
Diagnostika, léčba a urologické komplikace kloakálních malformací
Diagnosis, therapy and urological complications of cloacal malformations
Major Statement: A review article about the diagnostic tools and management of persistent cloaca malformation that belongs to one of the most complex new-born anomalies with a need of the life-long follow-up by pediatric and general urologist. The majority of cloaca patients undergo multiple reconstructive procedures.
Cloacal malformation is the most severe form of anorectal malformation in girls. The complex treatment is possible only in specialized pediatric centers and life-long follow-up is needed. Females with persistent cloaca suffer also from many associ‑ated anomalies, especially urinary tract and spinal malformations. Correct anatomical and functional reconstruction of persistent cloaca belongs to one of the most challenging tasks in pediatric surgery and urology. The goal of the complex therapy is to help patients achieve urinary and fecal continence, regu‑lar menstruation at puberty and possibility of good quality of sexual life in adulthood. In this review article based on the current literature and our own experi‑ence we describe the spectrum of cloaca malforma‑tions and associated anomalies, possible techniques of reconstruction and expected outcomes after the treatment by multiple specialist team. For achieving urinary continence these patients often need several urology procedures. However, despite the complex treatment half of the cloaca girls will develop chronic kidney disease before achieving adulthood and there‑fore detailed and long-term follow-up by dedicated pediatric and general urologist is mandatory.
Keywords:
Cloacal malformation – hydrocolpos – mobilisation of urogenital sinus.
Authors:
Jan Trachta 1; Martin Kynčl 2; Richard Škába 1
Authors‘ workplace:
Klinika dětské chirurgie 2. LF UK a FN Motol, Praha
1; Klinika zobrazovacích metod 2. LF UK a FN Motol, Praha
2
Published in:
Ces Urol 2019; 23(1): 19-29
Category:
Review article
Overview
Hlavní stanovisko práce: Přehledový článek o diagnostice a terapii komplexní vrozené vady, která má pro pacienty celoživotní následky a vy‑žaduje trvalé sledování jak dětským, tak dospělým urologem. Velká část pacientek s kloakou potřebuje opakované rekonstrukční operace.
Kloakální malformace je závažnou formou anorek‑tální malformace u dívek, která vyžaduje komplexní péči ve specializovaných centrech a celoživotní sle‑dování. Dívky s perzistující kloakou mají navíc vyso‑kou incidenci přidružených vrozených vad, nejčastěji uropoetického traktu, páteře a míchy. Anatomická a funkční rekonstrukce kloakální malformace patří
mezi nejtěžší operace v dětské chirurgii a urologii vůbec. Cílem terapie je zajistit dívkám kontinenci moči a stolice, volný odtok menstruační krve, možnost pohlavního styku a kvalitního sexuálního života v do‑spělosti. V tomto přehledovém článku popisujeme na základě literatury a vlastních zkušeností diagnostiku různých typů kloakální malformace a přidružených vad, možnosti rekonstrukčních technik a očekávané výsledky multioborové péče. Pro dosažení kontinen‑ce moči jsou někdy nutné i vícečetné urologické rekonstrukční výkony. Vzhledem k tomu, že se přes veškerou lékařskou péči přibližně u poloviny paci‑entek rozvine před dosažením dospělosti chronické ledvinné selhání, vyžaduje jejich léčba a sledování ze strany dětských a následně dospělých urologů velké osobní a odborné nasazení.
Klíčová slova:
Kloakální malformace – hydrocolpos – mobilizace urogenitálního sinu
ÚVOD
Kloakální malformace (KM) je jedna z nejvážnějších vrozených vad vůbec. Je to samostatná forma anorektální malformace (ARM) postihující výhradně dívky. U novorozených pacientek nacházíme společné vyústění močové trubice, pochvy a rekta do jednoho vývodu, kloakálního kanálu, který ústí na perineu. Anus není vytvořen a otvorem mezi neúplně vytvořenými stydkými pysky odchází stolice i moč (Obrázek 1). Incidence KM je 1 : 50 000 živě narozených a v České republice se tak narodí jedna dívka s touto vadou ročně (1, 2, 3). Jakkoliv je to vada vzácná, znamená pro postižené dítě řadu operací a celoživotní sledování, mimo jiné i dětským a posléze dospělým urologem.
Fig. 1. A neonate with a single perineal orifice between
the labia minora; day 2 after delivery and creation
of sigmoidostomy, vesicostomy, and vaginostomy
for hydrocolpos.
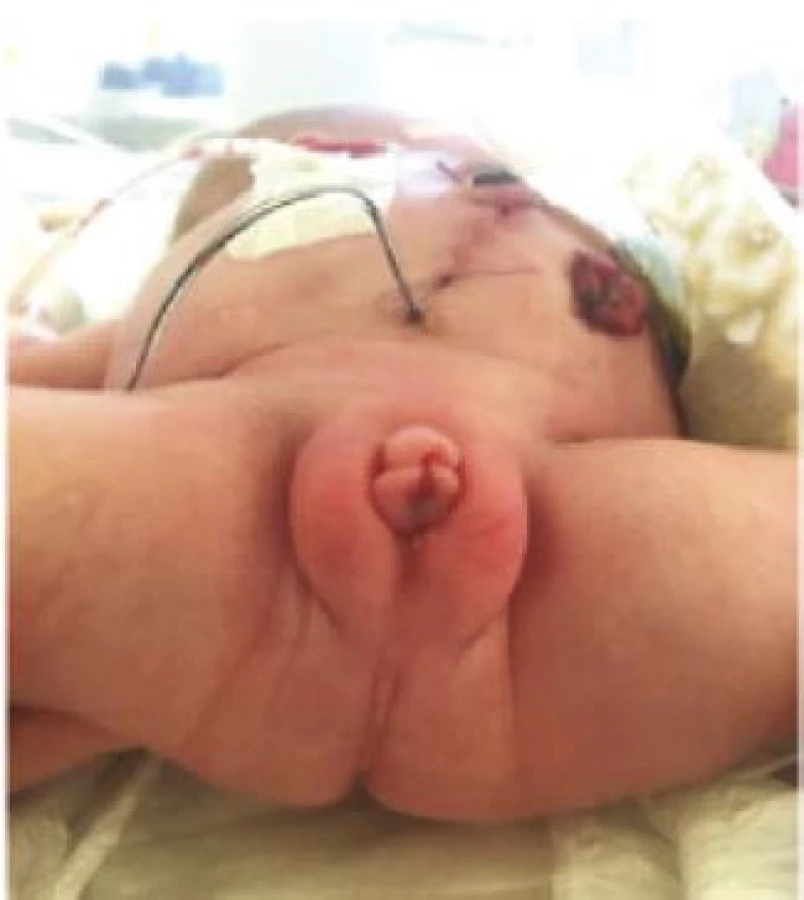
KM neboli perzistující kloaka by neměla být zaměňována za exstrofii kloaky, což je jiný typ vady, kterému dominuje defekt břišní stěny a genitálu, široký rozestup symfýzy, omfalokéla a protruze terminálního ilea skrze rozpolcené cékum vrostlé mezi oddělené poloviny měchýře. KM se zejména velmi často pojí s dalšími vrozenými vadami. Nejčastěji s vadami uropoetického traktu.
