
Novorozenecký screening cystické fibrózy a diagnostika CFSPID
Cystic fibrosis newborn screening and CFSPID diagnostics
Cystic fibrosis (CF) is a serious autosomal recessive disorder. Newborn screening (NBS) for CF allows for early diagnosis of CF before clinical symptoms occur. The patient´s benefits from NSCF are longer life expectancy and better quality of life.
NBS for CF in Czech Republic was introduced in 2009, based on experience from other developed countries. Results from newborn screening can be either negative, in which case the CF diagnosis is unlikely, or positive, which calls for further testing to confirm or exclude CF diagnosis. In some cases of positive NBS for CF, further tests results are inconclusive. Therefore, the term CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis) has been established in Europe.
From the Czech Republic 59 children were classified as CFSPID during the period between 2009 and 2018. Seven children from this group were later classified as CF patients.
The aim of this study is to describe the CFSPID group in the Czech Republic. Health status of 38 CFSPID patients could have been obtained. The majority of CFSPID patients are in good health status and do not display clear signs of CF. Even those children who have been reclassified as CF patients (except two patients) are in good health status without classical symptoms of the disease. Therefore, there is no reason to stress the families of CFSPID children, though it is highly recommendable to take care of these children in a specialized CF centre and parents should be informed about possible symptoms of cystic fibrosis.
Keywords:
Cystic fibrosis – newborn screening for cystic fibrosis – CFSPID
Authors:
J. Bartošová 1; D. Zemková 1; A. Holubová 2; R. Gaillyová 3; I. Valášková 3; A. Holčíková 4; M. Malá 4; V. Skalická 1
Authors‘ workplace:
Pediatrická klinika 2. LF UK a FN Motol, Praha
1; Oddělení klinické genetiky 2. LF UK a FN Motol, Praha
2; Oddělení lékařské genetiky, FN Brno
3; Klinika dětských infekčních nemocí, FN Brno
4
Published in:
Čes-slov Pediat 2019; 74 (7): 381-386.
Category:
Overview
