
Sluchové vady a poruchy dětského věku
Hearing loss and hearing impairment in chidhood
Acording WHO, a person who has hearing loss has hearing threshold greater than 40 decibels in the better ears in adult and a hearing loss greater than 35 in childern. Over 5% of the world´s population has hearing loss, there is around 466 milion people and 34 milion of these are chidren. In the Czech Republic are born aproximately 600–1200 childern with middle hearing loss and 100 with hard hearing loss.
Key words:
hearingloss, childhood, hearing impairment
:
D. Hošnová; M. Urík; I. Šlapák; J. Šenkyřík 1
:
Klinika dětské otorinolaryngologie, Lékařská fakulta Masarykovy univerzity a FN Brno
; Klinika dětské radiologie, Lékařská fakulta Masarykovy univerzity a FN Brno
1
:
Čes-slov Pediat 2018; 73 (7): 420-423.
:
Podle WHO je nedoslýchavá taková osoba, jejíž sluchový práh je horší než 40 dB HL u dospělých, resp. 35 dB HL u dětí na lepším uchu. Celosvětově je sluchově postiženo přes 5 % populace, tj. kolem 466 milionů obyvatel, z toho 34 milionů dětí. V České republice se ročně rodí přibližně 600–1200 dětí se středně těžkou sluchovou vadou a 100 dětí s těžkou sluchovou vadou. V přehledovém článku je shrnuta etiologie sluchových vad s ohledem na dobu jejich vzniku.
KLÍČOVÁ SLOVA:
nedoslýchavost, dětský věk, sluchová vada
ÚVOD
V minulosti byla sluchová vada diagnostikována často až kolem 18. měsíce věku, v době, kdy dítě začíná chodit a vzdaluje se tak od mluvčího, povětšinou matky. Dnes, díky novorozeneckému screeningu, je možné vrozenou sluchovou vadu zachytit již velmi brzy a takto sluchově postižené dítě rehabilitovat sluchadly v době ještě před vývojem řeči, tj. kolem 6. měsíce věku. Sluchové vady jsou v 60 % vrozené, ve 40 % jsou získané z období perinatálního nebo postnatálního [1].
VROZENÉ VADY SLUCHU
Vrozené sluchové vady vznikají buď na genetickém podkladě, nebo poškozením sluchového ústrojí v prvním trimestru těhotenství (zarděnky, spalničky, syfilis, CMV, ototoxické preparáty, RTG záření...), nebo je příčina nejasná [1]. Nejcitlivější k poškození je sluchové ústrojí v době svého vývoje, tj. kolem 20. dne od početí.
Poruchy vývoje ucha mohou postihnout sluchový aparát na všech úrovních, mohou a nemusí být spojeny s poruchou sluchu. Nejčastější vrozenou vadou je otapostáza, která se nepojí s poruchou sluchu, boltce ale mohou být výrazně malformovány nebo nevyvinuty vůbec, v tomto případě bývá často spojena s atrézií nebo stenózou zevního zvukovodu (obr. 1).
Fig. 1. Stenosis and atresia of the external auditory canal.
