
Vrozená vnímavost k mykobakteriálním onemocněním
Mendelian Susceptibility to Mycobacterial Diseases
Mendelian Susceptibility to Mycobacterial Diseases (MSMD) encompasses a newly emerged group of monogenic primary immunodeficiencies caused by defects in IL12-IL23/IFNγ mediated mononuclear phagocyte-Th1 communication pathway. Patients typically display various degree of selective impairment of immunity against mycobacteria (particularly against weakly virulent strains) and nontyphoid salmonellae; other aspects of host defence remain intact. Depending on the specific mutation, the clinical presentation ranges from mild adverse reactions to BCG vaccine to life-threatening disseminated mycobacterial infections and salmonellosis. The following case review reports on first two patients diagnosed with MSMD in the Czech Republic: a girl with IFNγ receptor mutation and a boy with STAT1 loss of function mutation.
Key words:
mendelian susceptibility to mycobacterial diseases, interleukin 12 (IL12), interferon gamma (IFNγ), STAT1, BCG, nontuberculous mycobacteria
Authors:
M. Bloomfield 1; E. Havránková 1; H. Houšťková 1; P. Kabíček 1; K. Křepela 1; A. Šedivá 2; J. Bustamante 3
Authors‘ workplace:
Pediatrická klinika IPVZ, 1. LF UK a Thomayerova nemocnice, Praha, přednostka doc. MUDr. H. Houšťková, CSc.
1; Ústav imunologie 2. LF UK a FN Motol, Praha, přednostka prof. MUDr. J. Bartůňková, DrSc.
2; Laboratoire de Génétique Humaine des Maladies Infectieuses, Institut National de la Santé et de la Recherche Médicale et Université Paris Descartes, France, vedoucí prof. J. L. Casanova, MD, Ph. D
3
Published in:
Čes-slov Pediat 2016; 71 (7-8): 340-344.
Category:
Tuberculosis
Kazuistika první pacientky byla přednesena na XI. českém pediatrickém kongresu 20. září 2014.
Overview
Vrozená vnímavost k mykobakteriálním onemocněním (Mendelian Susceptibility to Mycobacterial Diseases) je skupina monogenně podmíněných primárních imunodeficitů způsobených defekty v jednotlivých komponentech signalizační kaskády IL12-IL23/IFNγ zajišťující komunikaci mezi fagocyty a Th1 lymfocyty. Onemocnění se projevují selektivně sníženou obranyschopností proti mykobakteriím (především proti oportunně patogenním kmenům) a netyfoidním salmonelám, přičemž ostatní antimikrobiální imunita zůstává intaktní. Závažnost onemocnění se pohybuje od lokálních komplikací v místě inokulace BCG vakcíny až po fatální diseminované infekce. Předkládané kazuistiky popisují první dva pacienty diagnostikované v ČR: děvče s mutací receptoru pro IFNγ a chlapce s loss of function STAT1 mutací.
Klíčová slova:
vrozená vnímavost k mykobakteriálním onemocněním, MSMD, interleukin 12 (IL12), interferon gama (IFNγ), STAT1, BCG, atypické mykobakterie
ÚVOD
Základním pilířem humánní obranyschopnosti proti mykobakteriím je správně fungující interakce mezi mononukleárními fagocyty (makrofágy, monocyty, dendritické buňky) a Th1 lymfocyty, která je zajištěna signalizační osou IL12-IL23/IFNγ a několika dalšími přídatnými drahami (obr. 1). Infikovaný fagocyt produkuje IL12 a IL23, které se váží na své receptory na povrchu Th1 či NK buněk, jež reagují tvorbou IFNγ. Ten po vazbě na receptor na povrchu fagocytů spouští transfosforylaci Janusových kináz JAK1 a JAK2, následuje intracelulární přenos signálu pomocí transkripčního faktoru STAT1 (Signal Transducer and Activator of Transcription 1) do buněčného jádra [1]. Výsledkem je exprese IFNγ indukovaných genů směřující k destrukci internalizovaných mykobakterií (např. aktivací NADPH oxidázy produkující kyslíkové radikály) a k posílení proinflamatorní reakce (např. produkcí TNFα či další produkcí IL12).