Cystická fibróza (CF) je závažným nevyléčitelným monogenním onemocněním s autosomálně recesivní dědičností. Výrazným prvkem, jenž může přispět jak ke zkvalitnění, tak k prodloužení života nemocných, je časná diagnóza, nejlépe již v novorozeneckém věku. Novorozenecký screening cystické fibrózy umožňuje diagnostikovat pacienty s cystickou fibrózou v době, kdy ještě nejsou klinické příznaky plně vyjádřeny. Na základě časné diagnostiky pak mohou být pacienti s cystickou fibrózou léčeni již před vznikem ireverzibilních změn v organismu, což jejich život významně prodlužuje a také zlepšuje jeho kvalitu.
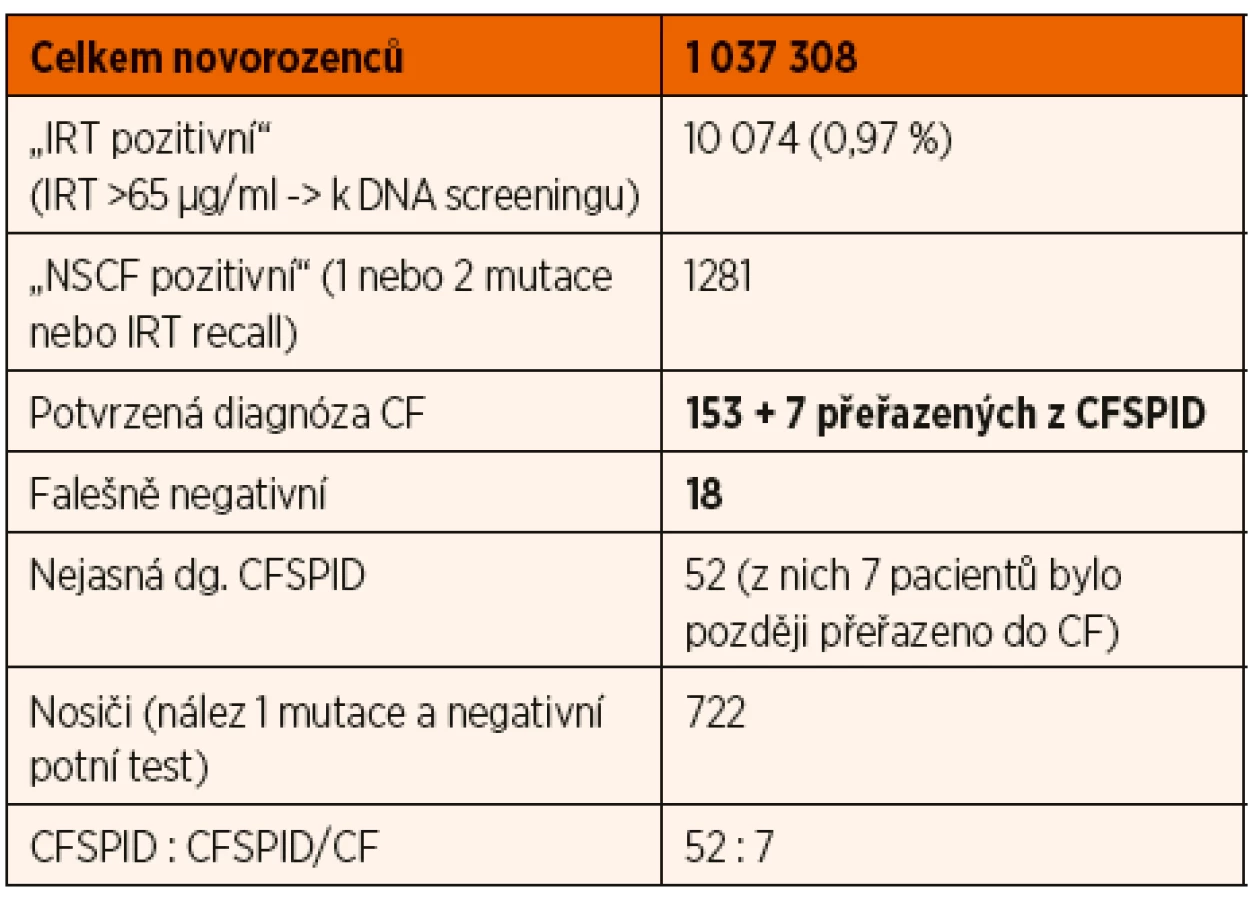
V České republice byl novorozenecký screening CF zahájen po vzoru řady dalších vyspělých zemí v roce 2009. Výsledek screeningu může být buď negativní a diagnóza cystické fibrózy je nepravděpodobná, nebo pozitivní, a pak je třeba vyloučit či potvrdit CF. Zvláštní skupinu tvoří určitý počet novorozenců, u nichž je screening pozitivní, ale výsledek dalšího diagnostického testování na CF je nejednoznačný. V Evropě se začal pro tuto skupinu používat termín CFSPID (Cystic Fibrosis Screen Positive, Inconclusive Diagnosis). Od zavedení screeningu bylo do konce roku 2018 v rámci celé ČR identifikováno a zařazeno 59 jedinců do kategorie CFSPID. Z nich 7 jedinců bylo z CFSPID později překlasifikováno a zařazeno mezi nemocné s CF. Práce upozorňuje na problematiku nové diagnostické jednotky CFSPID a uvádí první, prozatím dílčí výsledky sledování pacientů s touto diagnózou.
Hodnocení vývoje zdravotního stavu bylo možné u 38 sledovaných, kteří byli původně zařazeni do kategorie CFSPID. Většině pacientů s CFSPID se daří relativně dobře, nejeví jasné známky onemocnění CF. Dokonce i děti, které byly překlasifikovány a zařazeny mezi nemocné s CF, jsou kromě dvou v dobrém stavu, bez klasických projevů nemoci. Rozhodně není tedy vhodné rodiny CFSPID pacientů stresovat, na druhou stranu je ale potřeba doporučit sledování v centru CF a poučit rodiče o možných příznacích cystické fibrózy, které by se mohly objevit a při nichž by měli kontaktovat lékaře.
Klíčová slova:
cystická fibróza – novorozenecký screening cystické fibrózy – CFSPID
ÚVOD
Cystická fibróza (CF) je nejčastějším nevyléčitelným monogenním autosomálně recesivně dědičným onemocněním u evropských populací. Současná prevalence CF v České republice, vycházející z výsledků novorozeneckého screeningu do konce roku 2018, je 1 : 5827.
