
Čo sa môže skrývať za diagnózou atypickej cystickej fibrózy?
What disease can be hidden behind a diagnosis of atypical cystic fibrosis?
What disease can be hidden behind a diagnosis of atypical cystic fibrosis?
In our case report we present 11-year-old girl treated by pulmonologist for atypical cystic fibrosis with pulmonary manifestations. Since birth she has been suffering from recurrent respiratory infections, chronic wet cough and persistent accumulation of thick mucus. She has a mild obstructive ventilatory defect and bronchiectasis in the right middle lobe of the lung.
In this case report we want to point out the similarity of clinical signs of congenital diseases affecting mucociliary clearance and the importance and possibilities of differential diagnosis.
Key words:
mucociliary clearance, atypical cystic fibrosis, primary ciliary dyskinesia, high-speed video microscopy
:
L. Marušiaková 1; P. Ďurdík 1; I. Bacmaňáková 1; Z. Sňahničanová 1; L. Šofranková 1; O. Petrovičová 1; M. Kuchta 2; J. Buchanec 1
:
Klinika detí a dorastu, Univerzita Komenského v Bratislave, Jesseniova lekárska fakulta v Martine, Martin
prednosta prof. MUDr. P. Bánovčin, CSc.
1; Klinika detí a dorastu, Univerzita Pavla Jozefa Šafárika v Košiciach, Lekárska fakulta, Košice
prednostka doc. MUDr. I. Schusterová, PhD.
2
:
Čes-slov Pediat 2016; 71 (2): 80-86.
:
Case Report
V kazuistike predstavujeme 11-ročné dievča sledované pneumológom pre atypickú cystickú fibrózu s pľúcnymi prejavmi, ktoré od narodenia trápia opakované respiračné infekty, chronický vlhký kašeľ a pretrvávajúce zahlienenie. Dievča má ľahkú obštrukčnú ventilačnú poruchu a bronchiektázie stredného pľúcneho laloka vpravo.
Kazuistikou chceme poukázať na podobnosť klinických príznakov vrodených ochorení ovplyvňujúcich mukociliárny transport a dôležitosť a možnosti diferenciálnej diagnostiky.
Kľúčové slová:
mukociliárny transport, atypická cystická fibróza, primárna ciliárna dyskinézia, vysokorýchlostná videomikroskopia
ÚVOD
Cystická fibróza (CF) je jedným z najčastejších autozomálne recesívne dedičných ochorení v belošskej populácii s incidenciou cca 1 : 2000–4000 novorodencov [1]. Spôsobená je mutáciou v géne CFTR (CF transmembránový regulátor vodivosti), ktorá vedie k poruche transportu chloridov cez apikálnu plazmatickú membránu buniek [1, 2, 3]. Klasicky je CF charakterizovaná prítomnosťou respiračných príznakov (chronické bronchopulmonálne ochorenie, sinusitída, nazálna polypóza, bronchiektázie, kolonizácia typickými patogénmi), gastrointestinálnych príznakov (pankreatická insuficiencia spôsobujúca steatoreu a malnutríciu, mekóniový ileus, prolaps rekta, hepatopatia) a zároveň sú hladiny chloridov v pote zvýšené oproti normálu (nad 60 mmol/l). Ochorenie je diagnostikované už v detstve, a to na základe charakteristických klinických príznakov a/alebo pozitívnej rodinnej anamnézy a prítomnosti laboratórnych nálezov (zvýšená koncentrácia chloridov v pote a/alebo dokázaná prítomnosť 2 závažných mutácií v CFTR géne). K včasnému záchytu ochorenia prispieva aj zavedený novorodenecký skríning [4, 5, 6, 7].
Atypická CF je mierna forma CF, ktorá je často diagnostikovaná až v staršom detskom veku alebo v dospelosti. Pacienti s atypickou CF majú zvyčajne 1 závažnú mutáciu v géne CFTR a 1 zriedkavú mutáciu alebo poruchu v opakovaní trinukleotidov. Hladiny chloridov v pote sú u nich iba mierne zvýšené, hraničné (40–60 mmol/l) alebo normálne (do 40 mmol/l). Atypická CF v rôznej miere postihuje rôzne orgány. Na rozdiel od typickej formy CF môže byť prítomná dysfunkcia iba v jednom orgánovom systéme. Príznaky sú potom iba respiračné, alebo iba gastrointestinálne. Často sú oveľa miernejšie ako u typickej CF a môžu sa začať prejavovať až v dospelosti. U niektorých pacientov je diagnostikovaná len opakovaná sinusitída, nazálna polypóza, izolovaná obštrukčná azoospermia, chronická idiopatická pankreatitída alebo mierne pľúcne ochorenie (diseminované bronchiektázie). S postupujúcim vekom sa môžu príznaky rozvinúť až do obrazu typickej formy CF. Diagnóza atypickej CF je stanovená u pacientov s klinickými príznakmi z aspoň jedného orgánového systému, pričom sú hladiny chloridov v pote hraničné alebo normálne a zároveň sú u pacienta prítomné 2 mutácie v géne CFTR, z ktorých je aspoň jedna mierna. Pri hraničnej hodnote chloridov v pote a klinických príznakoch stačí potvrdenie 1 mutácie [4, 5, 6, 7].
V respiračnom systéme vedie CF k zhoršenému mukociliárnemu očisťovaniu. Porucha CFTR kanála má za následok poruchu sekrécie Cl-, tvorbu hyperviskózneho hlienu a významné zníženie hrúbky periciliárnej tekutiny, ktoré spôsobuje, že cílie sú „sploštené“ na povrchu buniek. Tým je sťažené očisťovanie hlienu z dýchacích ciest. Stagnujúci hlien a hlienové zátky upchávajú najmä malé dýchacie cesty. Vytvára sa tak vhodné prostredie na vznik opakovanej a chronickej infekcie vedúcej k postupnému ireverzibilnému poškodeniu pľúc vrátane bronchiektázií, emfyzému a fibrózy [3, 5, 8, 9].