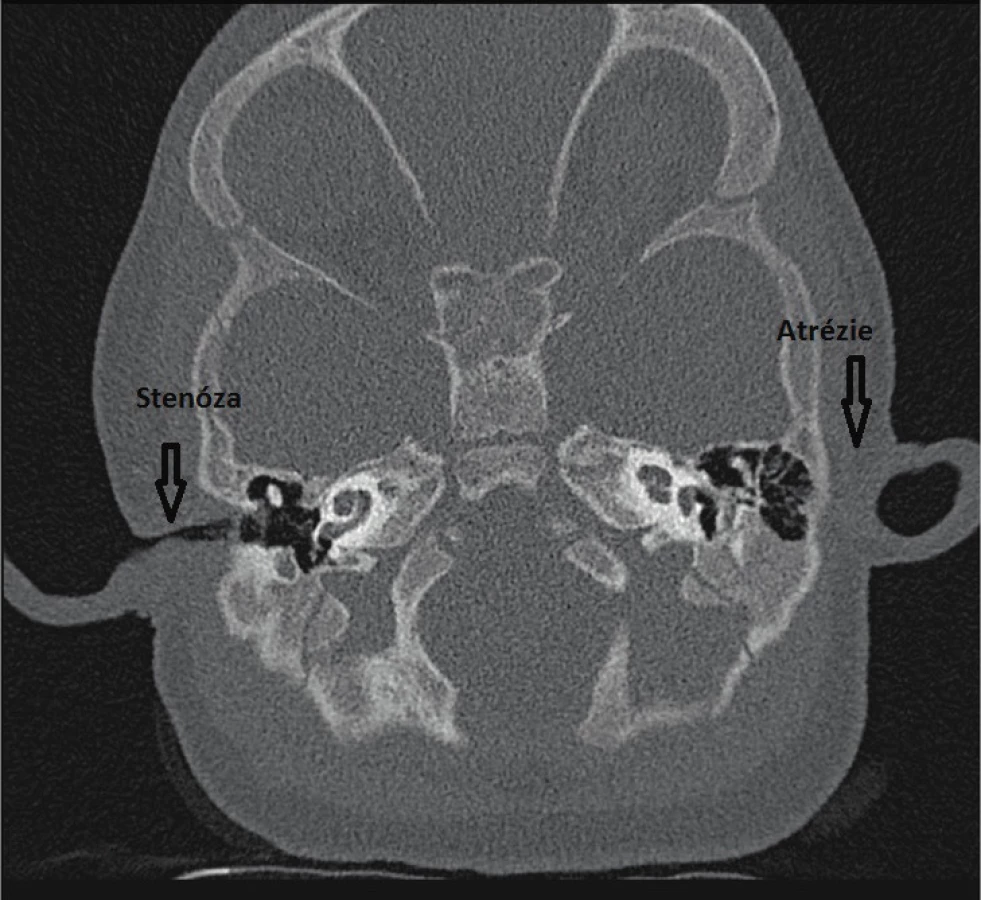
Na úrovni středního ucha se můžeme setkat s anomáliemi středoušních kůstek, malou středoušní dutinou nebo jejím úplným chyběním. Vrozené vady vnitřního ucha zahrnují poškození kostěného i blanitého labyrintu. Kostěné malformace, prokazatelné radiologicky (HRCT), představují asi 20 % z případů kongenitálních vad sluchu (obr. 2, 3, 4, 5, 6). Mnohem častěji se jedná o malformace membranózního labyrintu, a to v 80 % případů, které jsou prokazatelné jen histologicky. Spíše raritně se můžeme setkat s hypoplazií nebo aplazií sluchového nervu. Velmi často dochází ke kombinaci jednotlivých anomálií, ale mohou se vyskytovat i izolovaně. Často jsou součástí syndromů.
Fig. 2. Common cavity malformation of kochlea.