Onemocnění je způsobeno mutacemi genu CFTR (cystic fibrosis transmembrane regulator). Dosud bylo objeveno více než 2000 variant v tomto genu. V minulosti byla diagnóza stanovována převážně až na podkladě rozvinutých příznaků onemocnění. K těm patří nosní polypóza, recidivující sinusitidy a infekce dolních dýchacích cest a dlouhodobé zahlenění a kašel, dále prolapsy rekta a neprospívání a objemné mastné stolice při většinou dobré chuti k jídlu. Část pacientů může mít postižení jater se zvýšenými hodnotami jaterních testů a změnou echogenity jater při ultrazvukovém vyšetření. U kojenců a batolat se může onemocnění projevit metabolickou alkalózou s hyponatremií a hypochloremií. U novorozenců je třeba na diagnózu CF myslet při nálezu mekoniového ileu nebo mekoniové zátky, případně i jiné obstrukce střeva. U dospělých pacientů, především u mužů, může být prvním příznakem mírné formy CF neplodnost. Diagnóza je u symptomatických jedinců potvrzována patologickou hodnotou potního testu a následným průkazem dvou patogenních mutací genu CFTR při molekulárně genetickém vyšetření.
Novorozenecký screening cystické fibrózy (NSCF)
Vzhledem k tomu, že diagnóza byla dříve často stanovena až ve stadiu rozvoje komplikací CF, začalo se na sklonku 70. let minulého století v rozvinutých zemích uvažovat o zavedení postupů, jež by umožnily plošně diagnostikovat CF ještě před rozvojem klinických příznaků, nejlépe v rámci novorozeneckého screeningu. Studiemi bylo totiž prokázáno, že stanovení diagnózy CF během prvních 2 měsíců života (a z toho vyplývající časná léčba) vede k mírnějšímu postižení sino-bronchiálního systému, k lepšímu prospívání, a tím i k celkově příznivější prognóze onemocnění s delším přežíváním [1, 2].
V České republice byl celoplošný screening pro CF zaveden v roce 2009 (od 1. 10. 2009 v Čechách, resp. již od 17. 8. formou grantové studie, od 1. 12. 2009 na Moravě a ve Slezsku) a je součástí inovovaného celoplošného screeningu vybraných vrozených onemocnění [3]. V současné době se používá třístupňový model IRT/DNA/IRT. Prvním krokem NSCF je imunochemické stanovení imunoreaktivního trypsinogenu (IRT) ze suché kapky krve získané z patičky novorozence. Po případném zjištění koncentrace IRT nad stanovenou „cut off“ hodnotou (65 ng/ml) následuje ve druhém kroku molekulárně genetická analýza z téže suché kapky krve komerčním kitem obsahujícím 50 populačně specifických mutací genu CFTR se záchytem 91 % pro českou populaci (Elucigene CF-EU2v1, Elucigene Diagnostics). DNA analýza genu CFTR slouží u NSCF výhradně ke snížení falešné pozitivity při vyšetření IRT a jako indikace pro následný postup: buď a) ukončení vyšetřování, nebo b) po-zvání dítěte k provedení potního testu. U novorozenců s extrémně vysokou iniciální hodnotou IRT a žádnou detekovanou mutací genu CFTR je jako třetí krok NSCF proveden opakovaný odběr suché kapky krve ke stanovení IRT, tzv. „IRT recall“. Pokud je NSCF pozitivní, tj. je nalezena alespoň jedna mutace genu CFTR, nebo je hodnota druhého vzorku (IRT-recall) nad stanovenou cut off hodnotou, je dítě voláno k potnímu testu.
V případě pozitivního výsledku při stanovení koncentrace chloridů v potu (nad 60 mmol/l) je u dítěte a jeho rodičů provedeno diagnostické molekulárně genetické vyšetření genu CFTR z venózní krve. Pokud jsou přitom u dítěte nalezeny dvě patogenní mutace v pozici trans, je CF jasně prokázána a dítě je komplexně vyšetřeno v centru pro cystickou fibrózu, přičemž proběhnou potřebná vyšetření dítěte, edukace rodičů a zahájení léčby.
Pokud je výsledek potního testu pozitivní, ale při molekulárně genetickém vyšetření je nalezena pouze jedna z 50 nejčastějších mutací, následuje vyhledávání druhé, vzácnější mutace DNA analýzou metodou sekvenace a vyšetřením rozsáhlých intragenových přestaveb. Současně však, aby nedošlo k časové prodlevě, je okamžitě provedeno klinické a laboratorní vyšetření dítěte a je zahájena komplexní preventivní péče.
V případě hraničního výsledku potního testu jsou zjišťovány mutace genu CFTR a další postup pak závisí na výsledcích molekulárně genetického vyšetření.