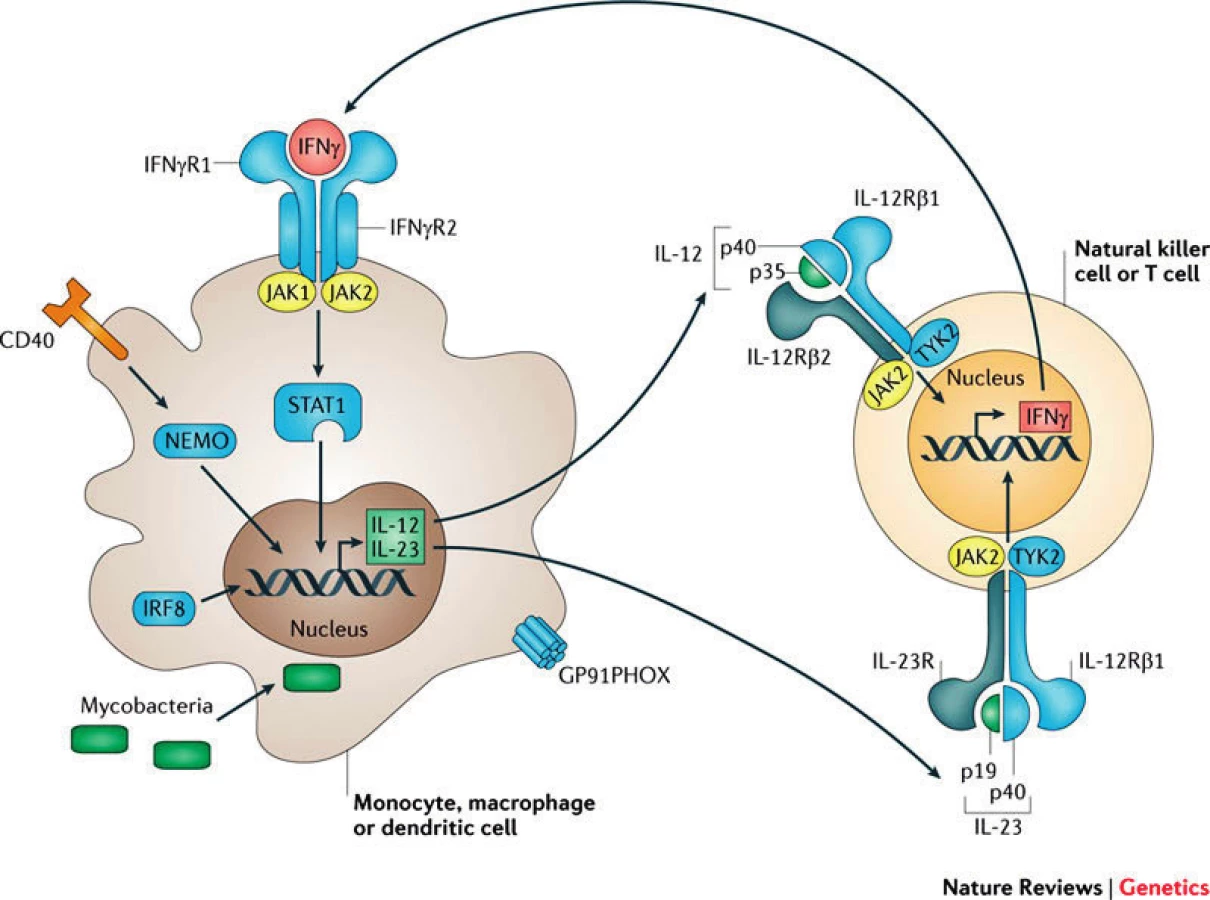
Poruchy v IL12-IL23/IFNγ signalizační kaskádě, podmiňující imunodeficity ze skupiny MSMD, jsou příčinou neefektivní obrany vůči mikroorganismům schopným intramakrofágového přežívání a mají za následek jejich lokální či systémovou diseminaci. Patognomické jsou infekce nízce virulentními, oportunními mykobakteriemi (např. M. avium, M. fortuitum, vakcinační kmen Bacillus Calmette Guérin – BCG), infekce M. tuberculosis, M. leprae probíhají fulminantně. Současně mohou být pacienti zvýšeně vnímaví k invazivním salmonelózám, kandidózám, listeriózám, klebsielózám, nokardiózám leishmaniózám a k některým virům [1, 3].
V současné době je známo 10 genů, jejichž mutace jsou příčinou 16 klinických podjednotek MSMD. Dědičnost může být autosomálně recesivní (AR), ale i autosomálně dominantní a X-vázaná [2]. Prevalence není známa, celosvětově je t.č. popsáno několik set případů, pacienti se často rekrutují z konsangvinních rodin. Zdaleka nejčastější jsou defekty receptoru pro IL12 a receptoru pro IFNγ, které mohou být částečné či úplné. Nejzávažnější jsou kompletní AR deficity receptoru pro IFNγ a kompletní STAT1 deficience [3].
Rutinní imunologická a hematologická vyšetření mohou být zcela normální. Diagnózu lze stanovit genetickým průkazem mutací a podpořit in vitro vyšetřením poststimulační sekrece cytokinů na odpovídající podněty, stanovením exprese povrchových receptorů či vyšetřením intracelulární signalizace. Plazmatické hladiny IFNγ mohou být nízké nebo normální, ale např. u kompletního deficitu IFNγR jsou násobně zvýšené [4]. K detekci mykobakteriální nákazy jsou využívány kultivační a PCR metody (senzitivita 40–60 %), validita vyšetření Mantoux II a Interferon Gamma Release Assay (Quantiferon TB Gold) je pochopitelně limitována imunopatologickou podstatou onemocnění. Schopnost tvorby specifických granulomů může být částečně zachována.
U pacientů s atypickými mykobakteriózami pomýšlíme vždy na imunodeficit, kromě MSMD jsou v pediatrické diferenciální diagnóze např. těžký kombinovaný imunodeficit (SCID), chronická granulomatózní choroba, ektodermální dysplazie s imunodeficiencí, idiopatická CD4+ lymfocytopenie či HIV infekce a lymfoproliferativní onemocnění.
