
Barthov syndróm – kazuistika
Barth syndrome – case history
Barth syndrome is a rare mitochondrial disorder linked to chromosome X. It is characterized by a wide range of clinical signs and laboratory findings, most typically heart disease, myopathy, neutropenia, growth failure and 3-methylglutaconic aciduria. It is considered to be a underdiagnosed disease with the risk of sudden death in childhood.
We report the first patient in Slovakia with proven Barth syndrome manifested by cardiac decompensation with the finding of left ventricular noncompaction cardiomyopathy and hepatic dysfunction with severe coagulopathy in the 3rd week of life. Based on the metabolic examination, clinical signs and a positive family history of two boys who died in infancy due to cardiac disorder, the diagnosis of Barth syndrome was genetically confirmed in the 7th week of life.
KEY WORDS
Barth syndrome, left ventricular noncompaction, 3-methylglutaconic aciduria, neutropenia, mitochondrial disorder
Authors:
S. Tárnoková 1; C. Šebová 1; K. Brennerová 2; J. Perečková 1; P. Kunovský 3; R. Riedel 4; M. Ďuranová 5; V. Bzdúch 2
Authors‘ workplace:
Centrum dedičných metabolických porúch Oddelenia laboratórnej medicíny, Národný ústav detských chorôb, Bratislava
1; Detská klinika LFUK a Národneho ústavu detských chorôb, Bratislava
2; Oddelenie JIS, Detské kardiocentrum, Národný ústav srdcových a cievnych chorôb, Bratislava
3; Detská klinika anesteziológie a intenzívnej medicíny, Národný ústav detských chorôb, Bratislava
4; Laboratórium lekárskej genetiky, Alphamedical s. r. o., Bratislava
5
Published in:
Čes-slov Pediat 2018; 73 (6): 384-388.
Category:
Overview
Barthov syndróm je zriedkavé mitochondriové ochorenie viazané na chromozóm X. Je charakterizované širokým spektrom klinických príznakov a laboratórnych nálezov, z ktorých najtypickejšie je postihnutie srdca, myopatia, neutropénia, rastová retardácia, 3-metylglutakónová acidúria. Považuje sa za poddiagnostikovanú klinickú jednotku s rizikom náhleho úmrtia v detskom veku.
V kazuistike prezentujeme prvého pacienta s dokázaným Barthovým syndrómom na Slovensku, u ktorého sa ochorenie manifestovalo v 3. týždni života kardiálnou dekompenzáciou s nálezom ľavokomorovej nonkompaktnej kardiomyopatie a hepatálnou dysfunkciou so závažnou koagulopatiou. Na základe metabolického vyšetrenia, klinických príznakov a pozitívnej rodinnej anamnézy – úmrtie dvoch chlapcov v dojčenskom období na ochorenie srdca, bola diagnóza Barthovho syndrómu potvrdená molekulovo-genetickým vyšetrením v 7. týždni života.
KĽÚČOVÉ SLOVÁ:
Barthov syndróm, ľavokomorová nonkompaktná kardiomyopatia, 3-metylglutakonová acidúria, neutropénia, mitochondriové ochorenie
ÚVOD
Barthov syndróm (BTHS; OMIM#302060), zriedkavé na X chromozóm viazané ochorenie charakterizované dilatačnou kardiomyopatiou, neutropéniou, myopatiou, rastovou retardáciou a 3-metylglutakónovou acidúriou (3-MGA-úria), prvýkrát komplexne opísali P. Barth a kol. v roku 1983 u rodiny s vysokou doj-čenskou úmrtnosťou chlapcov. U pacientov boli prí-tomné ultraštrukturálne abnormality v mitochon-driách myokardu, neutrofiloch kostnej drene, v menšej miere (0–9 %) v kostrovom svalstve. Vo svalových vláknach typu I bolo zvýšené množstvo tukových kvapôčok [1, 2].