Od zavedení NSCF v roce 2009 bylo do konce roku 2018 vyšetřeno celkem 1 037 308 novorozenců. Protokol IRT/DNA//IRT ve sledovaném období odhalil celkem 160 pacientů s CF (tab. 1). NSCF je ale pouze screeningovou metodou a tak se na jeho výsledky nelze stoprocentně spoléhat, neboť může být za určitých situací falešně negativní. Od jeho zavedení se podle příznaků manifestovalo 18 pacientů s negativními výsledky screeningu. Proto je na CF nutno stále myslet i u těch dětí, které NSCF prošly jako negativní, a v případě, že mají jakékoliv příznaky vzbuzující podezření na toto onemocnění, indikovat vyšetření potním testem.
CFSPID
Zavedení NSCF vedlo k záchytu novorozenců, u nichž je toto screeningové vyšetření pozitivní, ale s nejednoznačným výsledkem diagnostického testování na CF [4‒8]. V Evropě se začal pro tuto skupinu používat termín CFSPID (CF Screen Positive, Inconclusive Diagnosis).
Do skupiny CFSPID patří ti jedinci s pozitivním NSCF, kteří jsou asymptomatičtí a
- a) vykazují hraniční hodnotu koncentrace chloridů v potu (30–59 mmol/l) a současně žádnou či pouze jednu mutaci způsobující CF (mohou být ale přítomny až 2 varianty CFTR genu s nejasným klinickým významem),
nebo
- b) vykazují normální hodnotu chloridů v potu (pod 30 mmol/l) a současně dvě mutace genu CFTR, z nichž alespoň jedna má nejasný klinický význam.
Mezi mutace s nejasným klinickým významem patří např. R117H;7T, D1152H a 5T, vyšetřované v rámci komerčního kitu.
V roce 2015 byla publikována konsenzuální doporučení ohledně sledování jedinců s CFSPID [5], podle nichž byl v roce 2016 brněnskými kolegy (Holčíková, Homola, Gaillyová) navržen Standardní operační postup (SOP) pro CFSPID, který byl schválen v listopadu 2016 na celostátním multioborovém setkání specialistů na CF.
Ze SOP jednoznačně vyplývá, že CFSPID jedince nelze na základě výsledků NSCF považovat za nemocné, ale také nelze zcela vyloučit rozvoj příznaků CF v pozdějším věku.
Ze SOP vyplývá následující doporučení postupu při záchytu CFSPID jedince:
- sledovat,
- poučit rodinu,
- v případě potřeby překlasifikovat na jedince s CF a léčit.
Sledování by mělo probíhat v centrech pro cystickou fibrózu. Kontroly jsou vhodné 1x ročně do 6 let věku, a pokud jedinec nejeví příznaky onemocnění, tak poté až ve 12, 18, 24 a 30 letech.
Pokud není stanoveno jinak, má každá kontrola zahrnovat [5]:
- anamnézu s ohledem na možné příznaky CF; zvláštní pozornost je třeba věnovat kašli vyskytujícímu se déle než 2 týdny,
- klinické vyšetření,
- měření, vážení,
- vyšetření elastázy ve stolici,
- vyšetření plicních funkcí u spolupracujících dětí,
- kultivační vyšetření (krk, odsátý sekret, sputum),
- kontrolní potní test (provádí se v 1 a 2 letech věku [5], dále vždy při zvýšeném podezření na rozvoj CF).
Krevní odběry a zobrazovací metody jsou indikované pouze na základě příznaků [5]. Pokud do 30 let věku nedojde k rozvoji jakýchkoliv příznaků CF, sledování se ukončí.
Stejným způsobem je třeba sledovat i sourozence CFSPID dětí, pokud u nich vyšetřením koncentrace chloridů v potu a molekulárně genetickým vyšetřením zjistíme stejné odchylky (a to i když u nich byl NSCF negativní).
U rodičů by se neměly vzbuzovat zbytečné obavy, ale je nutné je poučit o ev. příznacích CF (zejména neprospívání, chronické řídké stolice, chronické respirační příznaky) a o nutnosti tyto příznaky došetřit v centru CF. Dále je třeba zdůraznit nevhodnost kouření ve společné domácnosti, potřebu běžného očkování a vhodnost pravidelného očkování proti chřipce, dále vhodnost zdravého životního stylu. Dětem s CFSPID a hraniční hodnotou koncentrace chloridů v potu se nemá ve stravě redukovat příjem soli, jak je obecně v populaci doporučováno.
Rodině CFSPID jedince je v případě záchytu minimálně jedné patogenní mutace doporučeno genetické poradenství.