Přibližně 60 % pacientek s KM (33 až 83 %) má přidruženou agenezi ledviny, podkovovitou ledvinu, zdvojený anebo ektopický ureter, ureterokélu, obstrukční megaureter nebo veziko-ureterální reflux a to jak primární, tak sekundární (4, 5). Dále se KM sdružují s vadami vnitřních pohlavních orgánů – septovanou nebo zdvojenou vagínou, septovanou nebo zdvojenou dělohou, případně agenezí pochvy a to asi u 40 % pacientek (5). Zdvojená pochva může být symetrická nebo asymetrická s jednou z pochev výrazně menších. Jestliže je jedna z pochev menší a slepá, může snadno uniknout popisu na zobrazovacích metodách a projeví se až v pubertě retrográdní menstruací do břišní dutiny, kdy menší množství krve z hypoplastické dělohy způsobuje pravidelné bolesti břicha. Z přidružených vad gastrointestinálního traktu je nejčastější atrézie jícnu a tracheoezofageální píštěl v 10 %, dále i atrézie jiných částí gastrointestinálního traktu nebo duplikatury (4). Důležitá je včasná diagnóza srdečních vad.
Některé pacientky se rodí s mnohačetnými vadami sdruženými v tzv. VATER nebo VACTERL syndrom (podle anglického označení jedno tlivých vad, které mají tendenci se vyskytovat společně: Vertebral, Anorectal, Cardiovascular, Tracheoesophageal fistula, Esophageal atresia, Renal/Radial, Limb anomalies). Prognosticky důležité pro vznik neurogenní dysfunkce dolních močových cest jsou přidružené skryté rozštěpové vady páteře a míchy (fixovaná mícha, sakrální malformace – nejvážnější je parciální anebo úplná ageneze sakra a vady označované v anglické literatuře jako „spina bifida“ – meningokéla, myelomeningokéla, lipomyelomeningokéla, aj.). Malformaci sakra má kolem 50 % pacientek s KM (3). Jestliže se s KM pojí i parciální nebo totální ageneze sakra, případně i ageneze lumbálních obratlů, spolu s hypoplastickým pánevním dnem, mluvíme o syndromu kaudální regrese. Syndrom kaudální regrese má nepříznivou funkční prognózu a derivace stolice není řešitelná jinak, než trvalou kolostomií.
Kloaka je u správně se vyvíjejícího embry normální přechodná fáze vývoje zadního střeva a alantois mezi třetím a pátým gestačním týdnem. V šestém týdnu se kloaka rozdělí vrůstající mesenchymální přepážkou zvanou urorektální septum na urogenitální sinus ventrálně a rektum dorzálně. U dívky se urogenitální sinus dále po kontaktu s fúzujícími Mulleriánskými vývody rozděluje ventrálně na měchýř, uretru a vestibulum vaginae a dorzálně dává základ distálním dvou třetinám pochvy, zatímco kraniální třetina pochvy a děloha se formují ze zmíněných Mulleriánských vývodů
Chyba v tomto embryonálním dělení může vést ke vzniku buď perzistující kloaky anebo ke vzniku perzistujícího urogenitálního sinu. Perzistující urogenitální sinus vídáme u novorozených dívek buď jako samostatnou patologickou jednotku anebo jako součást nejčastější formy poruchy pohlavního vývoje – kongenitální adrenální hyperplazie.
VYŠETŘENÍ A PÉČE O NOVOROZENCE S KM
KM lze diagnostikovat v prenatálním období v rámci sonografického screeningu, což se úspěšně daří asi u poloviny plodů s KM (8). Diagnóza KM by měla být zvažována u plodů ženského pohlaví v případě bilaterální dilatace ledvinných kalichů a pánvičky, obtížně zobrazitelném měchýři a mono či bilobární cystické struktuře v malé pánvi (6, 7). 30 až 50 % plodů s KM má totiž významný hydrocolpos, tj. rozšířenou, distendovanou pochvu, močí, stolicí a hlenem, která utlačuje hrdlo močového měchýře a působí sekundární dilataci horních močových cest (5, 8). Ascites může mít plod v důsledku retrográdního toku moči z měchýře přes pochvu, dělohu a tubu až do dutiny břišní. K upřesnění prenatální diagnózy a anatomických poměrů vnitřních orgánů plodu, stejně tak jako prognózy plodu po narození, doporučujeme MRI plodu, které je indikováno obvykle mezi 20. a 26. týdnem těhotenství.
Vzhledem k ojedinělosti a závažnosti této vrozené vady považujeme za vhodné, aby MRI plodu bylo provedeno již na specializovaném pracovišti, schopném tuto vadu řešit postnatálně. Ve stejném centru, kde bude MRI plodu provedena, jsme pak schopni zajistit i adekvátní prenatální konzultaci s jedním nebo oběma rodiči a naplánování porodu. Součástí prenatální konzultace je i diskuze o možnosti umělého přerušení těhotenství. KM patří dle přílohy zákona č. 66/1986 Sb. O umělém přerušení těhotenství mezi závažné dědičné choroby a vývojové vady diagnostikované u plodu metodami prenatální diagnostiky, u kterých může lékař nabídnout matce interupci do 24. týdne těhotenství. Vadu ovšem považujeme za postnatálně korigovatelnou, a proto naše doporučení k přerušení těhotenství z medicínských důvodů není nijak silné. Pakliže je vysloveno podezření na KM až po porodu, je vhodné odeslat novorozence schopného převozu do specializovaného centra ihned.
Pro co nejlepší postnatální operační korekci je vhodné, aby byl operatér zběhlý v řešení všech těchto vad jak po teoretické, tak i praktické stránce a pacientky řešil v týmu, kde je zastoupen dětský proktolog, urolog a gynekolog. Ve specializovaných centrech je k dispozici kromě výše uvedených specialistů i neonatologická, kardiologická, nefrologická, neurochirurgická, endokrinologická a genetická péče. Ideálně pak dokáže neonatologický tým zorganizovat i psychologickou podporu rodičů a v případě zájmu i jejich propojení s dalšími rodinami s dětmi s podobným onemocněním.
