renal tubular dysgenesis and fetal liver injury in neonatal hemochromatosis. Pediatr Res 2010; 67 (2): 188–193.
8. Bale PM, Kan AE, Dorney SF. Renal proximal tubular dysgenesis associated with severe neonatal hemosiderotic liver disease. Pediatr Pathol 1994; 14 (3): 479–489.
9. Morris S, Akima S, Dahlstrom JE, et al. Renal tubular dysgenesis and neonatal hemochromatosis without pulmonary hypoplasia. Pediatr Nephrol 2004; 19 : 341–344.
10. Johal JS, Thorp JW, Oyer CE. Neonatal hemochromatosis, renal tubular dysgenesis, and hypocalvaria in a neonate. Pediatr Dev Pathol 1998; 1 : 433–437.
11. Uematsu M, Sakamoto O, Obuta T, et al. A further case of renal tubular dysgenesis surviving the neonatal period. Eur J Pediatr 2009; 168 : 207–209.
12. Rand EB, Karpen SJ, Kelly S, et al. Treatment of neonatal hemochromatosis with exchange transfusion and intravenous immunoglobulin. J Pediatr 2009; 155 : 566–571.
13. Rennie JM, Roberton NRC (eds). Textbook of Neonatology. 3rd ed. Edinburgh: Churchill Livingstone, 1999 : 1409.
14. Feldman AG, Whitington PF. Neonatal hemochromatosis. J Clin Exp Hepatol 2013; 3 : 313–320.
15. Schneider BL, Stechell KD, Whitington PF, et al. Delta 4-3-oxosteroid 5 beta-reductase deficiency causing neonatal liver failure and hemochromatosis. J Pediatr 1994; 124 (2): 234–238.
16. Schreiber R, Gubler M-C, Gribouval O, et al. Inherited renal tubular dysgenesis may not be universally fatal. Pediatr Nephrol 2010; 25 : 2531–2534.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2014 Issue 6
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Dextromethorphan in the hands of a teenager – cheap and legal ticket on a „trip“
- Neonatal hemochromatosis associated with renal tubular dysgenesis
-
Onychomadesis po onemocnění ruka-noha-ústa
(hand-foot-mouth disease) - Refeeding syndrome in childhood