Péče o pacienty vyžaduje širokou mezioborovou spolupráci pediatrů, imunologů, ftizeologů, infektologů, chirurgů, ortopedů, mikrobiologů, histologů a dalších specialistů. Terapeuticky se uplatňují dlouhodobé kombinace antituberkulotik, u některých deficitů adjuvantně interferon gama, u kompletních signalizačních blokád je indikována transplantace kostní dřeně, která je ovšem zatížena riziky spojenými s imunosupresí u pacienta s aktivní mykobakteriální infekcí a také vysokou mírou rejekce [5, 6].
Prognóza závisí na funkčním dopadu konkrétní mutace: u pacientů s parciálními defekty je při adekvátní léčbě příznivá a je zajímavé, že v zemích, kde není zavedena plošná BCG vakcinace, je např. u parciálního defektu IFNγR1 medián výskytu první oportunní mykobakteriózy až 13,4 let [7]. Naopak kompletní deficity se projevují časně a jsou bez transplantace kostní dřeně i přes intenzivní léčbu většinou fatální.
KAZUISTIKA 1
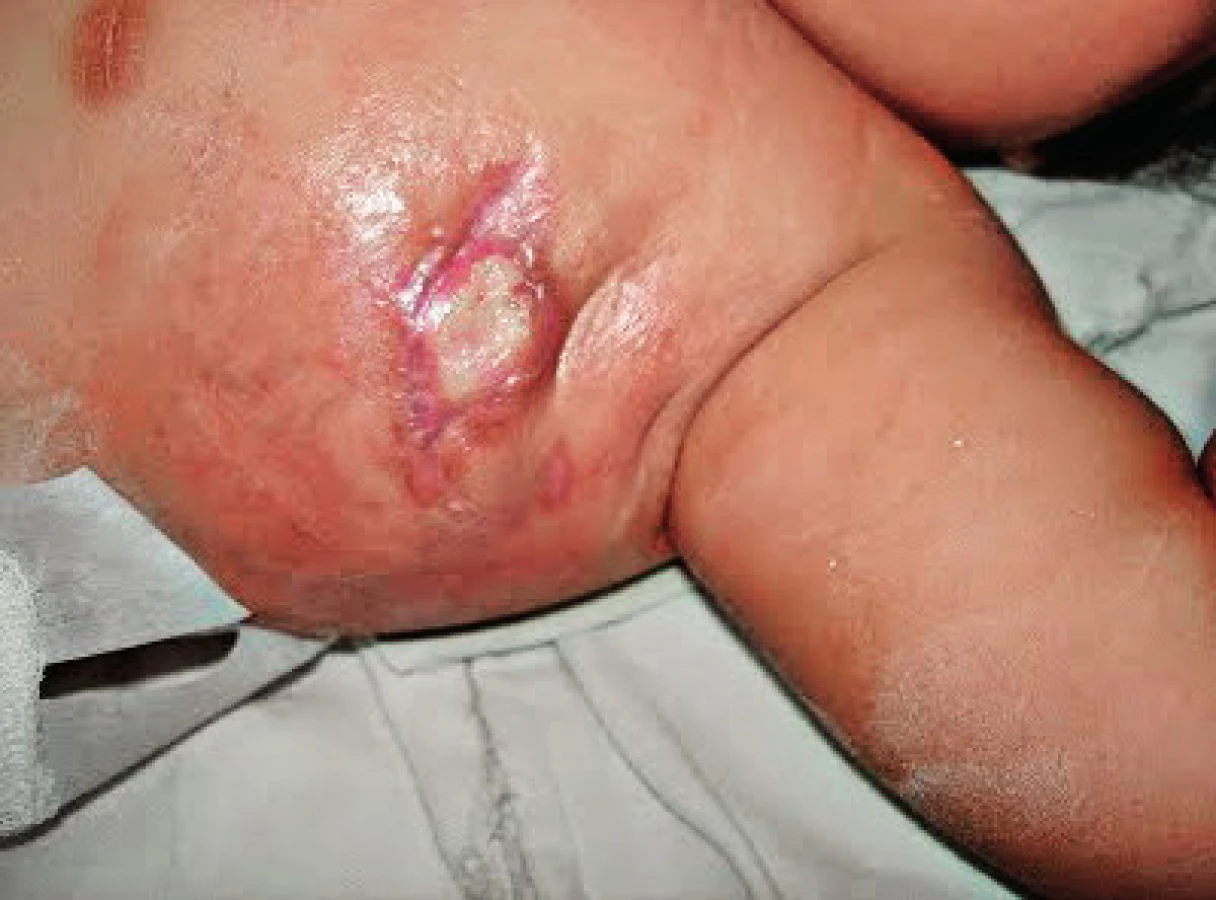
Pacientka, narozená zdravým, nepříbuzným rodičům, byla ve 3. měsíci života hospitalizována pro komplikace BCG vakcinace (dánský kmen 9. postnatální den) s rozvojem hnisavé léze v místě inokulace a axilární lymfadenitidou (obr. 2). Byla subfebrilní až nízce febrilní, sedimentace erytrocytů 68/109, CRP 120 mg/l, v krevním obraze byla patrná leukocytóza (22x109/l s převahou neutrofilů) a anémie (Hb 78 mg/l), Mantoux II byl 35 mm/72 hod s bulózní reakcí. Z exstirpované uzliny byly prokázány acidorezistentní tyčky (ART), histolog popsal specifickou granulomatózní reakci, etiologii potvrdilo PCR průkazem genu M. bovis a následná pozitivní kultivace M. bovis. Elevaci CRP, jinak netypickou pro BCGitidu, je možno vysvětlit současnou infekcí Klebsiella pneumoniae, která je dalším z častých patogenů pacientů s MSMD.