PŘEKLASIFIKOVÁNÍ Z CFSPID NA CF
K překlasifikování CFSPID na CF se přistupuje, pokud se objeví příznaky CF (po vyloučení jiných příčin), pokud se zvýší množství chloridů v potu, či pokud se prokážou 2 patogenní mutace [9]. K přehodnocení na CF stačí průkaz insuficience zevní sekrece pankreatu. U respiračních symp-tomů je zhodnocení, zda ještě patří nemocnost k běžné populační nemocnosti, obtížné. Je známo, že i u zdravých dětí cca od 3 do 6 let věku jsou obvyklé a frekventní nekomplikované katary horních cest dýchacích (KHCD), zejména navštěvují-li kolektivní zařízení.
Z hlediska CF jsou podezřelé respirační infekty, které dlouho perzistují, jsou provázeny dlouhodobým zahleněním a vyžadují opakovaně antibiotika. Velmi podezřelý je také kultivační záchyt Pseudomonas aeruginosa ze sekretu dýchacích cest. Pokud dojde k překlasifikování na CF, často pak průběh bývá mírnější než u klasické CF. V případě překlasifikování na CF by měla léčba probíhat individualizovaně, cíleně na příznaky.
VÝSLEDKY SLEDOVÁNÍ PACIENTŮ S CFSPID V ČESKÉ REPUBLICE
Od zavedení NSCF bylo do konce roku 2018 v rámci celé ČR identifikováno a zařazeno 59 jedinců do kategorie CFSPID (tab. 1), na pracovišti FN Motol bylo pod označením CFSPID vedeno 35 jedinců, na brněnském pracovišti jich bylo 24. Sedm jedinců bylo z původní kategorie CFSPID později přeřazeno do skupiny s CF. Při vyhledávání anamnestických a klinických dat CFSPID jedinců bylo zjištěno, že část z nich není pravidelně sledována (většinou proto, že se na doporučenou ambulantní kontrolu nepřihlásili).
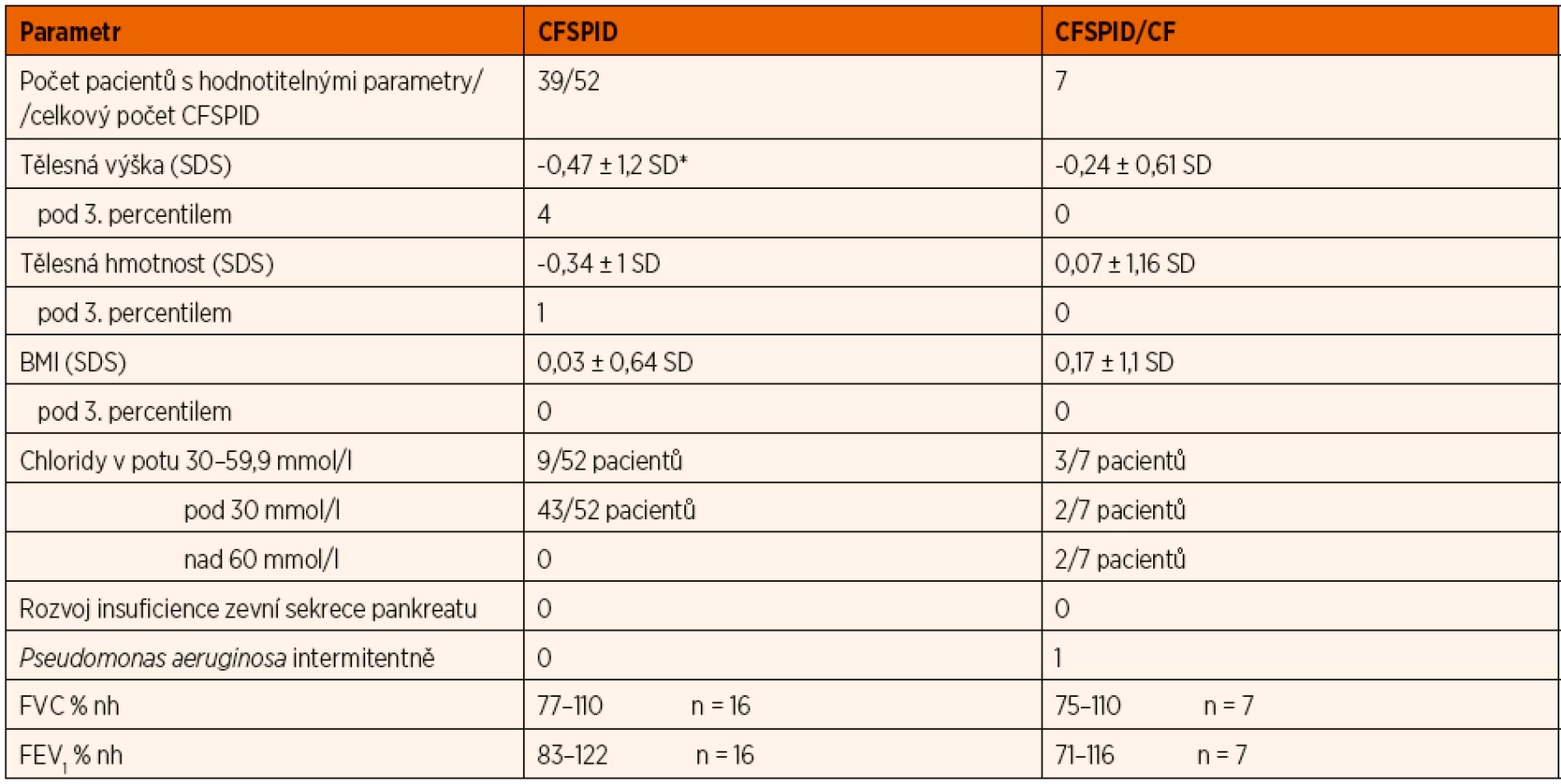
Hodnocení parametrů výživy bylo možné u 38 jedinců s CFSPID a všech 7 pacientů s CFSPID přeřazených postupně do CF skupiny. U plicní funkce to bylo pouze 16 pacientů s CFSPID a všech 7 pacientů s CFSPID/CF. Jejich charakteristiku uvádí tabulka 2.