Cílem terapie je zajistit pacientce do života kontinenci moči a stolice, buď na základě spontánního vyprazdňování, nebo s pomocí čisté intermitentní katetrizace (ČIK) a pravidelných klyzmat. Dále možnost pravidelné menstruace v pubertě a pohlavního styku v dospělosti s možností otěhotnět. Vzhledem k tomu, že jsou popsány vzácné případy otěhotnění dospělých pacientek s KM a porodů zdravých plodů, není hysterektomie běžně indikována (4).
Při fyzikálním vyšetření po porodu je typický jediný otvor mezi labií – vyústění kloakálního kanálu a chybění řitního otvoru. Genitál má menší nebo nevyvinuté malé stydké pysky a relativně větší klitoris, což může budit dojem poruchy pohlavního vývoje. Celkový vzhled perinea je důležitý pro odhad vnitřní anatomie a funkční prognózy. „Příznivé“ perineum má ve střední čáře viditelnou kožní rýhu, dorzálně zakončenou důlkem, tvořeným subkutánně dobře vyvinutým zevním svěračem anu. „Nepříznivé“ perineum má hladkou, vypjatou kůží bez rýhy a důlku a přechází v oploštěnou gluteální krajinu, jež je známkou parciální nebo kompletní ageneze sakra. Při vyšetření si všímáme případného vyklenutí stěny břišní dilatovanou pochvou (hydrocolpos) anebo dilatovaného rektálního pahýlu. Sekundární respirační insuficience urychluje nutnost drenáže hydrocolpos a derivace kolon.
Dítě s KM obvykle není nutné ihned po porodu operovat. Naopak je s výhodou výkon odložit a ponechat novorozenci prvních 24 až 48 hodin k adaptaci na zevní prostředí, pacientku dovyšetřit a k operaci připravit. Pacientce by se měla zavést kanyla k intravenóznímu podávání tekutin a širokospektrých antibiotik. Brzy po porodu je vhodná echokardiografie k vyloučení hemodynamicky závažných srdečních vad. V případě, že nelze volně zavést nazogastrická sonda do žaludku, je třeba provést nástřik sondy kontrastní látkou a potvrdit diagnózu přidružené atrézie jícnu. Je-li dítě snímkováno, pak je spolu s předozadním snímkem hrudníku a břicha indikován i laterální snímek k zobrazení celé páteře a vyloučení anomálie obratlů.
Peňa a Levitt v souboru 490 pacientek s KM (5) doporučují provést co nejdříve i ultrazvuk břicha a retroperitonea právě k vyloučení hydrocolpos a sekundární obstrukce močových cest. Hydrocolopos utlačující hrdlo a trigonum měchýře je nejčastější nerozpoznanou patologií, mylně považovanou za přeplněný močový měchýř (obr. 2). Pacientky tak nezřídka podstupují zbytečnou derivaci močových cest – vezikostomii, ureterostomie nebo nefro stomie – tam, kde stačí derivovat hydrocolpos. Na druhou stranu u pacientek s dlouhým kloakálním kanálem a přidruženou malformací lumbosakrální páteře, která je nepřímou známkou neurogenní dysfunkce, nemusí vést úspěšná derivace hydrocolpos ke spontánní mikci a úpravě dilatace horních močových cest. V tomto případě je třeba derivaci močových cest provést a založit punkční epicy stostomii nebo, u novorozenců s dysplazií ledvin a elevací plazmového kreatininu, vezikostomii. Je ovšem řada pracovišť, která primárně preferují přechodnou vezikostomii u všech KM, vzhledem k možným komplikacím spojeným s punkční epicystostomií, jako je neprůchodnost katétru, jeho dislokace anebo bakteriální osídlení a větší riziko infekce močových cest.
Fig. 2. Hydrocolpos and common channel cloaca.
(Adapted from Levitt MA, Peña A. Cloacal malformations:
lessons learned from 490 cases. Semin Pediatr
Surg. 2010; 19(2): 128–138)
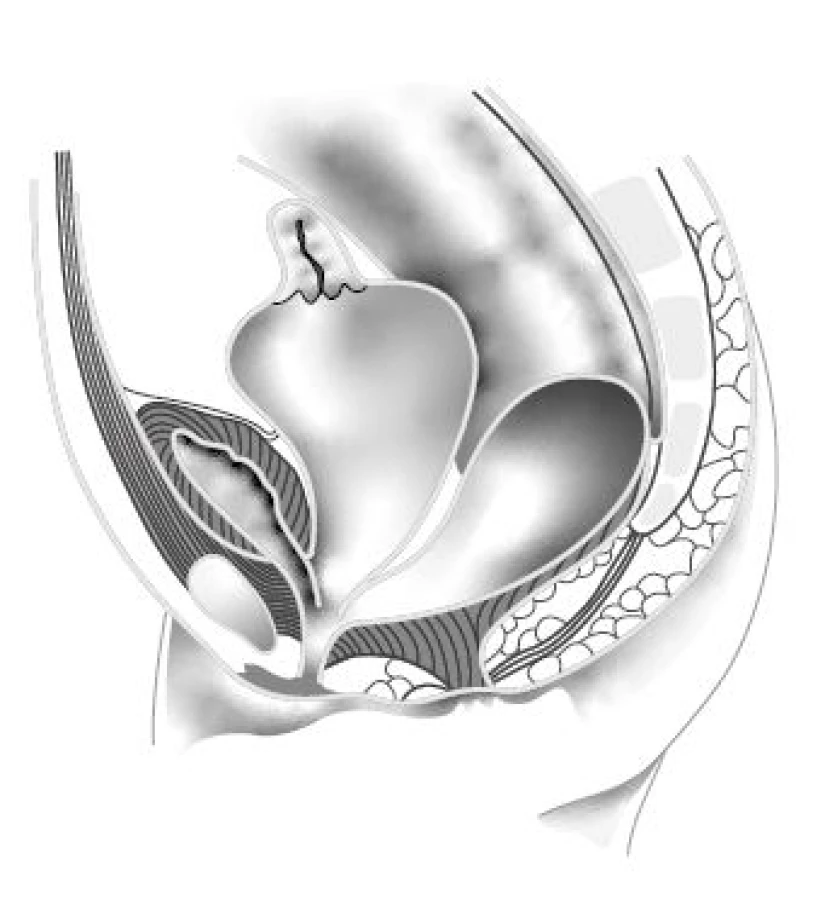
Levitt ve zmíněném článku dále píše: „Důvod proč dilatovaná pochva retinuje hlen, moč a jinou tekutinu, je záhada, protože kloakální kanál není nikdy atretický. Pravděpodobně vytváří poševní ústí v místě kloakálního kanálu chlopňový mechanizmus, který při větší náplni neumožňuje odtok retinované tekutiny.“ A dodává, že většina pacientek s hydrocolpos má dilatovanou pochvu z větší části septovanou a s dvěma nasedajícími dělohami (5). Jestliže se nerozpoznaná, včas nederivovaná hydrocolpos infikuje, vzniká pyocolpos, ohrožující dítě na životě. Součástí postnatálního UZ vyšetření by měl být také UZ lumbosakrální páteře a míchy, který první tři mě‑síce věku dokáže přes málo osifikované obratle výborně znázornit míšní morfologii.