Okrem CF existujú aj iné ochorenia, pri ktorých je porušený mukociliárny transport. Porucha môže byť vrodená alebo získaná, pričom je buď porušený ciliárny pohyb, alebo je problém vo vrstve hlienu (tab. 1) [10, 11]. Hlavnými symptómami porušeného mukociliárneho transportu sú produktívny kašeľ a dyspnoe. U pacientov s vrodenou poruchou sú typické aj recidivujúce sinopulmonálne infekcie začínajúce vo včasnom detstve a postupný vznik bronchiektázií [3, 10]. Keďže sú príznaky porušeného mukociliárneho transportu pri rôznych ochoreniach veľmi podobné, v určitých prípadoch nie je stanovenie správnej diagnózy jednoduché. V kazuistike uvádzame prehľad a závery vyšetrení realizovaných u našej pacientky v rámci diferenciálnej diagnostiky. Prípad prezentovanej kazuistiky je súčasťou riešeného projektu so súhlasom Etickej komisie JLF UK číslo EK 1572/2014.
![Charakteristika rôznych ochorení ovplyvňujúcich mukociliárny transport [10].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a5129913617b465c5b7d37b62313d292.png)
KAZUISTIKA
Predstavujeme prípad 11-ročného dievčaťa sledovaného pneumológom pre atypickú cystickú fibrózu s pľúcnymi prejavmi. Dieťa z V. gravidity, pôrod v 40. týždni tehotenstva, včasná popôrodná adaptácia bola primeraná. Po niekoľkých hodinách života však u neho došlo k zhoršeniu prekrvenia kože, vzniku difúznej cyanózy, rozvoju dyspnoe a nutnosti podpornej oxygenoterapie. Dieťa bolo výrazne zahlienené a vlhko kašľalo. RTG snímka hrudníka odhalila obraz pravostrannej pneumónie s atelektázou. Na antibiotickej a symptomatickej liečbe sa jeho klinický stav postupne upravil. V ďalšom priebehu však trpelo opakovanými respiračnými infekciami (rinitídy – serózne aj purulentné, bronchitídy, bronchopneumónie), chronickým vlhkým kašľom (najmä ráno a počas dňa, ale aj ponámahovo a mimo akútneho infektu), sťaženým dýchaním a pretrvávajúcim zahlienením.
Pre uvedené ťažkosti bola pacientka od 2 mesiacov veku sledovaná detským pneumológom s diagnózou dermorespiračný syndróm, recidivujúce infekcie dýchacích ciest (RIDC) a chronická mukopurulentná bronchitída. Zahájená bola bronchodilatačná, mukolytická a antialergická liečba. Pre pozitívny záchyt baktérií z horných dýchacích ciest (Streptococcus pneumoniae, Moraxella catarrhalis, Haemophilus influenzae, Staphylococcus aureus) bola opakovane preliečená antibiotikami. Kožné testy na inhalačné alergény boli u nej negatívne. Od predškolského veku ju trápili recidivujúce rinosinusitídy a otitídy, v dôsledku ktorých trpela prechodne obojstrannou prevodovou poruchou sluchu a opakovane podstúpila paracentézu a zavedenie ventilačných trubičiek. Pre uvedené ťažkosti bolo u pacientky realizované CT vyšetrenie prinosových dutín, ktorým bola zistená aplázia frontálnych sinusov a chronická hypertrofická pansinusitída s deštrukciou mediálnej steny oboch maxilárnych sinusov.
Pre nechutenstvo a neprospievanie bola pacientka v 6 rokoch života vyšetrená v gastroenterologickej ambulancii. Laboratórne vyšetrenia (hepatálne testy, amyláza, pankreatická elastáza, imunoglobulíny, sérológia na celiakiu) boli u nej opakovane v norme a stav bol uzavretý ako asténia a nechutenstvo ako sprievodný stav pri respiračnom ochorení. Vzhľadom na opakujúce sa infekcie dýchacích ciest, chronický kašeľ a neprospievanie mala vo veku 7 rokov vykonanú 24-hodinovú pH-metriu s pozitívnym nálezom, avšak antirefluxná liečba (inhibítory protónovej pumpy, prokinetiká, diéta, režimové opatrenia) bola bez významného klinického zlepšenia. Kontrolné 24-hodinové pH-metrie po vysadení antirefluxnej liečby boli už opakovane negatívne.
Pacientka opakovane podstúpila HRCT vyšetrenie pľúc. V 5. roku života bol u nej zistený centrilobulárny emfyzém prevažne v oblasti dolných pľúcnych lalokov, vo 8. roku atelektáza stredného laloka pľúc vpravo so vzdušnými zdeformovanými bronchami. O rok neskôr došlo k progresii uvedeného CT nálezu, prítomné boli už varikózne bronchiektázie stredného laloka vpravo.
Pre podozrenie na cystickú fibrózu (opakované bronchitídy a sinusitídy, chronický kašeľ a stavy zahlienenia, neskôr bronchiektázie) mala opakovane vyšetrený chloridový potný test (v 1. roku života, dvakrát v 5. roku a dvakrát vo 8. roku života), vždy však s negatívnym výsledkom. Vzhľadom na pretrvávanie klinických ťažkostí, aj napriek negatívnemu potnému testu, bolo u pacientky vo veku 9 rokov realizované genetické vyšetrenie na cystickú fibrózu, ktorým bola identifikovaná mutácia F508 del v jednej kópii. Následne bola sledovaná v centre pre CF ako atypická CF s pľúcnymi prejavmi, kde jej bola ordinovaná komplexná liečba (mukolytiká, bronchodilatanciá, inhalačné kortikoidy, inhalácie hypertonickými soľnými roztokmi, dychová rehabilitácia, makrolidy). V tom istom roku bol u pacientky elektrónovou mikroskopiou zistený závažný stupeň patologickej transformácie epitelu nosovej sliznice, pričom riasinky respiračného epitelu ani bazálne telieska neboli prítomné. Pre ťažký stupeň lézie riasinkového epitelu bola odoslaná za účelom ďalších vyšetrení na Kliniku detí a dorastu Jesseniovej lekárskej fakulty UK a Univerzitnej nemocnice Martin.