Z 52 pacientů, kteří zůstávají v kategorii CFSPID, mělo 43 při prvním vyšetření koncentraci chloridů v potu v rozmezí do 30 mmol/l, 9 sledovaných mělo hraniční hodnotu (30–60 mmol/l) koncentrace chloridů v potu. Čtyři sledovaní s původní hodnotou koncentrace chloridů v potu pod 30 mmol/l se při kontrolním vyšetřením dostali na hraniční hodnotu.
Čtyřicet devět sledovaných jedinců s CFSPID má dvě mutace genu CFTR, z nichž jedna má nejasný klinický význam. Další 3 pacienti mají hraniční koncentraci chloridů v potu a jen jednu identifikovanou patogenní mutaci genu CFTR (R1162X, N1303K a F508del). Všichni sledovaní pacienti s CFSPID mají zachovalou funkci zevní sekrece pankreatu.
Stav výživy všech sledovaných CFSPID i těch, kteří později byli přeřazeni do skupiny CF, se neliší od zdravých dětí. Pouze pacienti s CFSPID mají poněkud nižší tělesnou výšku (význ. na 5% hladině). Domníváme se, že je to způsobeno relativně malou skupinou pacientů a poněkud vyšším zastoupením dvojčat s nižší porodní hmotností.
Mezi pacienty s CFSPID a CFSPID/CF nejsou významné rozdíly ani ve funkci plic. Počet pacientů v CFSPID skupině, kteří mají spolehlivě provedené vyšetření plicní funkce, je ale malý. Podílí se na tom jednak fakt, že pacienti nechodí na doporučované vyšetření, a jednak jejich relativně nízký věk.
Pokud se provádělo mikrobiologické vyšetření, jednalo se většinou o výtěry z krku a nosu (vzhledem k tomu, že děti nebyly zahleněné a nevykašlávaly). U vyšetřovaných dětí se vyskytoval ojediněle Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, ev. Moraxella. Chronická kolonizace či infekce nebyly prokázány u žádného ze sledovaných CFSPID dětí. Jeden sledovaný chlapec, s normální hodnotou chloridů v potu a genotypem F508del/R117H;7T, měl 1x přechodně zvýšené jaterní testy s hodnotami ALT 0,89 µkat/l, AST 1,26 µkat/l, sonografický nález mírné difuzní léze parenchymu jater s pozdější normalizací a jednou se u něj objevila elevace amylázy (2,94 µkat/l) a lipázy (6,17 µkat/l). Chlapec je bez gastrointestinální symptomatologie.