V závažných případech KM bývá na UZ vidět kromě dilatace horních močových cest i redukce parenchymu ledvin, setření kortiko-medulární diferenciace a dysplastické změny parenchymu v podobě drobný cyst. Součástí monitorace takových novorozenců je opakované vyšetřování renálních funkcí a to s vědomím, že u donošených je první týden po porodu hladina plazmatického kreatininu zvýšená i kreatininem mateřským, u nedonošených i dva až tři týdny po porodu. U novorozenců s hypodysplastickými změnami ledvinného parenchymu a vyšším kreatininem volíme přechodnou vezikostomii jako nejúčinnější a nejbezpečnější derivaci močových cest.
Po objasnění základní anatomie je cílem terapie novorozence s KM derivace gastrointestinálního traktu kolostomií a derivace uropoetického traktu punkční epicystostomií nebo vezikostomií tam, kde je indikována. Derivace přítomného hydrocolposu je možná třemi způsoby. Obdobou punkční epicystostomie, tzn. transabominální zavedení silnějšího katétru (Cystofix 10F, Foley a jiné typy). U velmi objemných retencí lze vyšít pochvu jako nástěnnou stomii do střední čáry mezi pupek a symfýzu (obdoba vezikostomie).
Třetí možnost derivace hydrocolpos preferuje Rink et al. Místo transabdominální derivace zaučuje matku dítěte v čisté intermitentní kate trizaci kloakálního kanálu (9). ČIK až do definitivní operace preferuje Rink proto, že u většiny pacientek zaváděná cévka vklouzne přirozeně nikoliv do měchýře, který je spolu s uretrou dislokován ventrálně, ale právě do dilatované pochvy, kde se tekutý poševní sekret hromadí. Nácvik ČIK má pak výhodu i v tom, že si ji rodiče osvojí jako součást rutiny v péči o dítě, což se ukazuje jako velmi užitečný návyk pro pravidelnou evakuaci v případě neurogenního měchýře s trvale poškozenou schopností evakuace moči. Oponenti tvrdí, že ne vždy se katétr dostane správně do dilatované pochvy a její intermitentní derivaci tak považují za nespolehlivou (10).
Založení kolostomie u pacientů s ARM a její typ prodělal za posledních 60 let vývoj od „loop“ transversostomie, která je technicky nenáročná, až po dělenou sigmoideostomii dle Peňi, která je v současnosti standardem. Vyšít přívodnou kličku stomie v místě, kde colon descendens opouští retroperitoneální závěs, je výhodné pro prevenci prolapsu a zároveň zbývá dostatečná délka distálního pahýlu, vyšitého ke kůži jako mukózní píštěl, k tomu, aby mohl operatér během definitivní rekonstrukční fáze provést stažení dilatovaného rekta na hráz a bez napětí všít neoanus do svaloviny zevního svěrače.
Důvodem preference dělené stomie před typem „loop“ je i bezpečné vyloučení kontaminace odvodné kličky stolicí a zvýšené riziko infekcí močových cest přes společný kloakální kanál (11, 12). U transverzostomie je také popsán častější výskyt hyperchloremické acidózy a následné neprospívání kojenců z důvodu nepozorovaného retrográdního toku moči do distálního pahýlu kolon přes rektokloakální píštěl. Do zadržované moči se střevní sliznicí secernuje bikarbonát a vý‑měnou resorbují chloridové anionty (9).
DEFINITIVNÍ REKONSTRUKCE
Novorozenec s KM zajištěný funkční kolostomií, derivací hydrocolpos a urotraktu, může být propuštěn domů s plánem definitivního výkonu. Před rekonstrukční operací je třeba doplnit endoskopické vyšetření s nástřikem kloakálního kanálu kontrastní látkou – tzv. genitogram a MRI s náplní všech tří oddílů malformace fyziologickým roztokem pro jejich lepší zobrazení.
Výhodou MRI je zobrazení přidružených malformací páteře a míchy a zachycení případné hypoplazie svalstva pánevního dna, které hraje zásadní roli v kontinenci stolice. Navíc software dnešních MRI přístrojů umožňuje 3D rekonstrukci orgánů malé pánve, která je přehlednější než klasický genitogram kontrastní látkou. MRI navíc nevyžaduje nutně celkovou anestezii a do tří až čtyř měsíců věku ho provádíme technikou „feed and wrap“, tzn. nakojení těsně před vyšetřením, těsné zabalení kojence do zavinovačky a opatrné připoutání ke stolu pásy tak, aby 30 až 40 minut vyšetřování bez hnutí prospal.
Tato vyšetření lze provést během samostatné hospitalizace anebo po přijetí k definitivnímu výkonu. V načasování definitivní operace se jednotlivá centra liší. Jestliže dítě nepotřebuje přednostně korekci jiné vady, typicky srdeční, operuje Levitt et al. mezi prvním a třetím měsícem věku, (5) Rink et al. mezi šestým a dvanáctým měsícem věku, (9) naše pracoviště kolem půl roku věku (Obr. 3).