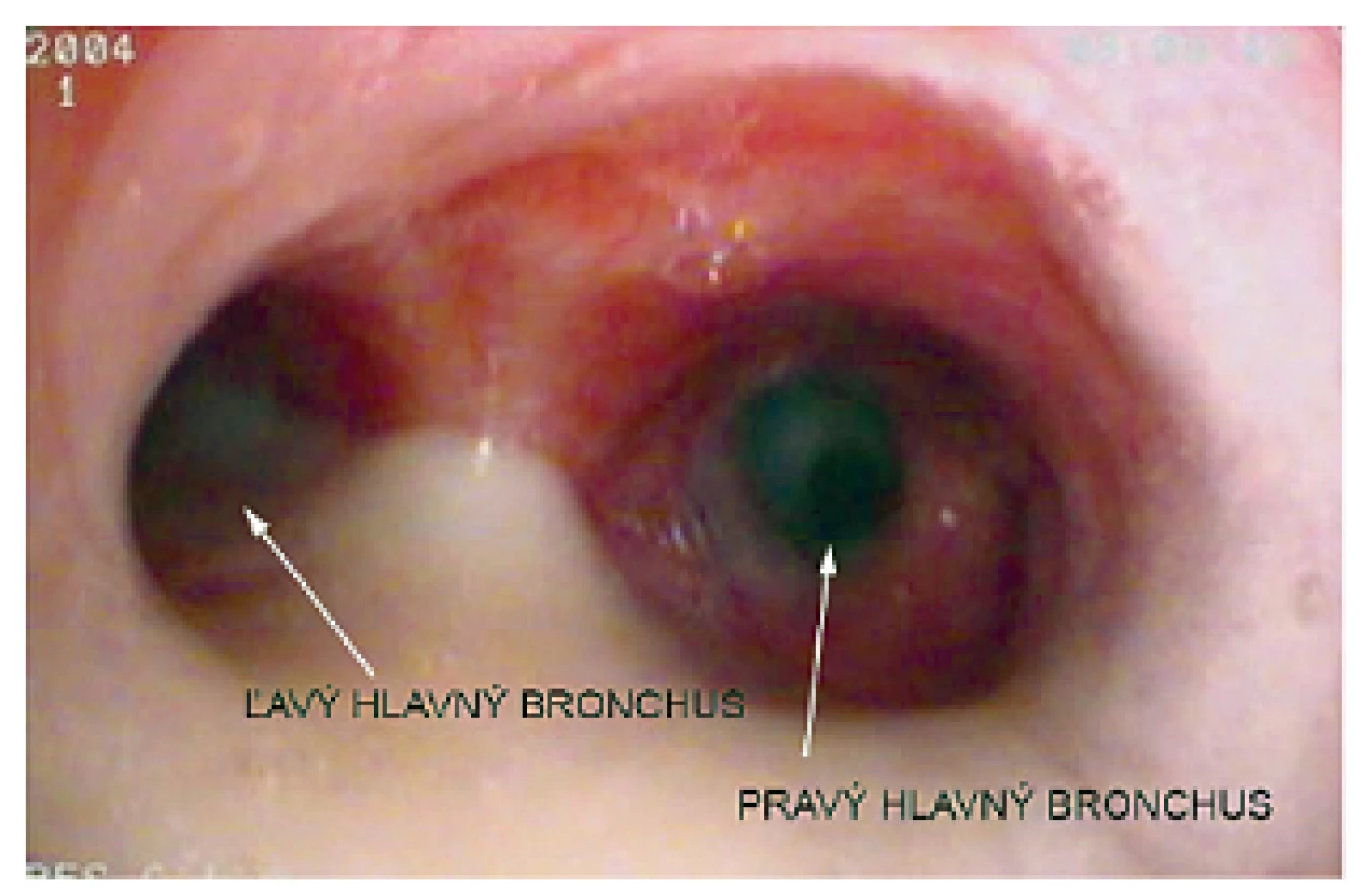
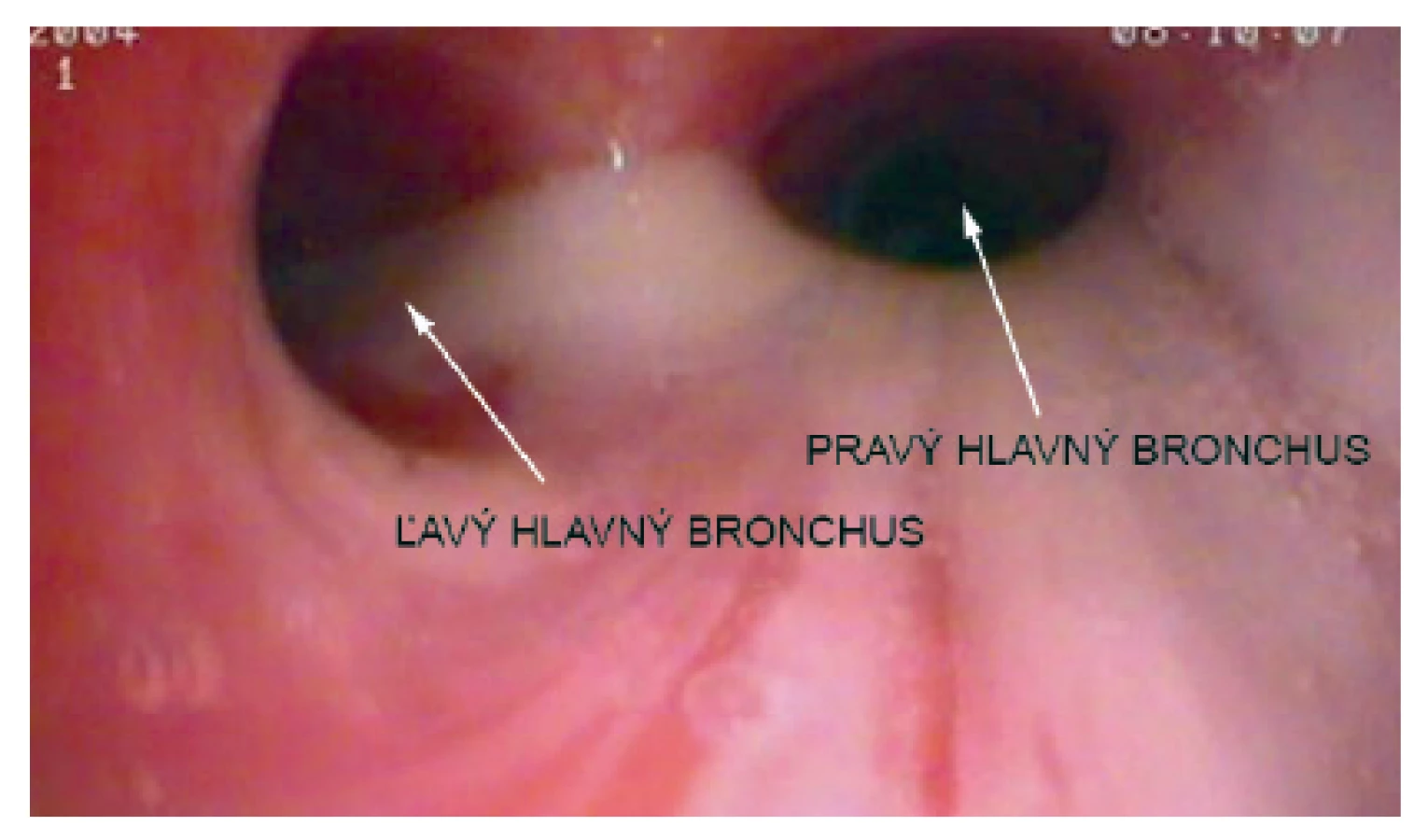
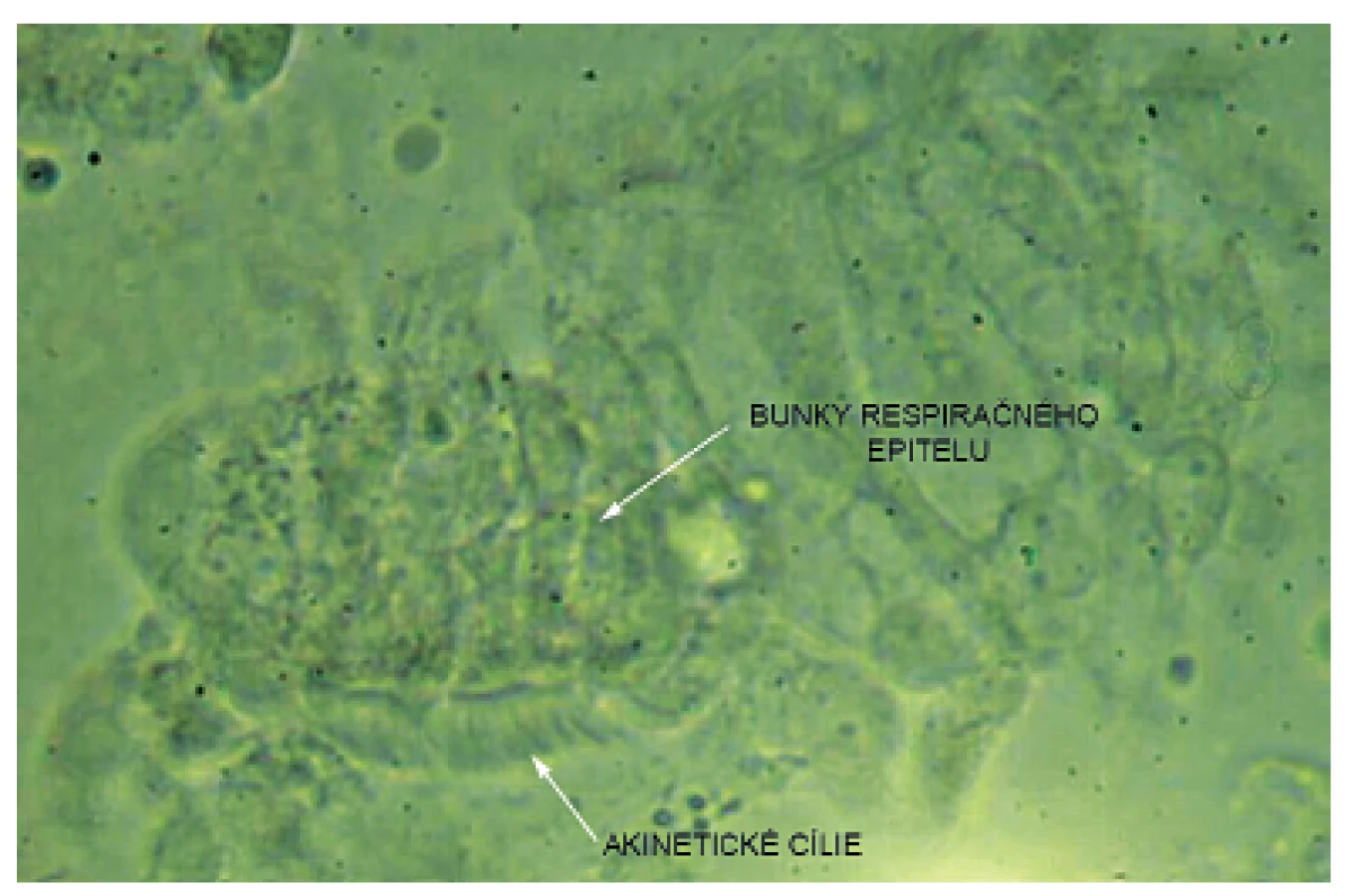
Pacientka prichádza na našu kliniku v novembri 2014 – vo veku 10 rokov. Klinicky u nej pretrvávajú stavy zahlienenia a opakované respiračné infekty (rinosinusitídy a bronchitídy). Vstupne realizujeme skríningové testy na primárnu ciliárnu dyskinéziu (PCD) a zisťujeme nízke hodnoty nazálneho oxidu dusnatého (pravá nosná dierka 25 ppb, ľavá nosná dierka 32 ppb) a predĺžený čas sacharínového testu (28 min. 30 s.). Medzi príznaky cílio-patií (t.j. ochorení spojených s dysfunkciou pohyblivých aj nepohyblivých cílií) patria tiež polycystické obličky, cysty pečene, biliárna atrézia, polysplénia/asplénia, vrodené ochorenia srdca a situs viscerum inversus. Preto u pacientky sonograficky vyšetrujeme brušné orgány – s fyziologickým nálezom a realizujeme kardiologické vyšetrenie, ktorým je vylúčená heterotaxia. Spirometricky u nej potvrdzujeme obštrukčnú ventilačnú poruchu ľahkého stupňa. Následne pacientka podstupuje bronchofibroskopické vyšetrenie v celkovej anestézii s nálezom výrazne hyperemickej sliznice dýchacích ciest a masívneho množstva hustého, belavožltého hlienu, ktorý v niektorých úsekoch parciálne alebo kompletne obturuje segmentálne