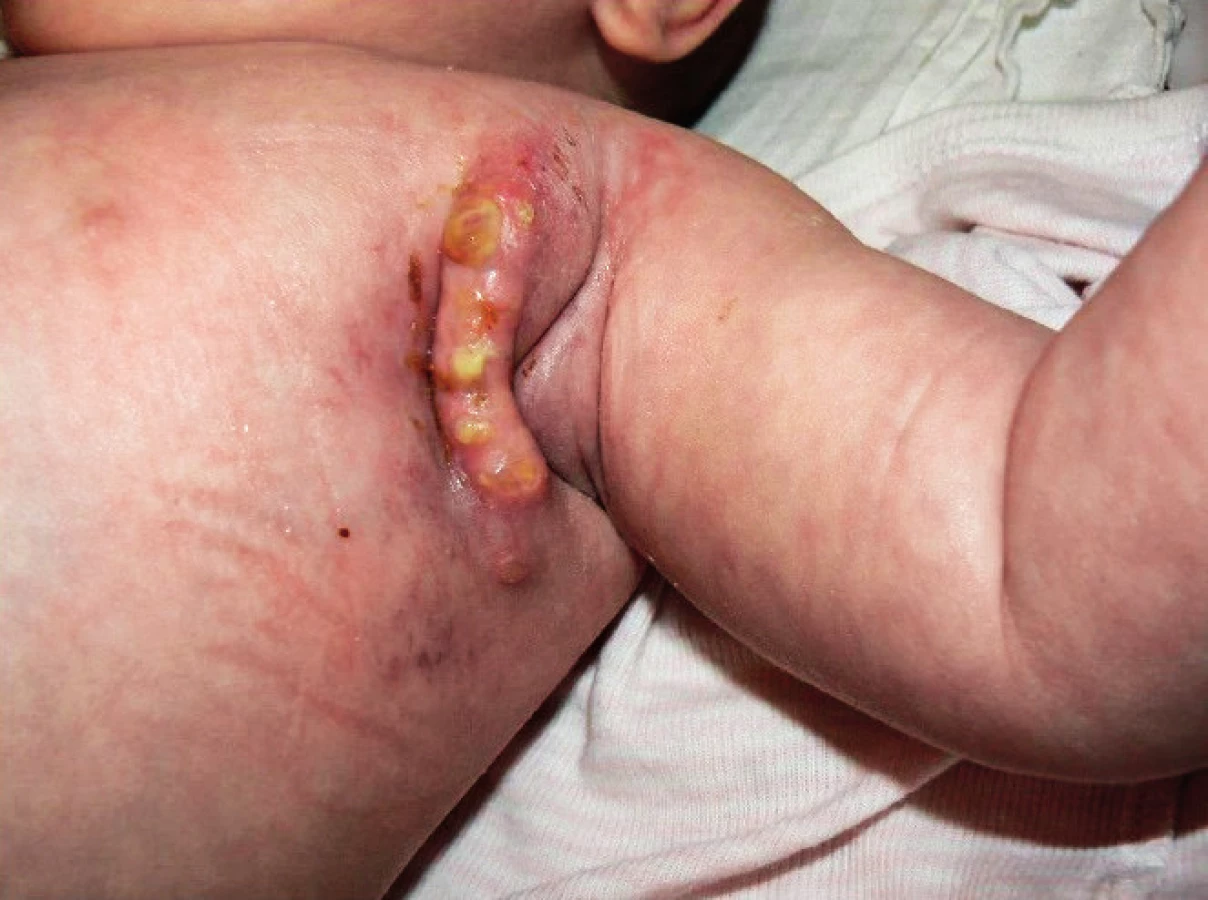
Kombinovaná antituberkulotická terapie trvala 15 měsíců a musela být opakovaně revidována pro nedostatečnou klinickou odezvu, protrahované hojení operační rány (obr. 3) či PET CT průkaz diseminace do plic v 11 měsících věku. Byly použity isoniazid, rifampicin, amikacin, clarithromycin, cykloserin a prednison.
Imunologické vyšetření vykazovalo zcela normální parametry humorální i buněčné imunity včetně Th17 populace, funkčních reakcí na mitogeny a PPD (purified protein derivate), normální byly fagocytární funkce i schopnost sekrece IFNγ lymfocyty. Cíleným genetickým vyšetřením nebyla prokázána zvažovaná mutace v MSMD kauzálních genech. Současně byla vyloučena HIV infekce.