Fig. 3. An algorithm of examinations and surgeries at the Department of Paediatric Surgery of the Motol
University Hospital
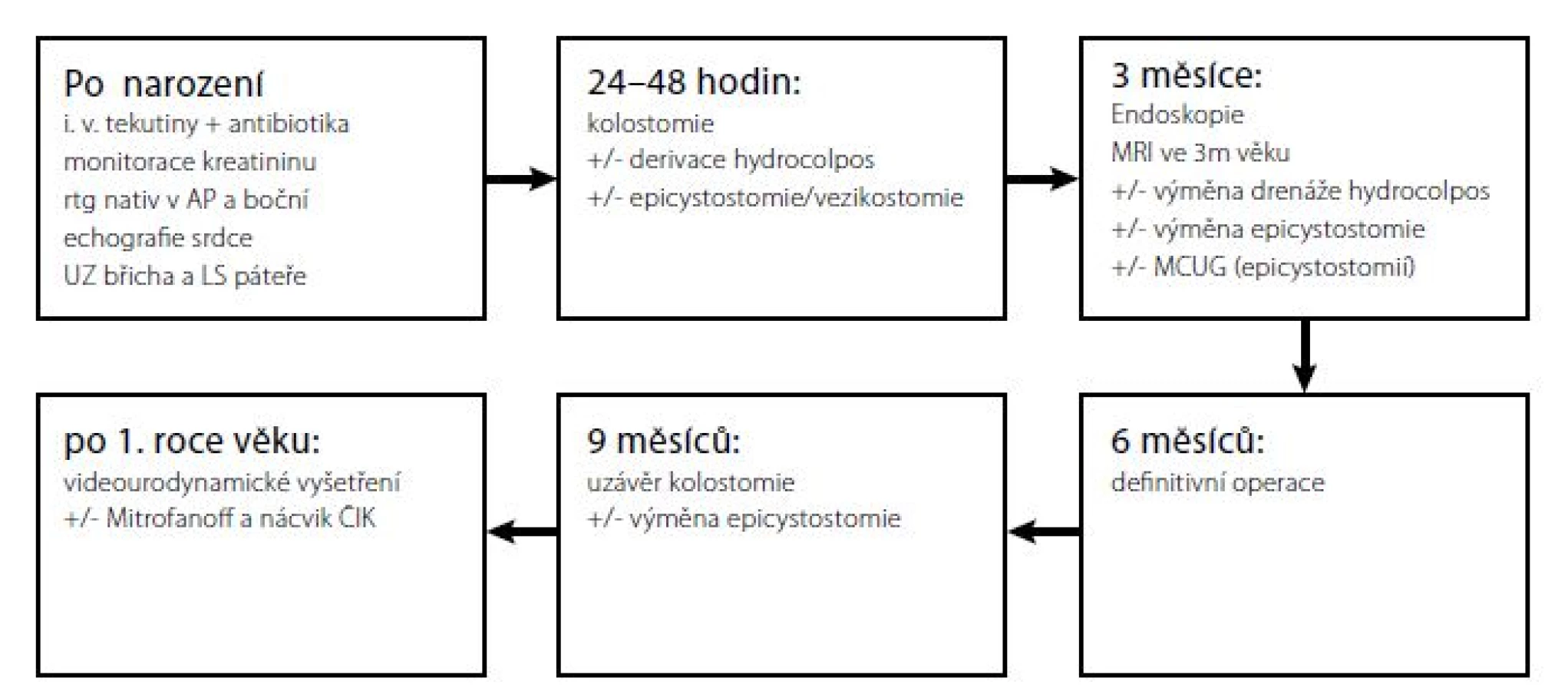
Posun v načasování operace přináší v poslední době publikované a hojně diskutované anesteziologické práce, zbývající se negativním vlivem opakovaných, dlouhých a hlubokých celkových anestezií na tvorbu neuronálních sítí v mozku dítěte. Tvorbu neuronálních sítí podle zatím ne zcela ověřených dat celková anestezie narušuje a to zejména první měsíce po narození, takže děti mají následně horší kognitivní schopnosti (13, 14). Proto respektujeme tendenci odložit všechny větší plánované operace u kojenců po šestém měsíci věku.
Endoskopické vyšetření je u dívek s KM zásadní ve volbě a plánování správného definitivního výkonu. Levitt a Peňa, kteří položili základ moderní diagnostiky a terapie ARM, včetně KM, navrhli na základě svých zkušeností s 490 pacientkami rozdělit KM na tzv. „nízké“ s kloakálním kanálem kratším než 3 cm, které lze operovat pouze z perineálního přístupu a mají lepší funkční prognózu a na tzv. „vysoké“ s kloakálním kanálem delším než 3 cm, které vyžadují kombinovaný přístup z perinea a dutiny břišní a jejich funkční prognóza je horší. Nejzávažnější typ KM je s kloakálním kanálem delším než 5 cm a zdvojenou pochvou ústící do hrdla nebo trigona měchýře. Délka kloakálního kanálu se určí změřením části cystoskopu, která sahá od vnějšího ústí kanálu ke spojení uretry a pochvy. Vyústění rekta do kloakálního kanálu rektální píštělí může být obtížné identifikovat – i proto je vhodné doplnění genitogramu nebo MRI.
KM byla ještě v 50. letech považována za vadu inoperabilní a neslučitelnou se životem (2). Řešení KM se systematicky začali věnovat Raffensperger a Hendren (15, 16). V roce 1982 Peňa publikoval poprvé svou inovativní operaci anorektálních malformací, kterou nazval PSARP – Posterior Sagittal Ano-Recto Plasty (17). V případech KM jí rozšířil na PSARUVP – Posterior Sagital Ano-Recto-Urethro-Vaginal Plasty (18). Princip spočívá v rekonstrukci rekta, vaginy i uretry tzv. zadním sagitálním přístupem, aniž by narušil jejich inervaci a cévní zásobení z laterálních stran, čímž lze dosáhnout dobrého funkčního výsledku.
Pacientka je umístěna na břicho s podloženou pánví a dolními končetinami v mírné abdukci. Chirurg na začátku výkonu lokalizuje transkutánně elektrostimulační sondou podkožně uloženou svalovinu análního svěrače, která se stimulací viditelně kontrahuje a označí si polohu svěrače markerem (značkovací tužkou). Řez probíhá v sagitální rovině z místa kostrče až po ústí kloakálního kanálu. Po protnutí kůže, subkutánního tuku a svěračového komplexu anu, který se embryonálně zakládá bez ohledu na přítomnost zadního střeva, otevře chirurg rektální pahýl, ústící v podobě píštěle do kloakálního kanálu. Píštěl resekuje a rektální pahýl uvolní tak, aby ho bylo možno stáhnout na hráz.
V případě krátkého kloakálního kanálu do 3 cm následuje „en bloc“ uvolnění a stažení pochvy (respektive pochev, je-li pochva zdvojená nebo septovaná) a uretry na hráz a rekonstrukce perinea. Původně Peňa od sebe odpreparovával pochvu a uretru a stáhnul je na hráz odděleně, což bylo technicky extrémně náročné a zatížené častými komplikacemi – jizvením pochvy a uretry v důsledku narušeného cévního zásobení nebo uretrovaginálními píštělemi (18). Tímto zjednodušením původní operace v roce 1997 dosáhl Peňa výborných kosme‑tických a funkčních výsledků. Z dnešního pohledu, tak jako všechny geniální myšlenky, jednoduchý a logický krok – jenom ho musel někdo poprvé udělat a popsat. Později se navíc ukázalo, že u většiny nízkých forem KM dokonce stačí pouze parciální mobilizace urogenitální sinu z dorzální strany a pochvu a uretru tak lze stáhnou na hráz rovněž. Kloakální kanál se v případě jak totální, tak parciální mobilizace podélně natne a položí jako chybějící vestibulum pochvy do rány ve střední čáře, takže okolí ústí uretry a pochvy je kryto růžovou „sliznicí“.