bronchy (obr. 1, obr. 2). Počas bronchofibroskopie vykonávame bronchoalveolárnu laváž (BAL), kde sú v prietokovej cytometrii elevované neutrofilné segmenty, ostatné bunkové elementy a subpopulácie lymfocytov sú v norme. Nález zvýšených neutrofilných segmentov v BAL je typický u pacientov s bronchiektáziami a odráža prebiehajúci chronický bakteriálny zápal. Následne odoberáme kefkovitým sterom z trachey a z nosa vzorku respiračného epitelu na vyšetrenie kinematiky cílií. Vo vyšetrovaných vzorkách je zachytený dostatočný počet ciliárneho respiračného epitelu, pričom vysokorýchlostnou videomikroskopiou nachádzame vo všetkých vzorkách iba akinetické cílie (obr. 3). V kultivácii z BAL je zachytený Streptococcus pneumoniae a Haemophilus influenzae. V kultivácii z tonzíl je prítomný Staphylococcus aureus a Haemophilus influenzae, z nosa Staphylococcus aureus a Streptococcus pneumoniae. Všetky baktérie sú rezistentné na makrolidy, ktoré vzhľadom k uvedeným nálezom z liečby vysadzujeme. Pacientka je preliečená cielenou antibiotickou liečbou ciprofloxacínom v trvaní 14 dní. Dopĺňame u nej genetické vyšetrenie, ktorým sú zatiaľ vylúčené najčastejšie mutácie spôsobujúce vznik PCD (DNAI1 a DNAH5). Prítomnosť ďalších mutácií plánujeme vyšetriť v blízkej budúcnosti.