Za BTHS sú zodpovedné mutácie v géne pre taffazin (TAZ, pôvodne označovaný ako G4.5), ktorý obsahuje 11 exónov [3] a je lokalizovaný na Xq28 [4, 5]. TAZ gén kóduje fosfolipidovú transacylázu, ktorá má dôležitú úlohu v remodelácii kardiolipínu [6, 7]. Kardiolipín tvorí súčasť vnútornej mitochondriovej membrány, je potrebný pre stabilizáciu mitochondriového respiračného reťazca a jeho strata vedie k dysfunkcii respiračného reťazca [1, 8, 9]. Mutácie v géne TAZ sú spojené s BTHS, ale prispievajú aj k ľavokomorovej nonkompaktnej kardiomyopatii (LVNC) a kardiomyopatii charakterizovanej trabekulizáciou myokardu [10, 11, 12]. V roku 2012 bolo známych už viac ako 120 rôznych mutácii v TAZ géne [13]. Nie je dokázaná korelácia medzi genotypom a fenotypom ochorenia, pričom veľká fenotypová variabilita môže byť aj medzi jedincami v rámci jednej rodiny [12]. Incidencia BTHS je odhadovaná (podľa registra BTHS www.barthsyndrome.org) na 1/300 000 až 400 000 narodených chlapcov, ale presný výskyt nie je známy a zdá sa, že ochorenie je poddiagnostikované [14].
KAZUISTIKA
Chlapec, v poradí tretie dieťa nepríbuzných rodičov kaukazskej rasy, sa narodil v 42. gestačnom týždni s pôrodnou hmotnosťou 3920 g, pôrodnou dĺžkou 52 cm. Popôrodná adaptácia bola dobrá. Otec anamnesticky udával bron-chiálnu astmu, jedna sestra alergiu, druhá sestra a matka pacienta boli zdravé. Po prepustení z pôrodnice bol plne dojčený, prospieval. Na 21. deň života postonkával, premodral, bol apatický. Pri vyšetrení praktickým pediatrom bol hypotonický, subcyanotický, laboratórne s hypoglykémiou 1,2 mmol/l. Dieťa bolo akútne hospitalizované na Detskej klinike anesteziológie a intenzívnej medicíny.
Vstupné laboratórne vyšetrenia odhalili metabolickú acidózu pH 7,06, PCO2 2,5 kPa, HCO-3 5,3 mmol/l, BE – 23,2, hyperlaktatémiu 12,9 mmol/l (norma 0,70–2,10 mmol/l), hyperamoniémiu 258 µmol/l (norma <60 µmol). V patológii dominoval výrazný hypokoagulačný stav s nehodnotiteľným PT-INR, nemerateľným APTT-R, výraznou hypofibrinogenémiou 0,3 g/l (norma 1,5–3,5 g/l), výrazne znížený plazminogén 15,2 % (norma 75–150 %), znížená aktivita AT 19,7 % (norma 40,0–80,0 %), pozitívny D-dimér >56,58 mg/l (norma 0,00–0,55 mg/l). Vstupný krvný obraz bol v norme. U dieťaťa sa rýchlo rozvíjal MODS, hemodynamická instabilita. V popredí klinického nálezu boli známky kardiálnej dekompenzácie (hepatomegália, ascites s postupným vývojom anasarky, USG známkami ischémie čreva). Echokardiografickým vyšetrením sa dokázala ľavokomorová nonkompaktná kardiomyopatia (obr. 1). Uvedený stav vyžadoval intenzívnu resuscitačnú terapiu, po ktorej sa klinický stav dieťaťa stabilizoval.
Fig. 1. Echocardiogram of patient with Barth syndrome in apical four-chamber view. In apex and lateral wall of left ventricle
depicted deep recesses (marked with arrows) characteristic of left-ventricular noncompactial cardiomyopathy (LA – left
atrium, LV – left ventricle).