Mnohem složitější a technicky náročnější je operace vysokého typu KM, kdy se musí chirurg po částečném uvolnění rektálního pahýlu a urogenitálního sinu přesunout dolní střední laparotomií do malé pánve a uvolnit výše jmenované orgány i z kraniální strany. Tuto abdominální fázi operace lze dnes i u kojence provést laparoskopicky nebo roboticky. V extrémních případech navrhuje Levitt odsekání kaudální poloviny symfýzy a zkrácení cesty ke stažení uretry a pochvy na hráz dorzálně od klitorisu (5, 20).
Na rozdíl od operace nízkého typu KM, která trvá řádově tři až čtyři hodiny, může operace vysokého typu KM s kombinovaným perineálním a abdominálním přístupem trvat i 10 až 12 hodin, což je pro kojence obrovská zátěž. Operatér pak stojí před rozhodnutím, zda celou operaci nezkrátit rozdělením na dvě fáze – stažení rekta a s odstupem několika měsíců až let i stažením pochvy (pochev) a uretry. Navíc mohou být pochvy i rudimentární nebo natolik vzdálené od perinea, že vyžadují částečnou nebo úplnou náhradu střevem, a to je další riskantní a časově náročný krok v algoritmu celé operace.
Nicméně i tak se Rink proti rozdělení operace na více fází ostře vyhrazuje. V nejnovějším vydání Cambellovy urologie píše: „Jakkoliv je chirurg v pokušení omezit se na pouhé stažení rekta na hráz („pull-through procedure“), je dnes jasné, že taková vícedobá operace kloaky dítě v konečných důsledcích poškozuje, a proto je absolutně kontraindikována. Optimální je odoperovat všechny anomálie (rekta, pochvy a uretry) v jedné době, což umožňuje nejlepší přístup k oddělení rekta od pochvy, který je v případě reoperace kompromitován jizvením v okolí již jednou mobilizovaného rekta“ (9). S tímto radikální přístupem je třeba polemizovat v případech KM s hypoplastickou vaginou ústící do močového ústrojí vysoko v oblasti hrdla močového měchýře. Zde je naopak s výhodou ponechat spojení pochvy a močového ústrojí intaktní, což není v rozporu s Rinkovými závěry, že chirurgické cíle u dítěte s KM jsou v průběhu prvního roku života následující: 1. derivace gastrointestinálního traktu kolostomií; 2. derivace urogenitálního traktu; 3. korekce potenciálně letálních urologických anomálií, poškozující funkci měchýře a ledvin; 4. definitivní rekonstrukce KM (9).
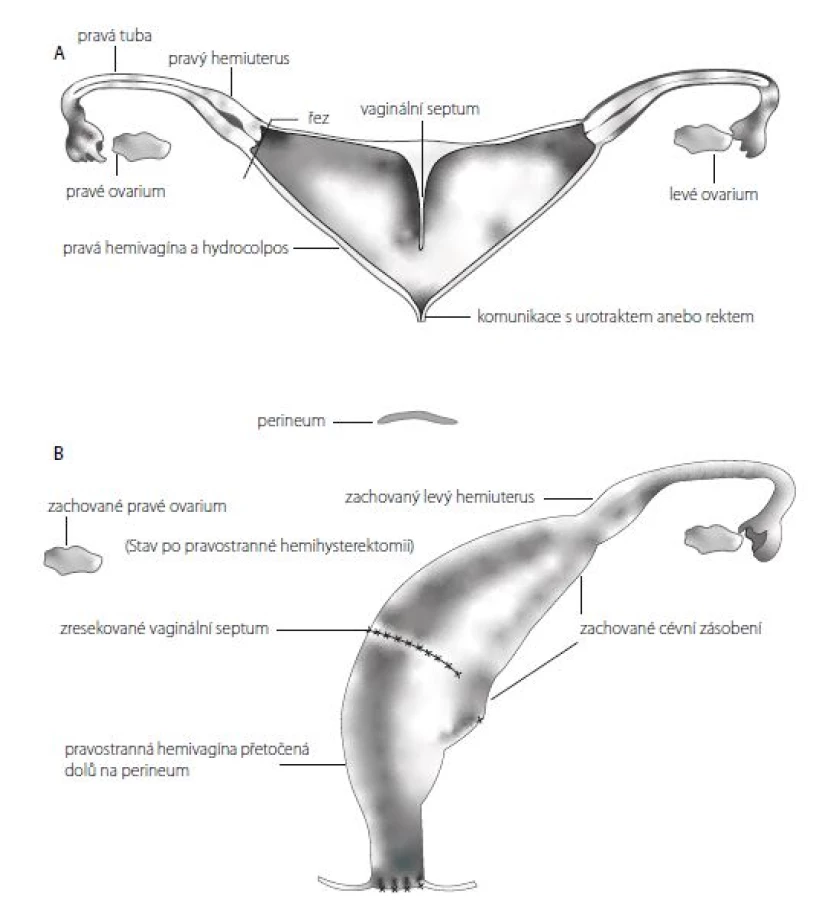
U chirurgicky problematických KM s agenezí pochvy nebo s pochvami ústícími do hrdla či trigona měchýře a velmi dlouhým kloakálním kanálem nad pět centimetrů popisuje Levitt dvě možností jak zajistit pacientce do budoucna možnost menstruace a pohlavního styku – tzv. „vaginal switch“ nebo náhradu pochvy tlustým, eventuálně tenkým střevem (5).
„Vaginal switch“ čili přetočení septované pochvy je možné v případě, že se částečně septovaná pochva otevírá do hrdla nebo trigona měchýře, je dostatečně dilatovaná a transverzální vzdálenost mezi dvěma nasedajícími hemi-dělohami je delší, než kraniokaudální délka pochvy. Jinými slovy přetočení pochvy a její stažení na hráz je možné jen u částečně zdvojené a hodně dilatované pochvy, která je „širší“ než „delší“. Jedna z děloh je excidována ze stěny septované pochvy a spolu s tubou odstraněna. Ovarium je ponecháno in situ. Otvor ve stěně pochvy po excizi dělohy je přetočen a stažen kaudálně na hráz jako poševní vchod. Cévní zásobení přetáčené a stahované pochvy je preparací zničeno a stažená část pochvy je tak živena pouze z části pochvy ponechané in situ (5) (Obr. 4A a 4B).
Fig. 4 A, B. Switching of a septate vagina and bringing it to the perineum, aka vaginal switch manoeuvre
(Adapted from Levitt MA, Peña A. Cloacal malformations: lessons learned from 490 cases. Semin Pediatr Surg.