Vzhľadom ku klinickým príznakom, pozitívnym skríningovým testom a nálezu akinetických cílií respiračného epitelu potvrdzujeme u pacientky diagnózu PCD. Odporúčame u nej aktívnu intenzívnu dychovú rehabilitáciu, imunizáciu a prevenciu infekcií, pravidelné sledovanie kultivačného záchytu z horných a dolných dýchacích ciest, preliečenie kolonizácií a pri respiračných infekciách cielenú antibiotickú liečbu. Vhodné sú u nej rehabilitačné pobyty, pravidelné kontroly otorinolaryngológom a dispenzarizácia v pľúcnej ambulancii.
DISKUSIA
Primárna ciliárna dyskinézia (PCD) je geneticky heterogénne, typicky autozomálne recesívne dedičné ochorenie, ktoré v dôsledku ultraštrukturálnych a funkčných zmien riasiniek (cílií) vedie k poruche ich motility a k nedostatočnému mukociliárnemu transportu [12, 13]. To umožňuje ľahšie prenikanie mikroorganizmov k bunkám respiračného epitelu a u pacientov dochádza k zvýšenému výskytu respiračných ochorení (rekurentné a chronické infekcie dýchacích ciest vedúce až k bronchiektáziám, chronická rinosinusitída) [14, 15, 16]. PCD je zriedkavé ochorenie, s incidenciou približne 1 : 10 000–20 000 živonarodených [17]. V súčasnosti je známych 32 génov zodpovedných za jeho vznik. Predpokladá sa, že 65 % pacientov má bialelickú mutáciu v jednom z nich [18]. Medzi najčastejšie príčiny PCD (18–30 % prípadov) patrí mutácia génov DNAI1 a DNAH5, ktoré kódujú komplex proteínov vonkajších dyneínových ramienok [16, 19, 20]. Klinické príznaky PCD sa začínajú prejavovať už v novorodeneckom období, sú však rôznorodé a nešpecifické. Preto je ochorenie všeobecne poddiagnostikované. Výskyt symptómov priamo súvisí s orgánmi, v ktorých je pohyb cílií súčasťou normálnej funkcie. U pacientov dominujú príznaky z horných a dolných dýchacích ciest [17, 21, 22]. U takmer polovice pacientov s PCD je prítomný situs viscerum inversus [16]. Triáda symptómov situs viscerum inversus, bronchiektázie a chronická sinusitída je známa ako Kartagenerov syndróm [17]. Typické klinické príznaky PCD sú zhrnuté v tabuľke 2.
![Klinické príznaky PCD podľa veku [16, 19, 23, 26].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/79cbb340d8d8ded9160906e901afe68e.png)
Diagnostika primárnej ciliárnej dyskinézie vyžaduje kombinovaný prístup navzájom komplementárnych diagnostických metód, z ktorých každá má svoje limitácie [21]. Mala by byť stanovená v špecializovaných centrách, a to na základe prítomnosti typických klinických príznakov, použitím skríningových (nazálny oxid dusnatý, sacharínový test, zhodnotenie mukociliárneho klírens rádioaerosolovou metódou) a diagnostických vyšetrení (vysokorýchlostná videomikroskopia, elektrónová mikroskopia, imunofluorescenčná analýza) a genetickej analýzy [23]. Na Slovensku prebieha diagnostika PCD na Klinike detí a dorastu Jesseniovej lekárskej fakulty UK a Univerzitnej nemocnice Martin.
Zo skríningových vyšetrení sme u našej pacientky vyšetrili sacharínový test a nazálny oxid dusnatý (NO). Princíp sacharínového testu spočíva v transporte sacharínu pohybom cílií z nosovej dutiny do orofaryngu, kde sa jeho prítomnosť prejaví sladkou chuťou [11]. U zdravého človeka trvá približne 10 minút. Vylúčenie poruchy chuti sa potvrdí položením kúska sacharínu priamo na jazyk vyšetrovaného [11, 24, 25]. Predĺžený sacharínový test je príznakom porušeného mukociliárneho transportu, najčastejšie v dôsledku porušenej kinematiky cílií alebo pri abnormalitách hlienu [23]. Vyšetrenie je pre nutnosť spolupráce, na základe ERS guideline z roku 2009, odporúčané až od veku 12 rokov [26]. U našej pacientky bol čas sacharínového testu 28 min. 30 s. Oxid dusnatý je kontinuálne produkovaný respiračným epitelom. Existuje viacero faktorov, ktoré regulujú jeho produkciu za fyziologických aj patologických stavov. Pacienti s PCD majú výrazne znížené hodnoty nazálneho NO (<105 ppb). Okrem PCD však existujú aj iné ochorenia, pri ktorých je nazálne NO znížené, no nie až tak ako u PCD – cystická fibróza, difúzna panbronchiolitída, nazálna polypóza a chronická sinusitída [23, 27, 28, 29]. Naša pacientka mala hodnoty nazálneho NO 25 ppb a 32 ppb.