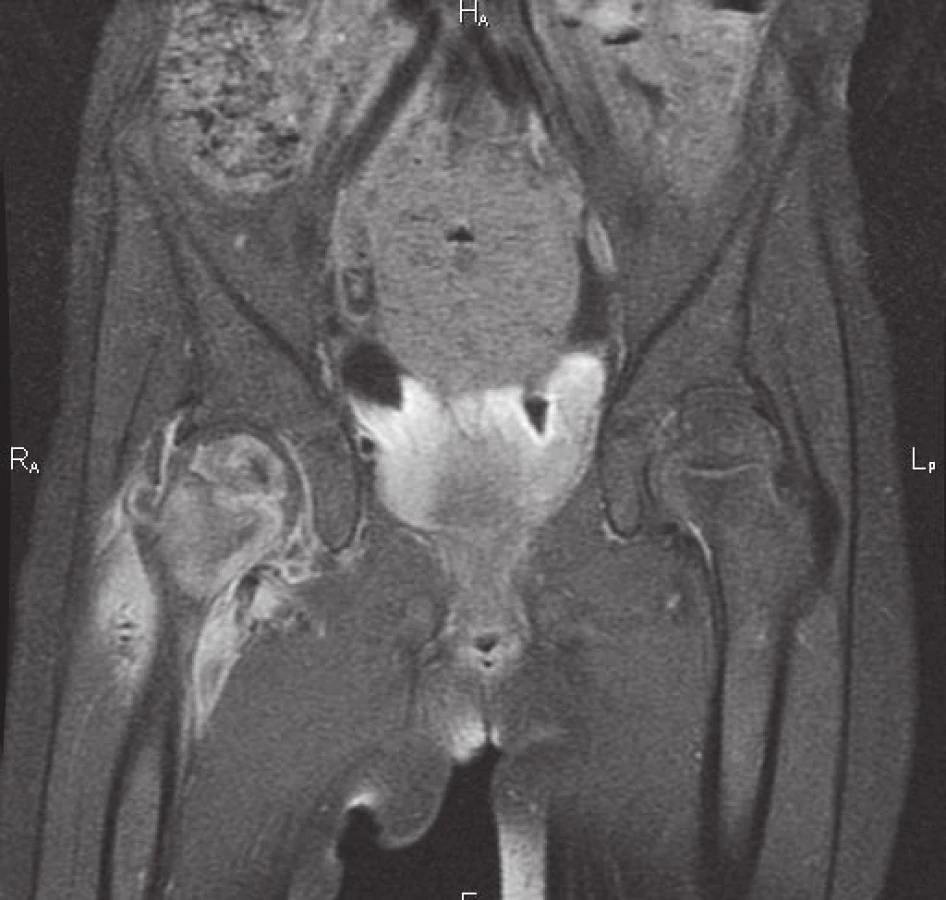
Po ukončení léčby byla v následujících 2 letech pacientka bez obtíží, nečetné běžné infekty probíhaly benigně, v době, kdy zbytek rodiny prodělával gastroenteritidu, byla jediná, kdo obtíže neměl. Ve 4 letech se dostavuje k vyšetření pro krční lymfadenitidu, bolest v pravé kyčli a 2 rezistence na kalvě. Laboratorně je patrná zvýšená sedimentace erytrocytů 60/120, v krevním obraze je 8,8x109/l leukocytů s převahou neutrofilů, CRP je 128 mg/l, PET CT ozřejmuje ložiska v levé plíci a slezině. Vyšetřením materiálu z okcipitálního abscesu se opět podařilo prokázat ART, histologicky byl popsán obraz nespecifické osteomyelitidy, PCR bylo negativní pro gen M. bovis, ale pozitivní pro nonTBC, kultivačně bylo ve 3. týdnu identifikováno M. avium subsp. avium. Komplexní imunologické vyšetření bylo opět prakticky v normě, při vysokém klinickém podezření na MSMD se však metodou sekvenace nové generace podařilo prokázat mikrodeleci 818del4 na 6. chromozomu podmiňující parciální defekt v R1 podjednotce receptoru pro IFNγ (receptorová porucha vysvětluje zachovanou schopnost sekrece IFNγ – viz obr. 1). Mutace je autosomálně dominantní, vznikla u pacientky de novo. Terapie byla vedena kombinací i.v. a p.o. antituberkulotik s předpokladem přirozené rezistence aviárního kmene, po potvrzení nepříznivého antibiogramu byla dále upravena (použity byly isoniazid, rifampicin, cycloserin, clarithromycin, amikacin). Na základě zjištěného imunodeficitu byl do terapie přidán rekombinantní s.c. interferon gama, jehož přínos zde spočívá v hyperstimulaci reziduálních funkčních IFNγ receptorů a zvýšení intracelulárního průniku makrolidových antibiotik [7]. Vzhledem k rozsahu nekrózy proximálního femuru, zřejmému na MRI (obr. 4), podstoupila pacientka chirurgický debridement. Po 2 měsících od zahájení léčby došlo dle PET CT k regresi původních ložisek, klesly zánětlivé ukazatele.
Celková délka terapie byla 31 měsíců, prvních 6 měsíců byla po operaci kyčle nutná imobilizace v masivní sádrové spice (obr. 5).