V rámci diferenciálnej diagnostiky sa indikoval selektívny skríning dedičných metabolických porúch, v ktorom dominoval nález signifikantnej 3-metylglutakónovej acidúrie 56,7 µmol/mmol krea (norma 0,0–0,9 µmol/mmol krea). Dodatočné doplnenie rodinnej anamnézy odhalilo úmrtie dvoch chlapcov zo strany matky (matkin strýko a matkin bratranec) v dojčenskom období na bližšie nešpecifikované ochorenie srdca (obr. 2). Vzhľadom na klinický, laboratórny nález a anamnestické údaje bolo vyslovené podozrenie na X chromozóm viazané ochorenie – Barthov syndróm. Cielené molekulovo-genetické vyšetrenie preukázalo patogénnu variantu c. 280-C - >-T (pArg94Cys) v TAZ géne v hemizygotnom stave. U pacientovej matky, matkinej matky a oboch pacientových sestier bola potvrdená prítomnosť mutácie v heterozygotnom stave (prenášačky ochorenia).
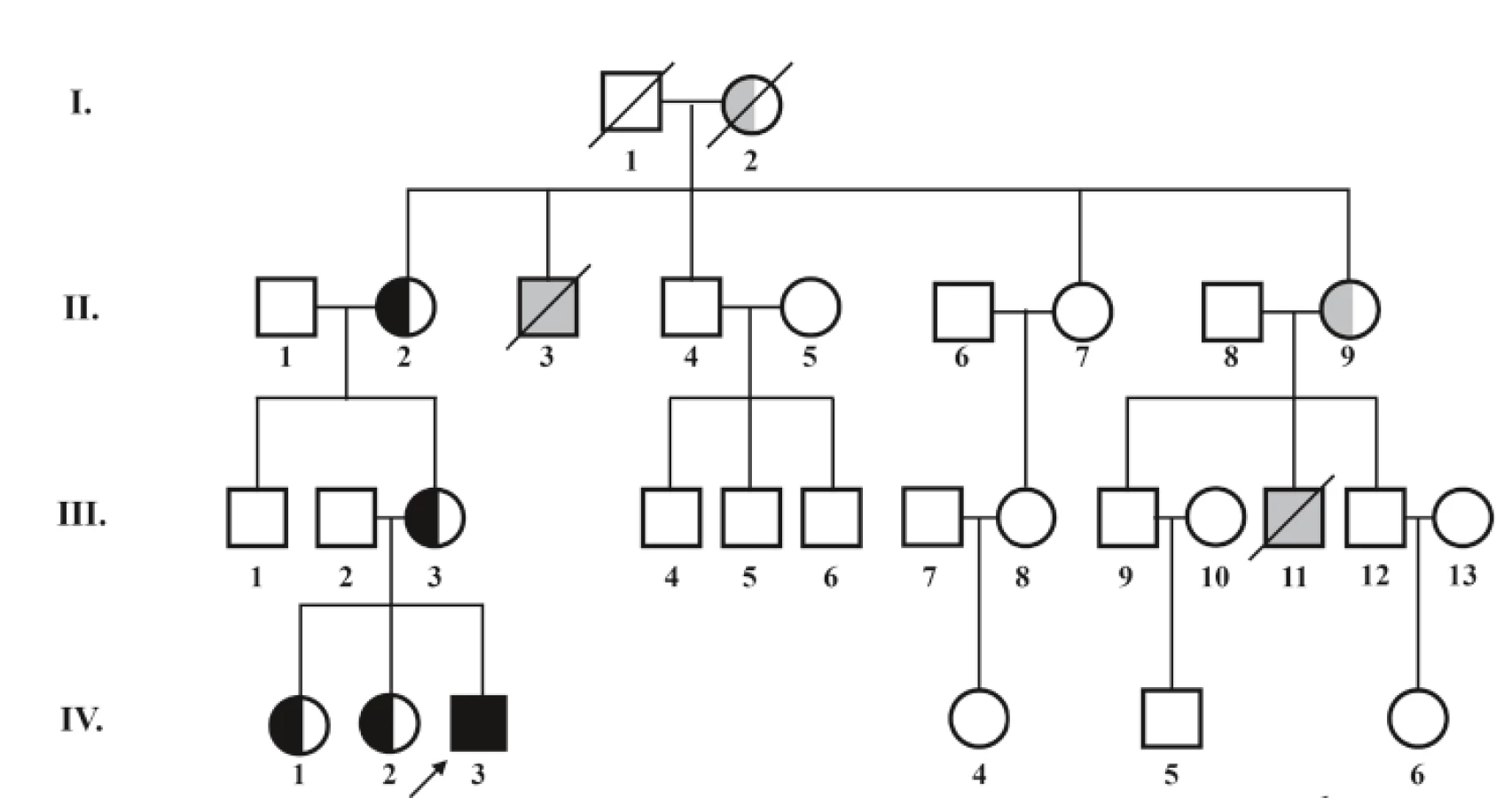
V súčasnosti má chlapec 14 mesiacov, telesnú hmotnosť 9,2 kg (medzi 3. a 10. percentilom) a výšku 76 cm (na 10. percentile). Je mierne hypotonický, motorický vývoj je spomalený: v tomto období sedí sám, ale sám sa neposadí, nestojí. Pretáča sa bez problémov z chrbátika na bruško. Kardiálne je kompenzovaný, funkcia ľavej komory sa zlepšila, echokardiografický nález je stabilizovaný. Je v sledovaní kardiológa, hematológa, imunológa, neurológa, metabológa. Má povolenú nezaťažujúcu rehabilitáciu. Pri manifestácii ochorenia bola prítomná výrazná hypocholesterolémia <1,30 mmol/l (norma 2,30–4,80 mmol/l) a naďalej pretrváva mierne znížená koncentrácia celkového cholesterolu při zníženej koncentrácii apoB.