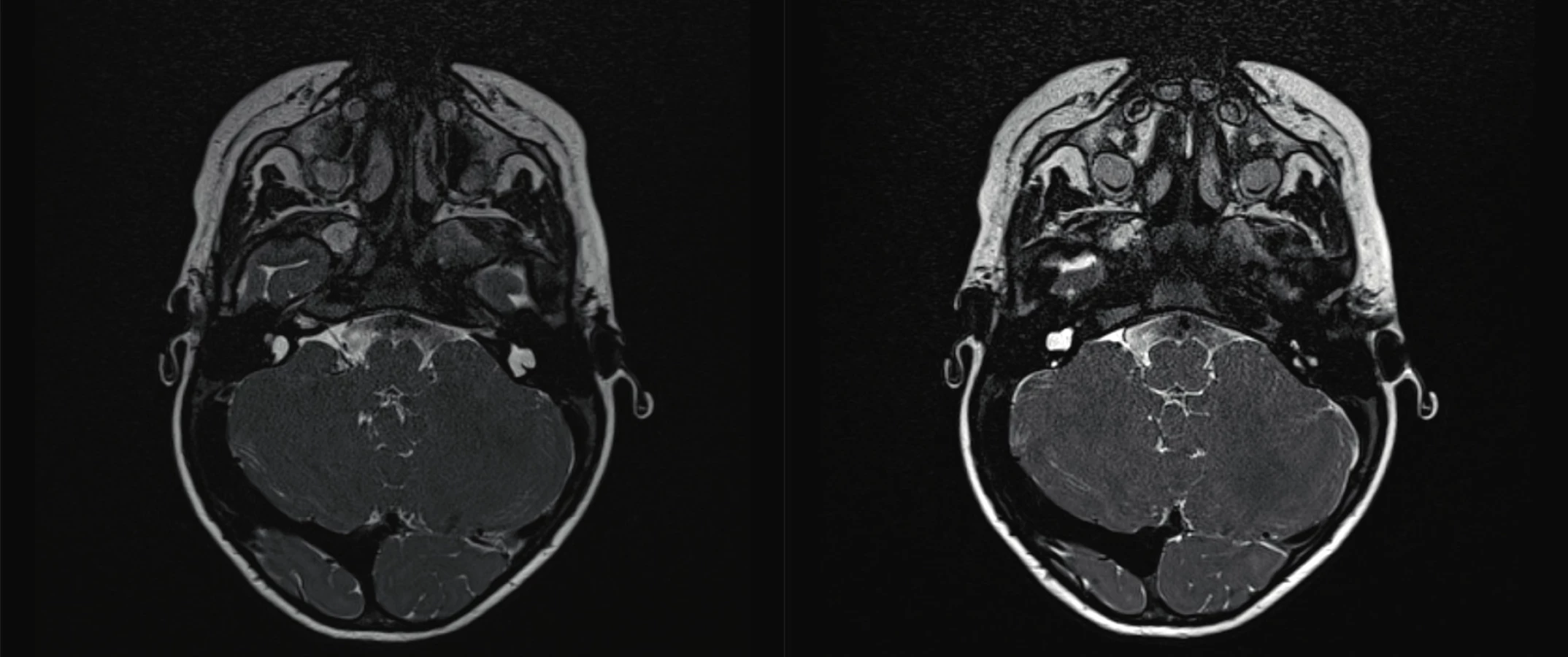
Fig. 3. Radiology reconstruction of common cavity malformation.

Fig. 4. Mondini malformation of kochlea.

Fig. 5. Enlarget vestibular aquaeduct.
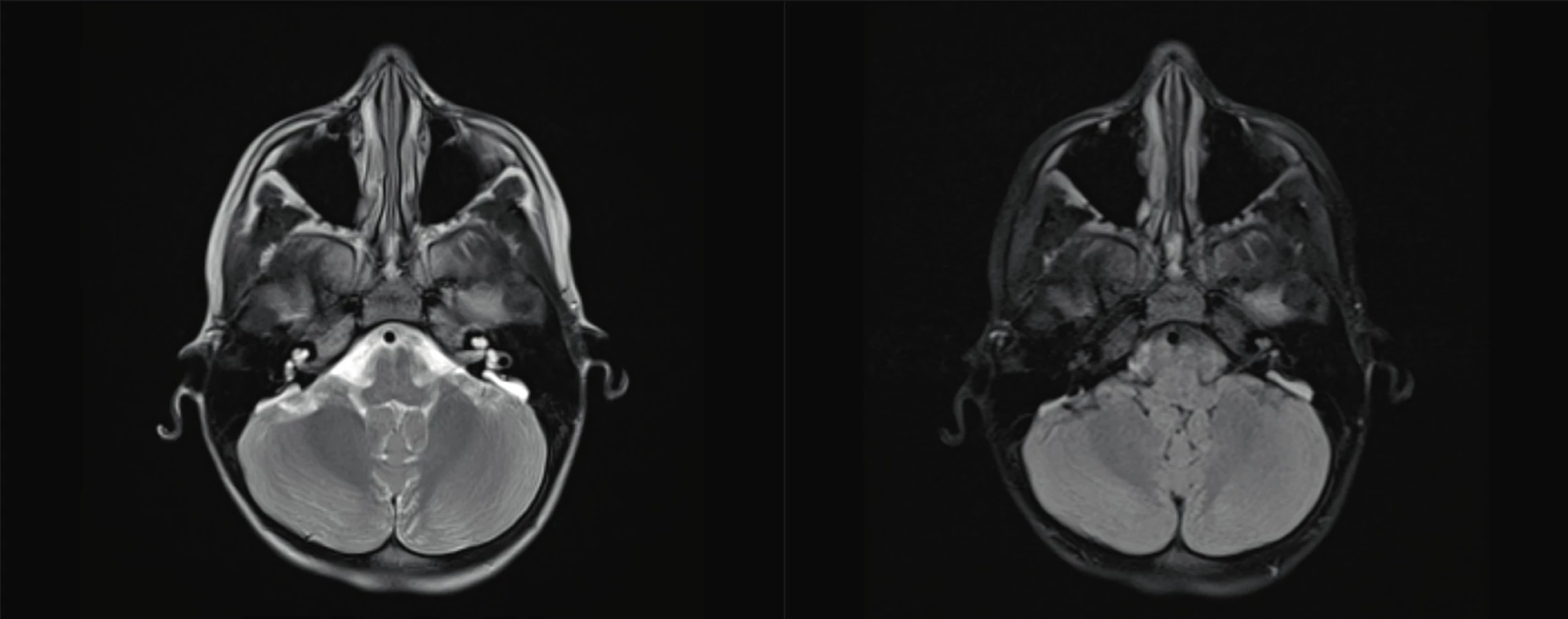
Fig. 6. Enlarget vestibular aquaeduct.

Z klinického hlediska je možno dělit sluchové vady na nesyndromové, kdy je poškození sluchu jediným manifestním příznakem, a syndromové, kdy je poškození sluchu součástí širšího postižení. Příčiny mohou být genetické i negenetické.
NESYNDROMOVÉ SLUCHOVÉ VADY
Genetické poškození se pojí s poruchou sluchu až v 50 % případů a může se dědit všemi typy mendelovské dědičnosti.
Autosomálně recesivní dědičnost
Zdaleka nejčastější je autosomálně recesivní (AR) typ dědičnosti, jenž se vyskytuje až u 75–80 % všech nesyndromových poruch sluchu. Pro nedoslýchavost AR dědičnou je typické časné, prelingválně vzniklé těžké senzorineurální sluchové poškození, většinou neprogredující.
Naprosto převažující mutací AR dědičnou je mutace v genu GBJ2 pro bílkovinu connexin 26 (Cx26 ) na 13. chromosomu – DFNB1. Způsobuje nesyndromovou poruchu sluchu. Connexin 26 je tkáňově specifický protein, který je součástí mezibuněčných spojení, kanálků, tzv. „gap junktion“ v plazmatické membráně vláskových buněk hlemýždě. Nepřítomnost nebo porušená funkce těchto spojení vede k narušené cirkulaci draslíkových iontů v průběhu repolarizace buněk, což je proces nezbytný pro správnou funkci hlemýždě a pro správný sluchový vjem. Mutace connexinu 26 tvoří až 50 % autosomálně recesivně dědičných poruch sluchu, u bílé populace až 80 %. Četnost heterozygotních přenašečů je v Evropě odhadována na 1 : 31. Charakteristická je těžká prelingvální porucha sluchu, většina pacientů podstupuje kochleární implantaci. V naprosté většině případů není spojena s žádnými dalšími příznaky. Zbylých 50 % známých AR mutací spojených se ztrátou sluchu se vyskytuje podstatně méně.