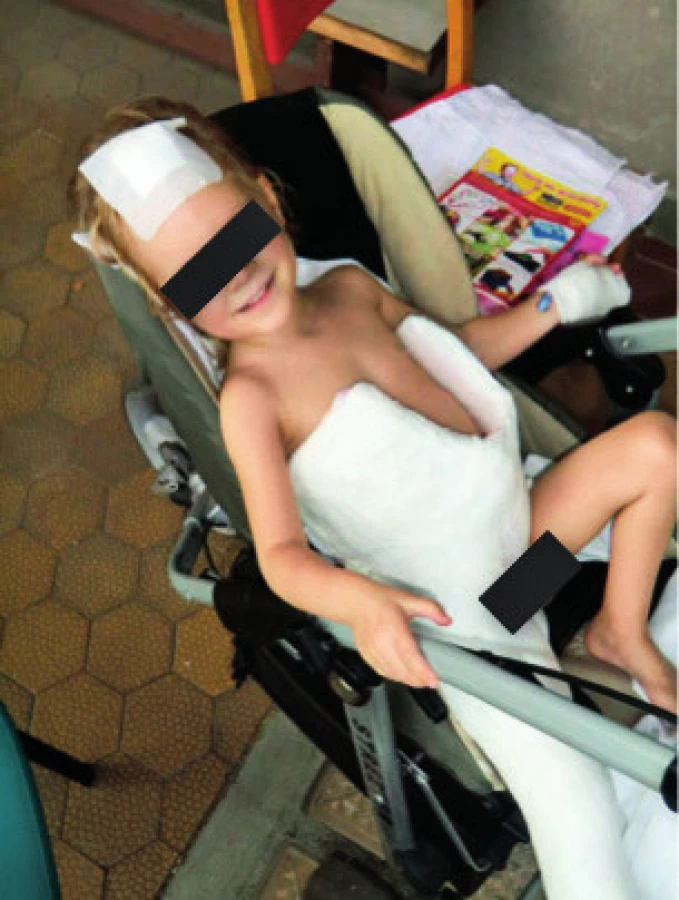
Komplikace léčby (např. nefro-, myelo-, oto-, oftalmotoxicita) se nevyskytly, po jejím ukončení byla pacientka schopná samostatné chůze. Zcela recentně, v 7 letech věku dítěte, si opětovná progrese krční lymfadenopatie vyžádala exstirpaci kolikvované uzliny, v níž byl prokázán kmen Mycobacterium abscessus/immunogenum, což je u pacientky již třetí atypická mykobakterióza. Dle PET MRI je infekce t.č. lokalizovaná, parametry zánětu i celkový stav dítěte jsou dobré, terapie je opět komplexní, je komplikována antibiotickou multirezistencí kmene a zahrnuje i interferon gama. I přes vysokou frekvenci infekčních komplikací u této pacientky je prognóza uvedené mutace relativně příznivá [8], transplantace kostní dřeně není indikována.
KAZUISTIKA 2
Pacient narozen zdravým nepříbuzným rodičům, s anamnézou protrahovaného hojení BCG jizvy u otce otce, byl poprvé léčen na naší klinice ve věku 10 týdnů pro BCGitidu (dánský kmen 4. postnatální den), která si vyžádala chirurgickou drenáž kolikvované axilární uzliny. V místě inokulace se postupně diferencovala dvě plošná zarudlá ložiska, jež byla 6 měsíců léčena isoniazidem s dobrým efektem. Po ukončení terapie přetrvávalo drobné zarudnutí, které po několika měsících začalo opět progredovat. K výraznému zhoršení lokálního nálezu došlo po varicele, jež jinak probíhala benigně. Ostře ohraničené ložisko na rameni dosahovalo velikosti přes 15 cm a objevil se papulózní exantém na trupu, hýždích a končetinách (obr. 6).
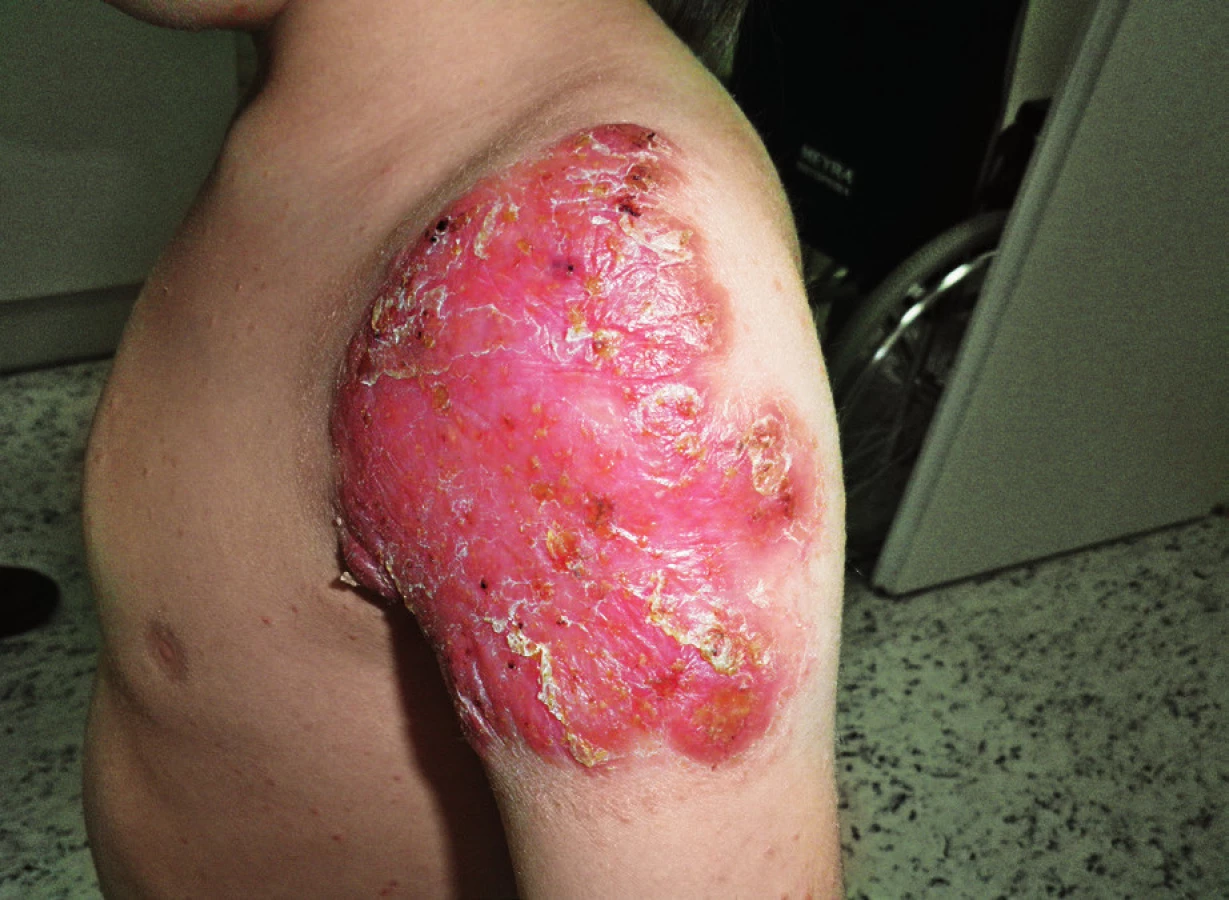