Pri poslednej kontrole vo veku 14 mesiacov bola hodnota celkového cholesterolu 2,57 mmol/l (norma 2,95–4,80 mmol/l) a apoB 0,39 g/l (norma 0,6–1,4 g/l). Neutropéniu 0,96x109/l (norma 1,80–5,40x109/l) sme zachytili na 22. deň života (2. deň hospitalizácie), najnižšia hodnota absolútneho počtu neutrofilov, ktorú sme u neho zaznamenali, bola 0,19x109/l. Do 4. mesiaca bola neutropénia intermitentná, od 4. mesiaca chronická. V 3. a 4. mesiaci prekonal opakované septické stavy, ktoré mu prechodne výrazne zhoršili kardiálnu kompenzáciu. Najmä pre tento fakt spolu s opakovane potvrdenou neutropéniou s absolútnym počtom neutrofilov <0,5x109/l imunológ a hematológ odporučili podávanie GCSF (granulocytové kolónie stimulu-júci faktor), liečba bo-la začatá v 6. mesiaci, dávka sa postupne nastavila na 30 µg subkutánne 1x v týždni. Antibiotická profylaxia Biseptolom sa podáva 3x do týždňa, a pri neutropénii 3x do týždňa Diflucan. Intenzívnejšia imunologická podpora bola zvolená pre potrebu minimalizovať septické komplikácie a tým zamedziť sekundárnemu zhoršeniu funkcie myokardu. Kardiológom je odporučená liečba Vasocardinom, Vero-spironom, Enapom, Digoxinom. Dieťa užíva aj podpornú vitamínoterapiu a koenzým Q10. V prvom polroku života pre nedostatočný príjem stravy bol intermitentne sondovaný, v terajšom období od 12. mesiaca života má dostatočný perorálny príjem.