Ako diagnostické vyšetrenie sme použili vysokorýchlostnú videomikroskopiu, ktorá je moderným vyšetrením ciliárneho pohybu, využívaným v rutinnej diagnostickej praxi vo viacerých európskych centrách [26]. Po odobratí zo sliznice horných a dolných dýchacích ciest sa bunky respiračného epitelu sledujú svetelným mikroskopom, pričom sa vysokofrekvenčnou kamerou zaznamenáva pohyb riasiniek. Nasnímané vzorky sa následne vyhodnotia pomocou špeciálneho počítačového softvéru, ktorý analyzuje viacero parametrov kinematiky cílií (napr. pohybový vzorec, frekvenciu kmitania) [23, 26, 30]. Elektrónovo-mikroskopické vyšetrenie ultraštruktúry je u nás nedostupné. V diagnostike PCD sa však už nepovažuje za „zlatý štandard“, keďže je známe, že niektorí pacienti s PCD môžu mať ultraštruktúru riasiniek normálnu [12, 19]. U našej pacientky sme vysokorýchlostnou videomikroskopiou dokázali prítomnosť akinetických cílií v horných aj dolných dýchacích cestách. Pohyblivosť cílií respiračného epitelu môže byť sekundárne znížená aj u CF – v dôsledku hyperviskózneho hlienu, nikdy však nie sú akinetické všetky cílie [8].
Keďže PCD aj CF spôsobujú poruchu mukociliárneho transportu, obe vedú k podobným respiračným symptómom [10, 31]. A pokiaľ je CF atypická, len s pľúcnymi prejavmi, je odlíšenie oboch ochorení o to náročnejšie. V tabuľke 3 uvádzame porovnanie CF a PCD, pričom nálezy u našej pacientky sú zvýraznené.
![Porovnanie cystickej fibrózy a primárnej ciliárnej diskinézie [3, 6, 8, 18, 23].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/b2f1d31deb55058f267d81607fc1a551.png)
V novorodeneckom období je u CF typický nález mekóniového ilea či protrahovaného novorodeneckého ikteru [6]. Neonatálna pneumónia bez predisponujúcich faktorov, ktorú prekonala naša pacientka, je typická skôr pre PCD [12, 20, 31]. U pacientov s PCD dominujú príznaky chronického zápalu postihujúceho horné dýchacie cesty (rinosinusitída – 100 % pacientov, otitis media – 95 % pacientov) aj dolné dýchacie cesty (chronický vlhký kašeľ – 84–100 % pacientov) [12, 19, 32]. U CF klinicky dominujú respiračné symptómy z dolných dýchacích ciest (chronické zápalové bronchopulmonálne ochorenie smerujúce k respiračnej insuficiencii a predčasnej smrti – medián prežívania sa uvádza 49,7 roka) [5, 33]. Chronický zápal však postihuje aj horné dýchacie cesty (rinitída, sinusitída). Bežný je vznik nazálnych polypov, ktoré sú patognomické pre CF u detí do 6 rokov [3, 5]. U PCD aj CF je prítomné zhoršovanie pľúcnych funkcií. Avšak u väčšiny pacientov s PCD je pomalšie ako u CF, a hoci niektorí z nich zomrú predčasne na respiračné zlyhanie, mnohí sa dožívajú normálnej dĺžky života [22, 32, 34]. Neúčinný mukociliárny transport môže byť do určitej miery kompenzovaný kašľom. V porovnaní s CF sú pacienti s PCD schopní lepšie vykašľať spútum. Je to spôsobené tým, že pri CF dochádza k dehydratácii hlienu a zmenšeniu vrstvy periciliárnej tekutiny, kedy sa glykoproteíny mucínu v hliene naviažu na glykokalyx epitelu. Tým sa zhoršuje očisťovanie hlienu mukociliárnym transportom aj kašľom [3, 10, 22]. Ako následok chronickej infekcie vznikajú u oboch ochorení bronchiektázie. U CF sa nachádzajú charakteristicky v horných pľúcnych poliach spolu s cystami s fibróznym lemom [3, 5, 35]. U PCD postihujú primárne stredné a dolné laloky pľúc, čo je dôležitým diferenciálnym znakom [12, 19, 26]. Naša pacientka má potvrdené bronchiektázie stredného pľúcneho laloka vpravo. U CF aj PCD je prítomná kolonizácia dýchacích ciest typickými patogénmi. K spektru baktérií typických pre CF patria Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa a komplex Burkholederia cepacia [3, 36]. Infekcia baktériou P. aeruginosa sa vyskytuje až u 60 % pacientov s CF starších ako 3 roky [3]. Najčastejšími baktériami izolovanými z dýchacích ciest u detí a adolescentov s PCD sú Haemophilus influenzae, Staphylococcus aureus a Streptococcus pneumoniae. Tieto baktérie boli prítomné v kultiváciách z horných a dolných dýchacích ciest aj u našej pacientky. Starší pacienti s PCD s rozvinutejším postihnutím dýchacích ciest bývajú infikovaní aj Pseudomonas aeruginosa a netuberkulóznymi mykobaktériami [19, 22, 26].
Diagnóza CF sa opiera najmä o charakteristické klinické príznaky, avšak nevyhnutnou súčasťou stanovenia diagnózy je jej potvrdenie pomocou laboratórnych nálezov. U pacienta s klinickými príznakmi musí byť zároveň zistená zvýšená koncentrácia chloridov v pote pri dvoch alebo viacerých potných testoch, alebo musia byť zistené 2 „klasické“ mutácie CFTR génu [37]. U našej pacientky bola zistená mutácia F508 del len v jednej kópii. Chloridový potný test mala opakovane negatívny. Medzi diagnostické kritériá PCD patria: 1. klinické príznaky, 2. vyšetrenie nazálneho NO, 3. vysokorýchlostná videomikroskopia, 4. elektrónová mikroskopia, 5. imunofluorescenčná analýza a 6. genetická analýza. Na diagnózu PCD musia byť splnené minimálne 3 z nich [23]. Naša pacientka má prítomné klinické príznaky, nízke hodnoty nazálneho NO a vysokorýchlostnou videomikroskopiou dokázané akinetické cílie.