Základní laboratorní vyšetření byla v normě, jen mírně zvýšené CRP a sedimentace erytrocytů, Quantiferon TB Gold negativní. V bioptickém materiálu z hlavní léze byla popsána smíšená zánětlivá infiltrace, přítomny byly ale i částečně formované granulomy se známkami kaseózní nekrotizace, nebyly prokázány ART, PCR průkaz mykobakteriální DNA byl negativní. Teprve 52. den od zahájení kultivace byl detekován růst dobře citlivého M. bovis. Vzhledem k dosavadní absenci jiných závažných infekcí bylo vysloveno podezření na MSMD. Parametry humorální i buněčné imunity byly stejně jako u předchozí pacientky v normě, včetně populací Th17 a Treg, fagocytárních funkcí a IFNγ sekrece. Sekvenací DNA byla prokázána heterozygotní loss of function mutace c.1921G>A, p.A641T v genu pro STAT1 na 2. chromozomu, přenašečem mutace je otec, matka je nosičkou funkčního typu alely. STAT1 je klíčový signální protein v přímé dráze signalizace přes receptor pro IFN gama. Jsou známy čtyři STAT1-defektní fenotypy, u pacienta předpokládáme parciální defekt se sklonem k mírnějšímu průběhu atypických mykobakterióz a salmonelóz. V současné době probíhá detailnější vyšetření funkčního dopadu mutace.
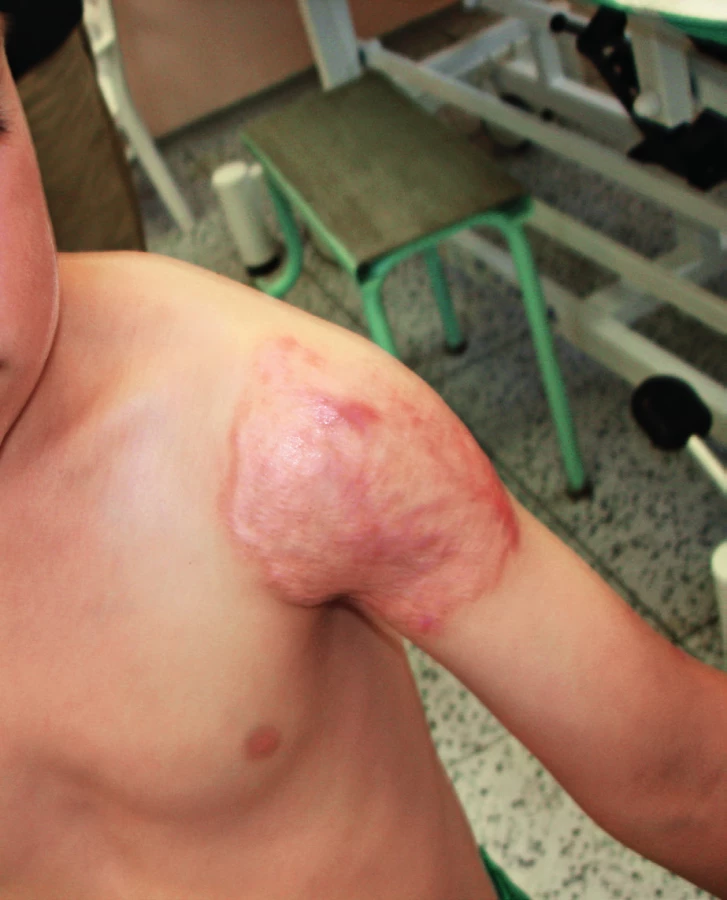
Terapie, spočívající v dlouhodobé antibiotické terapii, bývá efektivní. IFNγ může v případě nedostatečné odpovědi zlepšit průběh, transplantace kostní dřeně není indikována [9, 10, 11]. Pacient byl léčen 6 měsíců trojkombinací isoniazid, rifampicin, ethambutol a lokálně magistraliter mastmi se streptomycinem a rifampicinem, dalších 8 měsíců dvojkombinací antituberkulotik. Kožní nález regredoval, defekt se zhojil jizvou (obr. 7), vymizel i papulózní exantém, jenž byl hodnocen jako eozinofilní vaskulitida podmíněná idovou reakcí.