DISKUSIA
K najčastejším príznakom BTHS patrí kardiomyopatia, neutropénia, myopatia, rastová retardácia a 3-metylglutakónová acidúria [15]. Postihnutie srdca má charakter dilatačnej kardiomyopatie, ľavokomorovej nonkompaktnej kardiomyopatie, prípadne hypertrofickej kardiomyopatie, môže sa vyskytnúť aj endokardiálna fibroelastóza. Niektoré prípady si vyžadujú transplantáciu srdca [16, 17]. V štúdii Robertsa a kol. 69 zo 73 pacientov z registra Barthovho syndrómu udávalo v anamnéze kardiomyopatiu. Deväť zo 73 pacientov podstúpilo transplantáciu srdca, ktorá bola úspešná (údaj v čase realizácie štúdie). U mnohých pacientov kardiomyopatia často reaguje na štandardnú liečbu srdcového zlyhania a môže byť stabilná mnoho rokov, časom sa môže kardiologický nález dokonca aj zlepšiť [16]. Môžu sa u nich vyskytnúť srdcové arytmie ako dôsledok mitochondriovej dysfunkcie alebo pridruženého kardiálneho fenotypu, keďže komorové arytmie sú dobre zdokumentované pri dilatačnej, hypertrofickej aj nonkompaktnej kardiomyopatii [19, 20, 21]. U nášho pacienta bola prítomná ľavokomorová nonkompaktná kardiomyopatia (LVNC), čo je genetické ochorenie charakterizované excesívnymi a neobvyklými trabekuláciami ľavej komory (LV). Predpokladá sa, že ide o zastavenie vývoja srdca v tomto štádiu, takže sa nevytvorí plne kompaktný myokard. Klinicky a patologicky je typické špongióznym vzhľadom myokardu primárne LV, s abnormálnymi trabekulizáciami hlavne v hrote a midlaterálnej stene LV. Klasicky je prítomné okrem trabekulizácii a recesov aj zhrubnutie myokardu v dvoch zreteľných vrstvách – kompaktného a nekompaktného myokardu. Môže sa vyskytovať s dilatáciou alebo hypertrofiou LV, systolickou a/alebo diastolickou dysfunkciou, dilatáciou predsiení alebo s rôznymi formami vrodených chorôb srdca. Funkcia srdca, veľkosť a hrúbka myokardu sa môžu neočakávane meniť – tzv. undulujúci fenotyp. U postihnutých jedincov je riziko zlyhania LV, pravej komory alebo oboch. Srdcové zlyhanie môže vzniknúť na podklade zvýšenej fyzickej námahy alebo môže byť prítomné aj v kľude, ale mnoho pacientov môže byť asymptomatických. Ďalším život ohrozujúcim rizikom sú komorové arytmie a atrioventrikulárna blokáda, klinicky sa prezentujúce ako synkopy alebo náhla kardiálna smrť. Incidencia LVNC je približne 1 na 7000 živonarodených detí. Vyskytuje sa u novorodencov, malých detí a dospelých, s najhoršou prognózou u dojčiat, hlavne u tých s asociovaným systémovým alebo metabolickým ochorením [22].
Neutropénia u pacientov s BTHS má viacero foriem [23], môže byť závažná a chronická (<0,5x109/l) alebo cyklická. Väčšinou je však intermitentná a nepredvídateľná [15]. Závažná neutropénia spolu s kardiomyopatiou, mitochondriovou dysfunkciou, nízkou svalovou hmotou, tendenciou k hypoglykémii a laktátovej acidóze môžu zvýšiť pravdepodobnosť úmrtia na závažnú bakteriálnu infekciu. V diferenciálnej diagnostike pacientov – chlapcov so závažnými septickými stavmi by sa mal zvažovať aj BTHS [2].