ZÁVER
Primárna ciliárna dyskinézia je zriedkavé vrodené ochorenie cílií vedúce k porušenému mukociliárnemu transportu, ktoré je pre svoje rôznorodé a nešpecifické príznaky všeobecne poddiagnostikované. Špecifická liečba zlepšujúca pohyblivosť cílií v súčasnosti neexistuje, terapeutické možnosti sú len symptomatické. Včasná diagnostika a terapia sú však dôležité pre ďalší priebeh ochorenia a prevenciu komplikácií (bronchiektázie, zhoršenie pľúcnych funkcií). Problémom je nejednotný manažment ochorenia v jednotlivých krajinách Európy a najmä fakt, že pre niektoré odporúčania chýbajú dôkazy o účinnosti, alebo sú jednoducho prebraté z odporúčaní pre liečbu CF, čo je pri rozdielnej patofyziológii oboch ochorení nevhodné [16, 23].
Základ liečby u pacientov s PCD predstavuje fyzioterapia v kombinácii so správne vykonávanými cvičeniami, ktoré sú zamerané predovšetkým na zlepšenie očisťovacích mechanizmov dýchacích ciest nezávislých na funkcii cílií [10, 38]. Techniky respiračnej fyzioterapie (RFT) sa menia s vekom dieťaťa a jeho schopnosťou spolupráce, pričom od najútlejšieho veku je potrebné učiť deti zásadám správneho dýchania, pravidelnému striedaniu vdychu nosom a výdychu ústami a základným hygienickým návykom [23, 38, 39]. Pre zaistenie optimálnej priechodnosti dýchacích ciest sú zásadné drenážne techniky RFT s riadenou expektoráciou a kontrolou kašľa, medzi ktoré patria: aktívny cyklus dychových techník, autogénna drenáž, PEP (pozitívny exspiračný pretlak) systémy dýchania, inhalačná liečba (v kombinácii s drenážou) a respiračný handling (určený dojčatám a deťom do 2–3 rokov). Princípom respiračného handlingu je polohovanie a manipulácia s dieťaťom pri bežných činnostiach, pričom sa využíva technika kontaktného a reflexného dýchania. Terapia potláča nefyziologické pohyby, ktoré sú spôsobené respiračným ochorením a je dôležitá ako prevencia motorických odchýlok u dojčiat. Najlepšie výsledky sú dosiahnuté pri zahájení terapie do prvých šiestich mesiacov života dieťaťa [38, 40, 41, 42]. Pre efektívne očisťovanie dýchacích ciest je potrebné vykonávať RFT pravidelne a denne [23, 38].
V manažmente liečby pacientov s PCD sa ďalej odpo-rúča pravidelné sledovanie klinického stavu (kultivačné vyšetrenie spúta, monitorovanie respiračných funkcií, vyšetrenie sluchu) a včasná cielená agresívna antibiotická liečba respiračných infekcií. Podávajú sa perorálne antibiotiká vo vyššom dávkovaní po dobu niekoľkých týždňov (v závislosti od klinického stavu). Intravenózne antibiotiká sú vyhradené na liečbu infekcie vyvolanej Pseudomonas aeruginosa, pneumónie a exacerbácií ochorenia spojených s febrilitami. Neexistujú dôkazy, ktoré by podporovali profylaktické podávanie antibiotík [16, 23, 38]. Deti s PCD by mali byť očkované v štandardnej schéme podľa očkovacieho kalendára, obzvlášť sa odporúča očkovanie proti S. pneumoniae, H. influenzae typu b a chrípke. U detí by sa mal pravidelne sledovať aj rast, keďže sa u nich môže objaviť rastová retardácia [16, 43]. K ďalším možnostiam liečby patrí použitie bronchodilatancií, RhDNázy, kortikosteroidov a hypertonických soľných roztokov, avšak o ich účinnosti nie je dostatok dôkazov [16, 44].
Táto práca bola podporená projektom „Meranie kinetiky cílií respiračného traktu“ (ITMS 2622022019) podporeného zo zdrojov EU a Grantom UK/162/2015 „Sledovanie kinematiky cílií respiračného epitelu vo vzťahu ku genetickým mutáciám“.
Došlo: 25. 8. 2015
Přijato: 1. 12. 2015
MUDr. Peter Ďurdík, PhD.
Klinika detí a dorastu JLF UK a UNM
Kollárova 2
036 59 Martin
Slovenská republika
e-mail: peter.durdik@gmail.com, marusiakova.lucia@gmail.com
Sources
1. Balaščaková M, Piskáčková T, Holubová A, et al. Současné metodické postupy a přehled preimplantační, prenatální a postnatální DNA diagnostiky cystické fibrózy v České republice. Čes-slov Pediat 2008;63 (2): 62–75.
2. Ďurdíková V, Gajanová J, Babčanová E. Nefarmakologická terapia bronchopulmonálneho postihnutia u detí s cystickou fibrózou. Pediatria (Bratisl) 2008;3 (5): 273–276.
3. Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol Pathol 2007; 35 (1): 116–129.
4. Schram CA. Atypical cystic fibrosis: identification in the primary care setting. Can Fam Physician 2012; 58 (12): 1341–1345.
5. Kayserová H. Cystická fibróza. Via pract 2007; 4 (3): 128–132.
6. Vávrová V, et al. Cystická fibróza. 1. vyd. Praha: Grada Publishing, 2005.
7. Wilschanski M, Famini H, Strauss-Liviatan N, et al. Nasal potential difference measurements in patients with atypical cystic fibrosis. Eur Respir J 2001; 17 (6): 1208–1215.
8. Mall MA, Elborn JS. Cystic Fibrosis. ERS Monograph 2014; 64 : 1–47.
9. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010; 363 (23): 2233–2247.
10. Munkholm M, Mortensen J. Mucociliary clearance: pathophysiological aspects. Clin Physiol Funct Imaging 2014; 34 (3): 171–177.
11. Plaza-Valía P, Carrión-Valero F, Marín-Pardo J, et al. Saccharin test for the study of mucociliary clearance: Reference values for a Spanish population. Arch Bronconeumol 2008; 44 (10): 540–545.
12. Leigh MW, Pittman JE, Carson JL, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med 2009; 11 (7): 473–487.
13. Strippoli MP, Frischer T, Barbato A, et al. Management of primary ciliary dyskinesia in European children: recommendations and clinical practice. Eur Respir J 2012; 39 (6): 1482–1491.