Z častějších možno jmenovat mutaci DFNB4, mutace v genu SLC26A4 (PDS), jež je spojena s malformacemi vnitřního ucha – Mondiniho dysplazie, a nesyndromová EVA (rozšířený vestibulární akvadukt), které se ale mohou vyskytovat jako součást syndromů (obr. 5, 6) [1–3].
Autosomálně dominantní dědičnost
Autosomálně dominantně (AD) je přenášeno 20 % sluchových vad, porucha sluchu je typicky postlingvální, je mírnější a progresivní [2].
Ostatní typy dědičnosti
Asi 1 % sluchových vad je pojeno s X-recesivně vázánou dědičností a s dědičností mitochondriální, která je spojována se ztrátou sluchu po aplikaci aminoglykosidových antibiotik [2].
SYNDROMOVÉ SLUCHOVÉ VADY
Vady sluchu jsou často součástí vrozených syndromů (20–30 %), kdy porucha sluchu je pouze jedním z dalších příznaků, je součástí daného syndromu. Je popsáno asi 100 syndromů spojených s vadou sluchu. Porucha sluchu může být převodní (postižení zvukovodu, středního ucha), senzorineurální kochleární (postižení vnitřního ucha), ev. retrokochleární i smíšená.
Převodní vady vznikají spíše sekundárně na podkladě kraniofaciální malformace, jakožto důsledku chybného utváření obličejového skeletu v době svého vývoje, a to nejčastěji v přímé souvislosti s opakovanými akutními otitidami, chronickou sekretorickou otitidou nebo rozštěpovými vadami. Jde např. o Downův syndrom, Turnerův syndrom, Crouzonův syndrom, Apertův syndrom, Pierre--Robinův syndrom aj. Převodní porucha rovněž může být primární, spojená např. s atrézií zvukovodu a malformacemi středního ucha, např. Treacherův-Collinsův syndrom, Goldenharův syndrom, Osteogenesis imperfecta aj.
Senzorineurální vady jsou často spojovány s jinými než kraniofaciálními malformacemi a mnohdy je porucha sluchu prvním vodítkem k poznání závažného celkového postižení. Jedním z častěji se vyskytujících syndromů spojených se senzorineurální poruchou sluchu je Pendredův syndrom, AR dědičný, charakterizovaný prelingválně, ale i postlingválně vzniklou nedoslýchavostí až hluchotou, často v souvislosti s EVA nebo Mondiniho malformací vnitřního ucha (obr. 2).
V pubertě dochází k onemocnění štítné žlázy.
Velmi závažným syndromem je Usherův syndrom, tj. spojení nedoslýchavosti různého stupně a zrakového postižení různého stupně až do obrazu hluchoslepoty. Jervellův--Lange-Nielsenův syndrom, neboli long QT syndrom, je kromě senzorineurální vady spojen s prodlouženým QT intervalem na EKG. Často může být řadu let nepoznán a projevit se náhle kolapsovými stavy s fatálními následky [1, 3].
PERINATÁLNÍ POŠKOZENÍ SLUCHU
Hlavními příčinami podílejícími se na perinatálním poškození sluchu jsou porodní nezralost, nízká porodní hmotnost <1500 g, hypoxie, asfyxie, komplikace během porodu aj. Poškození sluchu může být kochleární i retrokochleární (poškození sluchového nervu). Ztráta sluchu je v těchto případech několikanásobně častější než u fyziologických novorozenců.
U pacientů s perinatální rizikovou anamnézou je nutné kromě vyšetření otoakustických emisí v rámci novorozeneckého screeningu provádět rovněž vyšetření metodou AABR (screening evokovaných sluchových potenciálů mozkového kmene) k vyloučení retrokochleární neuropatie. Tato diagnóza může být přehlédnuta, otoakustické emise jsou zde výbavné, odrážejí funkční zevní vláskové buňky vnitřního ucha. U pacientů s auditorní neuropatií kolísá ztráta sluchu od lehké nedoslýchavosti po zbytky sluchu. Výrazně je narušeno rozumění řeči, zvláště v hluku, a kontrastuje s lepší percepcí při vyšetření tónovou audiometrií [4].