Časté je zaostávanie rastu v prepubertálnom období [24]. Polovica pacientov zahrnutých do štúdie Spencera a kol. bola pod 5. percentilom pre výšku a/alebo hmotnosť pre daný vek. Niektorí pacienti s BTHS môžu mať oneskorený rastový špurt a svoju konečnú výšku dosiahnu až v neskoršom veku [16]. Thiels a kol. v roku 2016 prezentovali atypickú klinickú manifestáciu mutácií v TAZ géne u dvoch pacientov, ktorí boli vyšetrovaní pre zaostávanie rastu a miernu myopatiu. Na Barthov syndróm sa u nich spočiatku nemyslelo, keďže neboli prítomné ďalšie hlavné príznaky ochorenia. Aj z týchto prípadov vyplýva, že mutácie v TAZ géne sa nemusia prejaviť kompletným fenotypovým spektrom BTHS [24]. Wilson a kol. v štúdii u 22 pacientov s BTHS (vek od 4 mesiacov do 24 rokov) zistili vo vekovej kategórii pod 14,4 roka nižšie a nad 14,4 roka vyššie hladiny rastového hormónu v porovnaní so zdravými jedincami [25]. Pokiaľ nie je dokázaný centrálny nedostatok rastového hormónu, v terapii sa bežne nepodáva. U pacientov s BTHS boli zdokumentované znížené koncentrácie aminokyseliny arginínu v sére, čo môže viesť k zníženej proteosyntéze. Deplécia arginínu môže u pacientov s BTHS prispievať k spomalenému rastu [26, 27].
Väčšina pacientov s BTHS má aspoň mierne oneskorenie hrubej motoriky [16, 28]. U nášho pacienta je prítomná hypotónia a mierne oneskorenie motoriky, vo veku 14 mesiacov ešte nechodí a sám sa neposadí.
Zvýšená 3-MGA-úria je častým nálezom u pacientov s DMP, najmä pri mitochondriových ochoreniach. 3-MGA je intermediátom v katabolizme leucínu, v moči zdravých jedincov sa vyskytuje len v stopách (0–10 μmol/mmol krea). Známa je skupina ochorení s konštantne zvýšenou exkréciou 3-MGA, ktoré sa označujú rímskymi číslicami I–V v poradí, v akom boli opísané. Barthov syndróm predstavuje 3-MGA--úriu II. typu [29]. Pri 3-MGA-úrii I. typu (deficit 3-methylglutaconyl-CoA hydratázy, AUH defekte) je zvýšená exkrécia 3-MGA dôsledkom deficitu enzýmu hydratázy v katabolizme leucínu [30, 31]. Pri ostatných typoch 3-MGA-úrie je mechanizmus, ktorý vedie k zvýšenej tvorbe 3-MGA, neznámy a nesúvisí priamo s katabolizmom leucínu [29], môže byť dôsledkom hromadenia acetyl-CoA pri ochoreniach, ktoré vedú k inhibícii Krebsovho cyklu [32].
Spencer a kol. zistili u 6 z 25 pacientov s BTHS hypocholesterolémiu [16]. U nášho pacienta sme pri manifestácii ochorenia zaznamenali výraznú hypocholesterolémiu a naďalej u neho pretrváva mierne znížená koncentrácia celkového cholesterolu pri zníženej koncentrácii apoB.
U pacientov s BTHS sa môže pri kardiálnych, bakteriálnych komplikáciách a metabolickej dekompenzácii vyskytnúť závažná laktátová acidóza, hypoglykémia aj hyperamoniémia [33, 34, 35].
V roku 2013 Mazurová a kol. publikovali klinické, biochemické a genetické nálezy prvých štyroch českých pacientov s BTHS, ktorí pochádzali zo štyroch nepríbuzných rodín. Stredný vek manifestácie ochorenia bol 5,5 ± 3,8 mesiacov. Jedno dieťa zomrelo vo veku 2 rokov na náhle srdcové zlyhanie, ďalší traja mali od 3 do 13 rokov. Svalová hypotónia bola prítomná u všetkých štyroch pacientov, kardiomyopatia (dvaja pacienti dilatačná a jeden hypertrofická kardiomyopatia), rastová retardácia u troch a neutropénia u dvoch z nich. Charakteristickým laboratórnym nálezom bola intermitentne zvýšená exkrécia kyseliny 3-metylglutakonovej. Boli zistené tri nové hemizygótne mutácie v TAZ géne (c.584G>T; c.109 + 6T>C; c.86G>A). Autori navrhli zahrnutie BTHS do diferenciálnej diagnostiky kardiomyopatie v detskom veku, najmä pri súčasnom výskyte dilatačnej kardiomyopatie a 3-metylglutakónovej acidúrie [14].