2010; 19(2): 128–138)
Po odpojení septované pochvy z hrdla nebo trigona měchýře je nutné buď jeho trvalé uzavření, nebo lze ponechat dlouhý kloakální kanál jako přístupovou cestu pro ČIK s tím, že je nutná kontinentní plastika hrdla měchýře např. dle Kroppa nebo Pippi-Salleho (19). Uzávěr hrdla provádíme ve dvou vrstvách a pro riziko vzniku píštěle i překrýváme suturu hrdla měchýře omentem nebo fascií z přímého břišního svalu. Pacientka je ponechána na vezikostomii a v druhé době pak podstoupí vytvoření katetrizovatelného stomatu pro ČIK podle Mitrofanoffa, Montiho nebo Casaleho (21).
Pooperační derivace měchýře je jiná u nízkých typů KM po parciální nebo totální urogenitální mobilizaci. Tam, kde pacientka spontánně močila před operací, stačí v pooperačním období obvykle Foleyův katétr do mobilizované uretry až do zhojení hráze, tzn. na jeden až dva týdny. Jestliže měla pacientka již před operací epicystostomii, pak ji v celkové ane stezii na začátku výkonu vyměníme a po operaci ji ponecháváme nadále in situ vzhledem k výhodné možnosti pravidelného uzavírání a nácviku mikce. Přes epicystostomii lze měřit i postmikční rezidua a orientační intravezikální tlak v cm vodního sloupce po její vertikalizaci.
Při agenezi pochvy je u vysoké KM nutná její náhrada tlustým nebo tenkým střevem. Náhradu lze provést z dilatovaného rekta, ze sigmatu anebo colon descendens, jejichž úsek se v délce pochvy vytne a na cévní stopce stáhne na hráz. Kraniálně se slepě uzavře anebo napojí na rudimentární pochvu, jestliže ji pacientka má. Náhrada z tenkého střeva je možná v oblasti cca 15 cm orálně od Bauhinské chlopně, kde je ileum na nejdelším závěsu, ale i tak může být velmi obtížné úsek ilea stáhnout až na hráz bez narušení cévního zásobení. Nevýhodou náhrad jsou časté komplikace, dehiscence, jizvení a sekrece hlenu, která pacientku obtěžuje.
DLOUHODOBÉ VÝSLEDKY
Jestli pacientka s KM nakonec bude kontinentní v moči záleží na délce kloakálního kanálu (čím delší, tím větší riziko inkontinence), koaptaci hrdla měchýře, na přidružených vadách páteře a míchy (a tedy neurogenního měchýře) a iatrogenní denervaci a jizvení malé pánve, zejména při definitivním výkonu u vysokého typu KM Kontinence moči, spontánní nebo pomocí ČIK, je dosaženo v 54 až 95 % pacientek operovaných pro KM (2, 8, 22, 23) Levitt udává ve svém souboru kontinenci moči u 74 % z 225 pacientek s kloakálním kanálem kratším než 3 cm a u 28 % pacientek ze 175 s kloakálním kanálem delším než 3 cm (5).
V jediné publikované prospektivní studii vyšetřila Warne et al deset dívek s KM urodynamicky před a po definitivní rekonstrukční operaci Kontrolní skupinou bylo 20 kojenců s méně závažnými typy ARM Děvet z deseti pacientek s KM (90 %) mělo abnormální urodynamický nález už před operací, dominovala hyperaktivita detruzoru a netlumené kontrakce během plnící fáze. U pěti deseti (50 %) pak došlo ke zhoršení funkce měchýře po operaci. V kontrolní skupině ARM došlo ke zhoršení jen u jednoho z 20 pacientů po PSARP (5 %) (24).
V této studii měly všechny pacientky s KM, u nichž došlo ke zhoršení funkce měchýře po rekonstrukční operaci, původně dlouhý kloakální kanál nad 3 cm a u všech došlo ke změně z hyperaktivity detruzoru na atonický měchýř bez spontánní kontraktility. Tato změna podporuje dřívější pozorování Levitta a Peni, kteří popisují u většiny svých pacientek s KM po definitivní operaci atonický, nízkotlaký a vysoko objemový měchýř, který působí inkontinenci z přetékání a nikoliv v důsledku hyperaktivity detruzoru nebo insuficientního hrdla a svěrače (5). Jinými slovy většina pacientek s KM, které ztratily schopnost spontánní mikce, jsou ideálními kandidátkami na ČIK bez nutnosti dlouhodobého užívání anticholinergik. Ty pacientky, které mají přetrvávající nízkokapacitní měchýř s přetrvávající hyperaktivitou nereagující na anticholinergika, jsou v kombinaci s katetrizovatel‑ným stomatem indikovány k augmentaci měchýře tenkým nebo tlustým střevem.
Vzhledem k tomu, že se výskyt sakrálních malformací u pacientek s KM pohybuje kolem 50 % a Rink udává záchyt míšních anomálií u KM na MRI v 43 %, z čehož celá třetina pacientek měla fixovanou míchu (25), znamená to, že řada pacientek s KM má vrozenou neurogenní dysfunkci měchýře a stačí mírné poranění nervového zásobení měchýře a uretry, aby došlo k denervaci měchýře a jeho atonii – zejména při operaci vysokého typu KM (8).
Jiná podstatná věc je dlouhodobá funkce ledvin. Všechny pacientky s KM by měly být sledované nejen urologem, ale i nefrologem, a přestože tomu tak v dnešní době skoro vždy je, dospěje polovina z nich dříve nebo později k chronickému renálnímu selhání (8). Selhání je dané nejčastěji jednak vrozenou dysplazií nebo solitární ledvinou, jednak dysfunkcí dolních močových cest v kombinaci se sekundárním refluxem a infekcemi močových cest. Proto doporučují autoři se zkušeností s KM agresivní přístup v léčbě dysfunkcí dolních močových cest a častějším zaváděním ČIK (5, 8, 10).
Kontinence stolice dosáhne asi 60 % pacientek s KM. Ovšem pouze 28 % se vyprazdňuje spontánně a 30 až 40 % potřebuje laxativa a pravidelná klyzmata, aby dosáhly sociální kontinence (2, 12, 20). Levitt uvádí příznivější čísla u svých pacientek – volní defekaci u 66 % pacientek s nízkým typem KM a 36 % s vysokým typem KM (5).
Řada gynekologických problémů se manifestuje až s nástupem puberty. Warne et al. ve své studii 41 dospělých pacientek operovaných v dětství pro KM udávají nástup puberty s menses u dvou třetin pacientek a u 20 % amenorrhoeu pro hypoplastickou rudimentární dělohu. 32 % pacientek mělo menstruaci pravidelnou a 36 % (15 pacientek) mělo hematometru nebo hematocolpos a cyklickou bolest v podbřišku během puberty (26).