Deset z pravidelně sledovaných dětí s CFSPID vykazuje mírnou respirační symptomatologii – občasné nekomplikované infekty horních cest dýchacích či ojedinělé obstrukční bronchitidy v raném věku. Jeden sledovaný prodělal v roce a půl nekomplikovanou ambulantně léčenou bronchopneumonii (chloridy v potu 16 mmol/l, F508del/R117H;7T). Jedna dívka s koncentrací chloridů v potu 11 mmol/l a genotypem F508del/L333F byla ve 2 měsících věku hospitalizována a léčena intravenózní antibiotickou terapií pro pravostrannou pneumonii. Trpí recidivujícími obstrukčními bronchitidami a je léčena inhalačními kortikoidy.
PACIENTI PŘEKLASIFIKOVANÍ Z KATEGORIE CFSPID NA CF
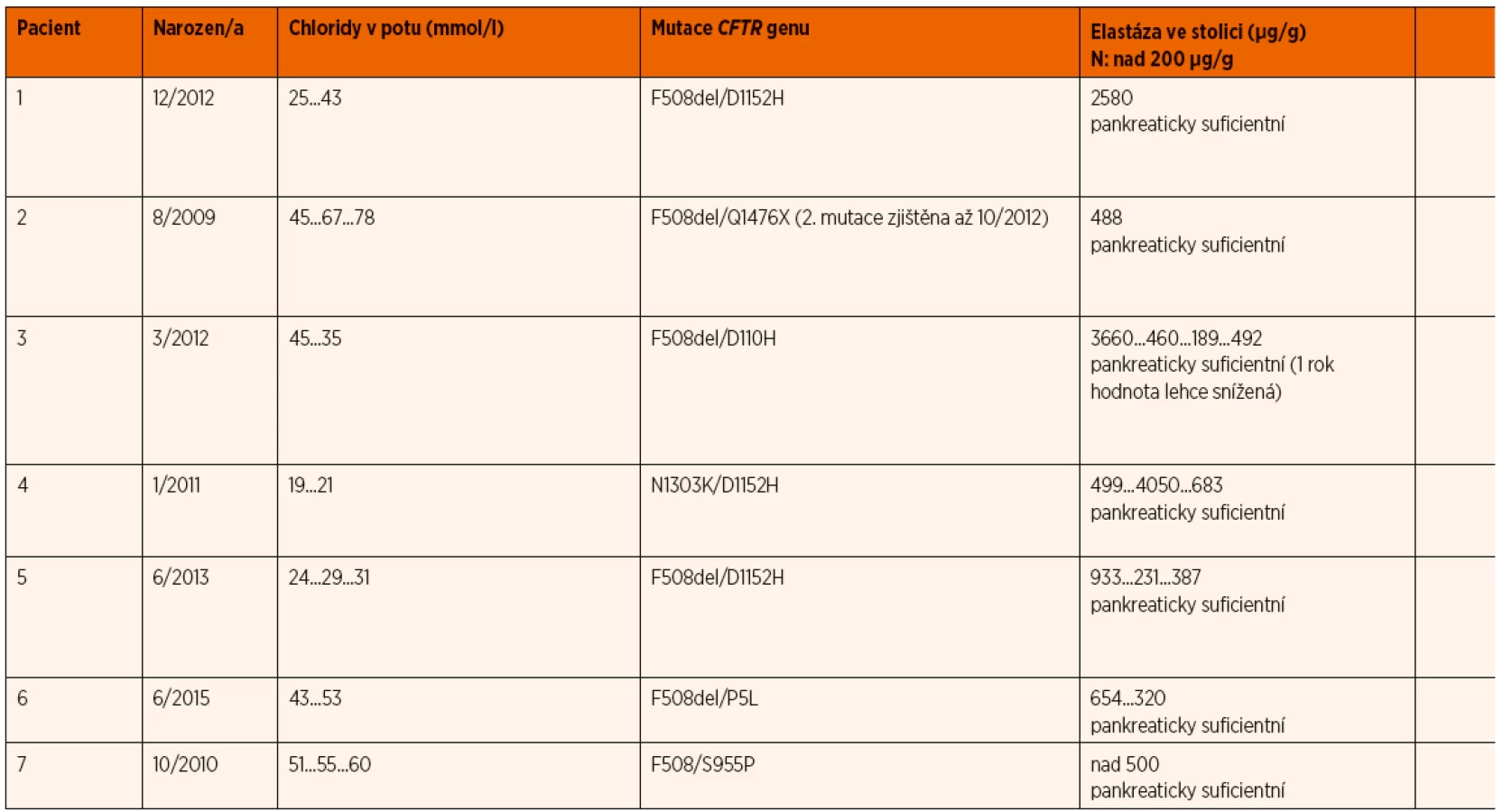
Tři sledované děti byly do kategorie CF přeřazeny z důvodu klinických příznaků a čtyři z důvodu nálezu druhé klinicky významné mutace genu CFTR.