Nami prezentovaný pacient sa manifestoval náhlou multiorgánovou dysfunkciou v 3. týždni života. V popredí boli známky kardiálnej dekompenzácie a hepatálnej dysfunkcie s výraznými zmenami v hemokoagulácii. Prítomné boli všetky hlavné klinické aj laboratórne nálezy BTHS. Yen a kol. v roku 2008 publikovali podobnú kazuistiku pacienta s BTHS, ktorý sa manifestoval akútnou metabolickou dekompenzáciou na 13. deň života, s rozvojom metabolickej acidózy, laktátovej acidózy, hyperamonémie, hypoglykémie a tiež koagulopatie v priebehu 8 hodín [34].
BTHS má jedinečnú patogenézu: je to jediné známe ochorenie u ľudí, kde je primárnou porucha remodelácia kardiolipínu – fosfolipidu, ktorý sa nachádza v membráne mitochondrií [36]. Keďže ochorenie postihuje mnohé orgánové systémy od intrauterinného obdobia až po dospelosť, malo by sa dostať do povedomia viacerých špecialistov: gynekológov, genetikov, pediatrov, kardiológov, neurológov, čo by zvýšilo možnosť skorej diagnostiky ochorenia [37].
ZÁVER
Vďaka tímovej spolupráci viacerých špecialistov bola diagnóza Barthovho syndrómu u pacienta potvrdená v 7. týždni života. Je to zatiaľ prvý známy pacient s BTHS na Slovensku. Rodine bola poskytnutá konzultácia klinického genetika a genetické poradenstvo s možnosťou vyšetriť aj ďalších členov širšej rodiny.
Skratky:
apoB – apolipoproteín B
3-MGA – 3-metylglutakónová kyselina
3-MGA-úria – 3-metylglutakónová acidúria
BTHS – Barthov syndróm
DMP – dedičné metabolické poruchy
GCSF – granulocytové kolónie stimulujúci faktor
LA – ľavá predsieň
LV – ľavá komora
LVNC – ľavokomorová nonkompaktná kardiomyopatia
Doc. MUDr. Vladimír Bzdúch, CSc.
Detská klinika LFUK
a Národného ústavu detských chorôb
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: bzduch@gmail.com
Sources
1. Barth PG, Valianpour F, Bowen VM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 2004; 126A (4): 349–354.
2. Barth PG, Scholte HR, Berden JA, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci 1983; 62 (1–3): 327–355.
3. Bione S, D‘Adamo P, Maestrini E, et al. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 1996; 12 (4): 385–389.
4. Adès LC, Gedeon AK, Wilson MJ, et al. Barth syndrome: clinical features and confirmation of gene localisation to distal Xq28. Am J Med Genet 1993; 45 (3): 327–334.
5. Bolhuis PA, Hensels GW, Hulsebos TJ, et al. Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28. Am J Hum Genet 1991; 48 (3): 481–485.
6. Koshkin V, Greenberg ML. Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem J 2002; 15; 364 (Pt 1): 317–322.
7. Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis 2007; 12 (5): 877–885.
8. Neuwald AF. Barth syndrome may be due to an acyltransferase deficiency. Curr Biol 1997; 7 (8): 465–466.
9. Brandner K, Mick DU, Frazier AE, et al. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell 2005; 16 (11): 5202–5214.
10. D‘Adamo P, Fassone L, Gedeon A, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61 (4): 862–867.
11. Brady AN, Shehata BM, Fernhoff PM. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn 2006; 26 (5):462–465.
12. Ronvelia D, Greenwood J, Platt J, et al. Intrafamilial variability for novel TAZ gene mutation: Barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol Genet Metab 2012; 107 (3): 428–432.
13. Clarke SL, Bowron A, Gonzalez IL, et al. Barth syndrome. Orphanet J Rare Dis 2013; 8 : 23.
14. Mazurová S, Tesařová M, Magner M, et al. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Medical Report 2013; 114 (3): 139–153.