14. Jeseňák M, Majtán J, Rennerová Z, et al. Immunomodulatory effect of pleuran (β-glucan from Pleurotus ostreatus) in children with recurrent respiratory tract infections. Int Immunopharmacol 2013; 15 (2): 395–399.
15. Jeseňák M, Bánovčin P, Rennerová Z, et al. Reproducibility of food atopy patch tests over time in the general child population. Int J Dermatol 2009; 48 (9): 941–946.
16. Djakow J, O‘Callaghan C. Primary ciliary dyskinesia. Breathe 2014; 10 (2): 122–133.
17. Kurkowiak M, Zietkiewicz E, Witt M. Recent advances in primary ciliary dyskinesia genetics. J Med Genet 2015; 52 (1): 1–9.
18. Marshall CR, Scherer SW, Zariwala MA, et al. Whole Exome Sequencing and Targeted Copy Number Analysis in Primary Ciliary Dyskinesia. G3 (Bethesda) 2015 Jul 2; 5 (8): 1775–1781. doi: 10.1534/g3.115.019851.
19. Knowles MR, Daniels LA, Davis SD, et al. Primary ciliary dyskinesia: Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 2013; 188 (8): 913–922.
20. Orosová J. Primárna ciliárna dyskinézia (syndróm imotílnych cílií, Kartagenerov syndróm). Pediatr prax 2011; 12 (1): 7–11.
21. Raidt J, Wallmeier J, Hjeij R, et al. Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur Respir J 2014; 44 (6): 1579–1588.
22. Sagel SD, Davis SD, Campisi P, et al. Update of respiratory tract disease in children with primary ciliary dyskinesia. Proc Am Thorac Soc 2011; 8 (5): 438–443.
23. Ďurdík P, Bánovčin P. Primary ciliary dyskinesia – current therapeutic approach. In: Jeseňák M. Advances in Respiratory Therapy Research. New York: Nova Science Publishers, Inc., 2015 : 157–176.
24. Delehaye E, Dore MP, Bozzo C, et al. Correlation between nasal mucociliary clearance time and gastroesophageal reflux disease: our experience on 50 patients. Auris Nasus Larynx 2009; 36 (2): 157–161.
25. Varechová S, Mikler J, Murgaš D, et al. Cough reflex sensitivity in children with suspected and confirmed gastroesophageal reflux disease. J Physiol Pharmacol 2007; 58 (Suppl 5): 717–727.
26. Barbato A, Frischer T, Kuehni CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J 2009; 34 (6): 1264–1276.
27. Walker WT, Jackson CL, Lackie PM, et al. Nitric oxide in primary ciliary dyskinesia. Eur Respir J 2012; 40 (4): 1024–1032.
28. Bánovčin P, Jeseňák M, Michnová Z, et al. Factors attributable to the level of exhaled nitric oxide in asthmatic children. Eur J Med Res 2009; 14 (Auppl 4): 9–13.
29. Jeseňák M, Bánovčin P. Atopy patch test in the diagnosis of food allergy in children with atopic dermatitis. Acta Medica (Hradec Králové) 2006; 49 (4): 199–201.
30. Koniar D, Hargaš L, Štofan S. Segmentation of motion regions for biomechanical systems. Procedia Engineering 2012; 48 : 304–311.
31. Kapellerová A. Primárna dyskinéza riasiniek – súčasnosť a perspektívy. Čes-slov Pediat 2002; (11): 611–614.
32. Djakow J, Svobodová T, Uhlík J, et al. Primární ciliární dyskineze část 1. – Patogeneze a klinický obraz. Alergie 2009; (1): 45–50.
33. Stephenson AL, Tom M, Berthiaume Y, et al. A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur Respir J 2015; 45 (3): 670–679.
34. Strapková A, Nosáľová G, Bánovčin P, et al. Zmeny reaktivity hladkého svalu dýchacích ciest po expozícii toluénu. Stud Pneumol Phtiseol 1995; 55 (4): 263–271.
35. Vávrová V, Zemková D, Skalická V, et al. Problémy v diagnostice cystické fibrózy – potřeba novorozeneckého screeningu: Přehledový článek o aktuálních problémech. Čes-slov Pediat 2006; 61 (12): 703–709.
36. Dřevínek P, Fila L. Role moderní mikrobiologické diagnostiky v péči o pacienty s cystickou fibrózou. Čes-slov Pediat 2008; 63 (2): 83–89.
37. Ratjen F, Döring G. Cystic fibrosis. Lancet 2003; 361 (9358): 681–689.
38. Djakow J, Svobodová T, Uhlík J, et al. Primární ciliární dyskineze část 2. – Diagnostika a léčba. Alergie 2009; (2): 38–44.
39. Smolíková L. Hygiena horních cest dýchacích – součást léčebné rehabilitace. Pediatr praxi 2002; (6): 262–267.
40. Smolíková L, Máček M. Respirační fyzioterapie a plicní rehabilitace. Brno: Národní centrum ošetřovatelství a nelékařských zdravotnických oborů, 2010.
41. Heroutová M, Fábry J. Fyzioterapia pri respiračných ochoreniach u detí. Pediatria (Bratisl) 2009; 4 (1): 47–52.
42. Ďurdíková V, Ďurdík P. Dychová rehabilitácia v liečbe a prevencii RIDC. In: Jeseňák M, Rennerová Z, Bánovčin P. Recidivujúce infekcie dýchacích ciest a imunomodulácia u detí. Praha: Mladá fronta, 2012 : 419–426.
43. Svobodová T, Djakow J, Zemková D, et al. Impaired growth during child-hood in patients with primary ciliary dyskinesia. Int J Endocrinol 2013; 2013 : 731423.
44. Jošková M, Fraňová S, Nosáľová G, et al. A beneficial influence of Provinol on the reduction of allergen induced hyperreactivity in guinea pigs. Bratisl Med J 2009; 110 (8): 454–458.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2016 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Shiga toxin-producing Escherichia coli infections in children
- What disease can be hidden behind a diagnosis of atypical cystic fibrosis?
- Nonketotic hyperglycinemia: a case of a serious congenital hypotonia diagnosed by magnetic resonance
- Schools in hospitals and other medical facilities