ZÍSKANÉ SLUCHOVÉ POŠKOZENÍ
V období postnatálním je dětský organismus vystaven působení celé řady faktorů, které mohou vyústit ve sluchovou poruchu. Pomineme-li příčiny způsobující převodní nedoslýchavost, jako jsou ušní maz, záněty zevního zvukovodu, středoušní záněty akutní či chronické, je dítě ohroženo vznikem náhlé percepční nedoslýchavosti. Jako nejčastější příčina vzniku náhlé nedoslýchavost je uváděna porucha mikrocirkulace, v dětském věku však spíše postižení vnitřního ucha nejčastěji virovou infekcí a autoimunitními onemocněními. Jako záchranná terapie se doporučuje infuzní kortikoidní terapie ev. s vazodilatancii, oxygenoterapie, hyperbarická komora nebo lokální aplikace kortikoidů přímo do vnitřního ucha aj. [5].
Velmi závažným stavem je purulentní labyrintitida jakožto následek purulentní meningitidy, kdy je poškození vnitřního ucha nevratné a labyrint se hojí osifikací. V těchto případech je nutná velmi časná kochleární implantace. V dětském věku je rovněž nutno počítat s poškozením sluchu ototoxickou terapií, aminoglykosidy, platinovými deriváty aj. Ze studií provedených u pacientů léčených cisplatinou plyne výraznější postižení sluchu u menších dětí ve srovnání s dospělými [6].
ZÁVĚR
Sluchová vada má pro člověka mnoho negativních dopadů. Je to především neschopnost nebo omezená schopnost komunikace se slyšícími. U dětí je to ohrožení rozvoje řeči a obtížné začlenění do společnosti vrstevníků. Nepoznaná nedoslýchavost může vést ke zhoršeným školním výsledkům, což může být pro dítě velmi frustrující, v extrémních případech může být dítě považováno za mentálně retardované. Ze všech těchto a řady dalších důvodů je nesmírně důležité odhalení sluchové vady a včasná terapie, ev. rehabilitace.
MUDr. Dagmar Hošnová, Ph.D.
Klinika dětské otorinolaryngologie
Lékařská fakulta Masarykovy univerzity
Fakultní nemocnice Brno
Jihlavská 20
625 00 Brno
e-mail: dagmarp@email.cz
Sources
1. Dršata J, Havlík R, a kol. Foniatrie – sluch. 1. vyd. Havlíčkův Brod: Tobiáš, 2015 : 1–384. ISBN 978-80-7311-159-5.
2. Pourová R. Molekulárně genetická vyšetření u českých pacientů a rodin s vrozenou ztrátou sluchu a bez mutací v GJB2 genu. Praha: Disertační práce, 2014. https://dspace.cuni.cz/bitstream/handle/20.500.11956/67039//IPTX_2013_2_11130_D0400002_303499_0_150454.pdf?sequence=1.
3. Dlouhá O, et al. Poruchy vývoje řeči. 1. vyd. Praha: Galén, 2007. ISBN 978-80-7492-314-2.
4. Zeleník K, et al. Těžká oboustranná nedoslýchavost u dítěte s výbavnými otoakustickými emisemi. Pediatrie praxi 2015; 16 (2): 121–123.
5. Dršata J, et al. Léčba percepční ztráty sluchu reohemaferézou – první zkušenosti. Otorinolaryng a Foniat (Prague) 2014; 63 (3): 163–169.
6. Hošnová D. Věk jako rizikový faktor ototoxicity způsobené cisplatinou. Otorinolaryng a Foniat (Prague) 2015; 64 (1): 3–7.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 7
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Possibilities of antibiotic treatment of acute otitis media
- Tubulointerstitial nephritis as a cause of acute renal insufficiency in children
- Hearing loss and hearing impairment in chidhood
- History and present of cochlear implantations in Czechia