15. Kelley RI, Cheatham JP, Clark BJ, et al. X-linked dilated cardiomyopathy with neutropenia, growth retardation, and 3-methylglutaconic aciduria. J Pediatr 1991; 119 (5): 738–747.
16. Spencer CT, Bryant RM, Day J, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006; 118 (2): e337–346.
17. Hanke SP, Gardner AB, Lombardi JP, et al. Left ventricular noncompaction cardiomyopathy in Barth syndrome: an example of an undulating cardiac phenotype necessitating mechanical circulatory support as a bridge to transplantation. Pediatr Cardiol 2012; 33 (8): 1430–1434.
18. Roberts AE, Nixon C, Steward CG, et al. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A 2012; 158A (11): 2726–2732.
19. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet 2010; 375 (9716): 752––762.
20. Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;124 (24): 2761–2796.
21. Brescia ST, Rossano JW, Pignatelli R, et al. Mortality and sudden death in pediatric left ventricular noncompaction in a tertiary referral center. Circulation 2013; 127 (22): 2202–2208.
22. Towbin A, Ballweg J, Johnsson J. Left ventricular noncompaction cardiomyopathy. In: Jefferies J., Chang A., Rossano J. Heart Failure in the Child and Young Adult. 1st ed. London: Academic Press, 2018 : 269–290.
23. Cantlay AM, Shokrollahi K, Allen JT, et al. Genetic analysis of the G4.5 gene in families with suspected Barth syndrome. J Pediatr 1999; 135 (3): 311–315.
24. Thiels CH, Fleger M, Huemer H, et al. Atypical clinical presentations of TAZ mutations: An underdiagnosed cause of growth retardation? JIMD Rep 2016; 29 : 89–93.
25. Wilson LD, Al-Majid S, Rakovski CS, et al. Higher IL-6 and IL6:IGF ratio in patients with Barth syndrome. J Inflamm (Lond) 2012, 21; 9 (1): 25.
26. Coman D, Yaplito-Lee J, Boneh A. New indications and controversies in arginine therapy. Clin Nutr 2008; 27 (4): 489–496.
27. Rigaud C, Lebre AS, Touraine R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis 2013; 8 : 70.
28. Barth PG, Wanders RJ, Vreken P. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome)-MIM 302060. J Pediatr 1999; 135 (3): 273–276.
29. Wortmann SB, Duran M, Anikster Y, et.al. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis 2013; 36 (6): 923–928.
30. Duran M, Beemer FA, Tibosch AS, et al. Inherited 3-methylglutaconic aciduria in two brothers--another defect of leucine metabolism. J Pediatr 1982; 101 (4): 551–554.
31. IJlst L, Loupatty FJ, Ruiter JP, et al. 3-Methylglutaconic aciduria type I is caused by mutations in AUH. Am J Hum Genet 2002; 71 (6): 1463–1466.
32. Su B, Ryan RO. Metabolic biology of 3-methylglutaconic acid-uria: a new perspective. J Inherit Metab Dis 2014; 37 (3): 359–368.
33. Christodoulou J, McInnes RR, Jay V, et al. Barth syndrome: clinical observations and genetic linkage studies. Am J Med Genet 1994; 50 (3): 255–264.
34. Yen TY, Hwu WL, Chien YH, et al. Acute metabolic decompensation and sudden death in Barth syndrome: report of a family and a literature review. Eur J Pediatr 2008; 167 (8): 941–944.
35. Donati MA, Malvagia S, Pasquini E, et al. Barth syndrome presenting with acute metabolic decompensation in the neonatal period. J Inherit Metab Dis 2006; 29 (5): 684.
36. Vreken P, Valianpour F, Nijtmans LG, et al. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 2000; 279 (2): 378–382.
37. Kulik W, van Lenthe H, Stet FS, et al. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem 2008; 54 (2): 371–378.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 6
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
- Komplexný pohľad na deficit vitamínu B12 v detskom veku
- Novorozenecký screening dědičných metabolických poruch v České republice
- Lyzozómové choroby – vývoj diagnostiky a liečby na Slovensku