Charakteristiku 7 dětí překlasifikovaných z kategorie CFSPID na CF uvádí tabulka 3.

DISKUSE
Označení CFSPID se začalo používat v roce 2015 v návaznosti na revizi evropského konsenzu pro nejasnou diagnózu. Protokoly NSCF se v jednotlivých zemích velmi liší. Kontinuální dlouhodobé sledování jedinců s CFSPID často chybí, nebo je intenzita jejich vyšetřování daleko nižší než u pacientů s CF.
Většině našich pacientů s CFSPID se daří relativně dobře, nejeví jasné známky onemocnění CF. Z dětí, které byly překlasifikovány na jedince s CF, trpí jeden chlapec (jenž má současně DiGeorgův syndrom) recidivujícími respiračními infekty horních i dolních cest dýchacích, které s věkem spíše ustupují, pravděpodobně i v souvislosti s pravidelným podáváním substituce imunoglobulinů. Další pacient, dívka, má recidivující katary horních cest dýchacích a ve věku deseti let na HRCT (výpočetní tomografie s vysokým prostorovým rozlišením) mírné zesílení stěny centrálních bronchů a zvýraznění intersticia. Ostatní jsou bez klasických projevů nemoci. Rozhodně není tedy vhodné rodiny CFSPID pacientů stresovat, na druhou stranu je jistě vhodné doporučovat trvalé sledování v CF centru a poučit rodiče o možných příznacích CF, které by se mohly objevit a při nichž by měli kontaktovat lékaře.
ZÁVĚR
Vyšetření NSCF je velkým přínosem k včasné diagnostice a tudíž okamžitému zahájení léčby CF. Ne vzácně je však jeho výsledek nejednoznačný. Skupina dětí označovaná jako CFSPID má přesně určená diagnostická kritéria. Tyto děti mají riziko rozvoje příznaků CF s nutností pravidelné léčby. Dlouhodobé sledování v CF centru je proto nezbytné a je třeba dbát na jeho dodržování.
Dále je potřeba stále zdůrazňovat, že je nutné pomýšlet na onemocnění CF i v době NSCF, protože výsledek NSCF může být falešně negativní. Při jakémkoliv podezření je tedy třeba vždy provést potní test.
Podpořeno projektem koncepčního rozvoje výzkumné organizace 00064203 FN Motol Ministerstva zdravotnictví ČR.
MUDr. Jana Bartošová
Pediatrická klinika 2. LF UK
a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: janbar@seznam.cz
Sources
1. Ren CL, Fink AK, Petren K, et al. Outcomes in infants with indeterminate diagnosis detected by cystic fibrosis newborn screening. Pediatrics 2015; 135 (6): 1387–1392.
2. Kharrazi M, Yang J, Bishop T, et al. Newborn screening for cystic fibrosis in California. Pediatrics 2015; 136 (6): 1062–1072.
3. Votava F, Kožich V, Chrastina P, et al. Výsledky rozšířeného novorozeneckého screeningu v České republice. Čes-slov Pediat 2014; 69 (2): 77–86.
4. Ooi CY, Castellani C, Keenan K, et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics 2015; 135 (6): 1377–1385.
5. Munck A, Mayell SJ, Winters V, et al. Cystic fibrosis screen positive, inconclusive diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros 2015; 14 (6): 706–713.
6. Groves T, Robinson P, Wiley V, et al. Long-term outcomes of children with intermediate sweat chloride values in infancy. J Pediatr 2015; 166 (6): 1469–1474.
7. Ren CL, Borowitz DS, Gronska T, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr 2017; 181S: 45–51.
8. Terlizzi V, Mergni G, Buzzetti R, et al. Cystic fibrosis screen positive, inconclusive diagnosis (CFSPID): Experience in Tuscany, Italy. J Cyst Fibros 2019; 18(4):484–490.
9. Levy H, Nugent M, Schneck K, et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin Genet 2016; 89 (5): 539–549.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2019 Issue 7
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Novorozenecký screening cystické fibrózy a diagnostika CFSPID
- Vyšetrenie rotačnej tromboelastometrie (ROTEM) v manažmente krvácania a koagulopatie v pediatrii
- Historie cystické fibrózy u nás – editorial
- Alergická bronchopulmonálna aspergilóza u detských pacientov s cystickou fibrózou