V současné době čekáme na výsledky analýzy DNA mladšího sourozence pacienta, který nebyl očkován BCG a je zatím zdráv, a rodiny otce.
ZÁVĚR
První potvrzený případ MSMD byl publikován v roce 1996, kdy u 4 dětí příbuzných rodičů z Malty došlo k rozsáhlým diseminovaným atypickým mykobakteriózám na podkladě kompletní AR mutace v genu pro IFNγR1 [12]. V následujících letech se MSMD etablovala mezi „nové“ primární imunodeficity s patognomicky úzkým spektrem mikrobiální vnímavosti způsobené monogenními mutacemi, jejichž objev má zásadní klinický i výzkumný dopad. Podobné specificky zaměřené deficity imunitní obrany byly identifikovány v případech chronické mukokutánní kandidózy, způsobené některou z poruch IL17 signalizace, HSV encefalitidy s deficity v TLR3 signalizačních drahách, u invazivních pneumokokových nákaz s poruchami v genech IRAK4 a MyD88, u invazivních dermatofytóz spojených s CARD9 deficiencí či u invazivních meningokokových infekcí s defekty v C5-9 složkách komplementu a lze je očekávat i u dalších, v současné době objevovaných deficiencí spojených se selektivní poruchou obrany vůči cílovému mikroorganismu [13, 14].
I přes značné pokroky se konkrétní mutaci podaří prokázat jen zhruba u poloviny pacientů s fenotypem MSMD, což naznačuje existenci dalších kauzálních či asociovaných genů. Znalost mutace je přitom klíčová při volbě preventivní a léčebné strategie, při prognostických úvahách i s ohledem na možnost prenatální diagnostiky. Při infekci atypickými mykobakteriemi, invazivní salmonelóze, či při komplikacích BCG vakcinace by měl pacient absolvovat podrobné imunologické vyšetření na specializovaném pracovišti, které ovšem, jak jsme ukázali, může být v normě. Při trvajícím podezření na imunodeficienci je nutné provést genetické vyšetření, které se u těchto velmi vzácných onemocnění provádí na specializovaných pracovištích v zahraničí. Výzvou do budoucnosti je design vhodných funkčních testů ke zjednodušení diagnostiky.
Uvedení pacienti jsou prvními potvrzenými případy MSMD v ČR. Prevalence onemocnění je nepochybně vyšší, proto lze předpokládat jejich budoucí nárůst. V této souvislosti na závěr poukazujeme na fakt, že vzhledem k ukončení celoplošné vakcinace proti TBC v ČR v roce 2010 lze očekávat posun prvních projevů snížené obranyschopnosti proti mykobakteriím z časného kojeneckého věku do vyššího dětského věku, neboť tito pacienti na sebe neupozorní projevy BCGitidy/BCGosy.
MUDr. Markéta Bloomfield
Pediatrická klinika IPVZ
1. LF UK a Thomayerova nemocnice
Vídeňská 800
140 59 Praha 4 – Krč
e-mail: marketa.bloomfield@ftn.cz
Sources
1. Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol 2014; 26 : 454–470.
2. Al-Muhsen S, Casanova J. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol 2008; 122 : 1043–1051.
3. Filipé-Santos O, et al. Inborn errors of IL-12/23 - and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 2006; 18 : 347–361.
4. Fieschi C, et al. High levels of interferon gamma in the plasma of children with complete interferon gamma receptor deficiency. Pediatrics 2001; 107: E48.
5. Moilanen P, Korppi M, Hovi L, et al. Succesful hemopoietic stem cell transplantation from an unrelated donor in a child with interferon gamma receptor deficiency. Pediatr Infect Dis J 2009; 28 : 658–660.
6. Roesler J, Picard C., Borgioni P, et al. Hematopoietic stem cell transplantation for complete IFN-gamma receptor 1 deficiency: a multi-institutional survey. J Pediatr 2004; 145 : 806–812.
7. Kanako T, Toshinao K, Yumiko N. Augmentation of antitubercular therapy with IFNγ in a patient withdominant partial IFNγ receptor 1 deficiency. Clin Immun 2014; 151 : 25–28.
8. Dorman S, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet 2004; 364 : 2113–2121.
9. Boisson-Dupuis S, Abel L, Casanova J. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol 2012; 24 (4): 364–378.
10. Chapgier A, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest 2009; 109 : 1502–1514.
11. Chapgier A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet 2006; 2: e131.
12. Newport M, Huxley C, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996; 335 : 1941–1949.
13. Casanova J. Severe infectious diseases of childhood as monogenic inborn errors of imunity. PNAS 2015; 112 (51): E7128–E7137.
14. Casanova J, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annual Rev Genomics Hum Genet 2013; 14 : 215–243.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2016 Issue 7-8
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Tuberkulózní meningitidy v České republice – konfrontace s diagnózou po 20 letech
- Aktuálna situácia v detskej tuberkulóze na Slovensku
- Výskyt tuberkulózy a mykobakterióz v České republice u dětí v letech 2000–2015
- Problematika dětské tuberkulózy