Nejčastější příčinou obstrukce odtoku menstruální krve byla stenóza perzistujícího urogenitálního sinu a u všech těchto pacientek bylo nutné chirurgické řešení. 57 % žen bylo pohlavně aktivních a měly poševní styk, přičemž polovina z nich potřebovala v pubertě nebo po ní chirurgickou úpravu poševního vchodu pro stenózu (26). Kvalitu sexuálního života a senzitivitu genitálu u dospělých pacientek s KM zatím nikdo nepublikoval, otěhotnění a porod zdravých dětí vaginální cestou nebo císařským řezem je publikován v jednotlivých případech (4).
ZÁVĚR
Přestože většina pacientek narozených s KM dosáhne kontinence v moči a stolici, polovina z nich trpí chronickým ledvinným selháním, často v důsledku komplexních rekonstrukčních operací a přetrvávající neurogenní i non-neurogenní dysfunkce dolních močových cest. Je zřejmé, že postupem léčby KM vzniká skupina pacientek, které by měly být trvale sledovány nejen dětským chirurgem a urologem, ale i dospělým gynekologem a urologem, kteří se věnují dlouhodobé péči o dospělé pacientky s neurogenním měchýřem a po rekonstrukčních operacích i tak komplexních vrozených vad, jako je kloakální malformace.
Došlo: 10. 10. 2018
Přijato: 25. 1. 2019
Kontaktní adresa:
MUDr. Jan Trachta
Klinika dětské chirurgie 2. LF UK a FN Motol
V Úvalu 84, 150 06 Praha 5
e‑mail: Jan.Trachta@fnmotol.cz
Střet zájmů: Žádný.
Prohlášení o podpoře: Tento článek nebyl nikým finančně podpořen.
Sources
- Šnajdauf J, Škába R. Dětská chirurgie. 1. vydání. Praha Galén 2005; 11–18.
- Warne SA, Wilcox DT, Ransley PG. Long‑term urological outcome of patients presenting with persistent cloaca. J Urol 2002; 168 : 1859–1862.
- Rink RC, Herndon CD, Cain MP, et al. Upper and lower urinary tract outcome after surgical repair of cloacal malformations: a three‑decade experience. BJU Int. 2005; 96(1): 131–134.
- Fernando MA, Creighton SM, Wood D. The long‑term management and outcomes of cloacal anomalies. Pediatr Nephrol. 2015; 30(5): 759–765.
- Levitt MA, Peña A. Cloacal malformations: lessons learned from 490 cases. Semin Pediatr Surg. 2010; 19(2): 128–138.
- Qi BQ, Williams A, Beasley S, et al. Clarification of the process of separation of the cloaca into rectum and urogenital sinus in the rat embryo. J Pediatr Surg 2000; 35 : 1810–1816.
- Cilento BG, Jr, Benacerraf BR, Mandell J. Prenatal diagnosis of cloacal malformation. Urology 1994; 43 : 386–388.
- Warne SA, Hiorns MP, Curry J, Mushtaq I. Understanding cloacal anomalies. Arch Dis Child. 2011; 96(11): 1072–1076.
- Rink R. Surgical management of disorders od sex development and cloaca and anorectal malformations.CambellWalsh ‑ Urology, 11th ed., Philadelphia, PA: Elsevier/Saunders 2016; 3498–3520.
- VanderBrink BA, Reddy PP. Early urologic considerations in patients with persistent cloaca. Semin Pediatr Surg. 2016; 25(2): 82–89.
- Peña A, Levitt MA. Imperforate anus and cloacal malformations. In: Ashcraft KW, Holcomb GW, Murphy JP, eds. Pediatric Surgery. 4th ed. Philadelphia, PA: Elsevier Saunders 2005 : 496–517.
- Levitt MA, Peña A. Pitfalls in the management of newborn cloacas. Pediatr Surg Int 2005; 21 : 264–269.
- Lee JH, Zhang J, Wei L, Yu SP. Neurodevelopmental implications of the general anesthesia in neonate and infants. Exp Neurol. 2015; 272 : 50–60.
- Armstrong R, Xu F, Arora A, Rasic N, Syed NI. General anesthetics and cytotoxicity: possible implica‑tions for brain health. Drug Chem Toxicol. 2017; 40(2): 241–249.
- Raffensperger JG, Ramenofsky ML. The management of cloaca. J Pediatr Surg. 1973; 8 : 647–657.
- Hendren WH. Further experience in reconstructive surgery for cloacal anomalies. J Pediatr Surg 1982; 17 : 695–717.
- Peña A, Devries PA. Posterior sagittal anorectoplasty: important technical considerations and new applications. J Pediatr Surg. 1982; 17(6): 796–811.
- Peña A. The surgical management of persistent cloaca: results in 54 patients treated with a posterior sagittal approach. J Pediatr Surg 1989; 24 : 590–598.
- Peña A. Total urogenital mobilization-an easier way to repair cloacas. J Pediatr Surg. 1997; 32(2): 263–267.
- Peña A, Levitt MA, Hong A, Midulla P. Surgical management of cloacal malformations: a review of
- patients. J Pediatr Surg. 2004; 39(3): 470–479.
- Elder JS, Pippi‑Salle JL. Bladder outlet surgery for congenital incontinence. In: Gearhart, Rink, Mou‑riquand: Pediatric Urology, 2nd ed., Elsevier 2005; 761–773.
- Hendren WH. Cloaca, the most severe degree of imperforate anus: experience with 195 cases. Ann Surg 1998; 228 : 331–346.
- Peña A. Anorectal malformations. Semin Pediatr Surg 1995; 4 : 35–47.
- WarneSA, Godley ML, Wilcox DT. Surgical reconstruction of cloacal malformation can alter bladder function: a comparative study with anorectal anomalies. J Urol 2004; 172 : 2377–2381.
- Yerkes YB, Rink R. Surgical management of female genital anomalies, disorders of sex development, urogenital sinus, and cloaca anomalies. In: Gearhart, Rink, Mouriquand: Pediatric Urology, 2nd ed., Elsevier 2005; 476–499.
- Warne SA, Wilcox DT, Creighton S, et al. Long‑term gynecological outcome of patients with persistent cloaca. J Urol 2003; 170 : 1493–1496.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2019 Issue 1
Most read in this issue
- Endometrióza močového měchýře jako příčina iatrogenní perforace močového měchýře
- Hematurie u dětí pohledem urologa
- Diagnostika, léčba a urologické komplikace kloakálních malformací
- Sebepoškozování urogenitálního traktu – je řešení pouze na urologovi?